
PFKFB3 Inhibition Sensitizes DNA Crosslinking Chemotherapies by Suppressing Fanconi Anemia Repair

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Tumor Samples from High-Grade Ovarian Cancer Patients
2.3. Plasmid and siRNA Transfections
2.4. Establishment of shRNA Cell Lines
2.5. Quantitative Real-Time PCR (qRT-PCR) Analysis
2.6. Small-Molecule Inhibitors and Cytostatic Compounds
2.7. Cell Growth Kinetics
2.8. End-Point Cell Viability Assays
2.9. Clonogenic Assays
2.10. Extracellular Flux Analysis
2.11. Flow Cytometry for Cell Cycle Progression of Replicating Cells
2.12. Immunoblotting and Whole Cell Protein Extraction
2.13. Cell Fractionation of Whole Cell Soluble and Chromatin Bound Proteins
2.14. Immunoprecipitations
2.15. Fiber Assay
2.16. Immunofluorescence Microscopy
2.17. Statistics
3. Results
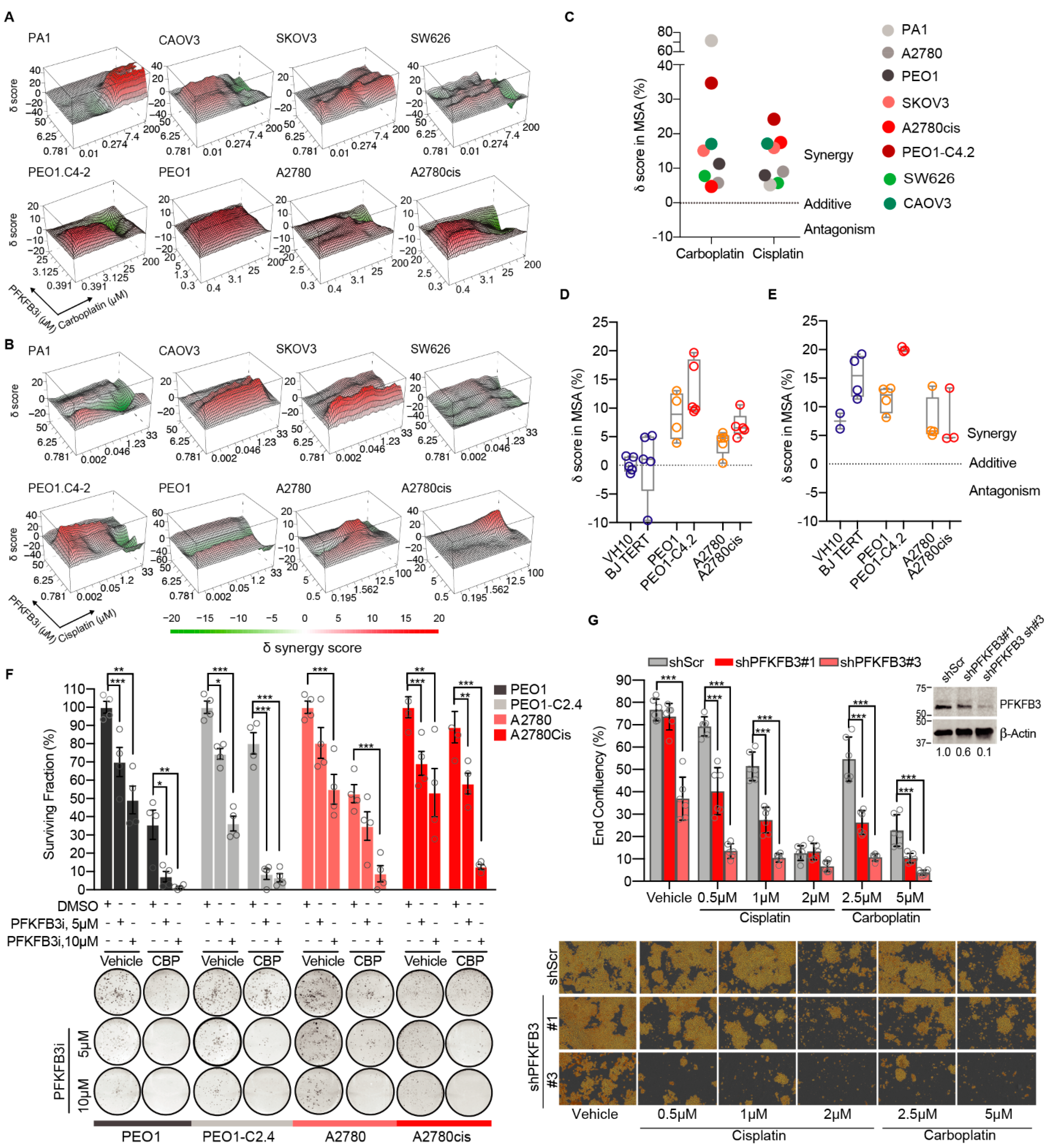
3.1. Targeting PFKFB3 Enzymatic Activity Results in a Non-Reversible Cancer-Specific Synergy with Platinum Drugs
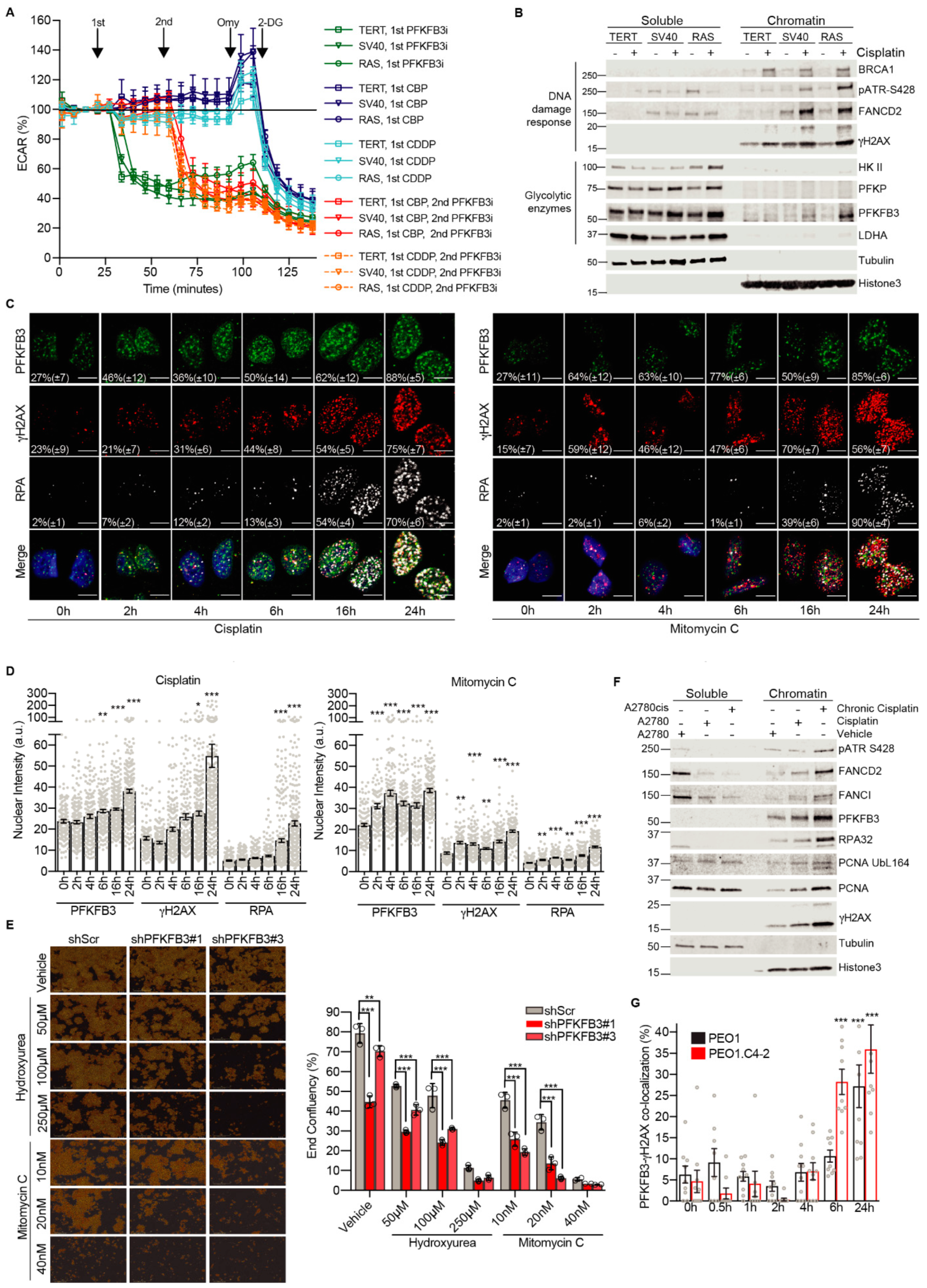
3.2. Platinum Drugs Do Not Induce a Preference for Glycolysis
3.3. Cancer-Specific PFKFB3 Chromatin Localization in Response to FA Pathway Activation Modulates Platinum-Tolerance of Resistant Cells
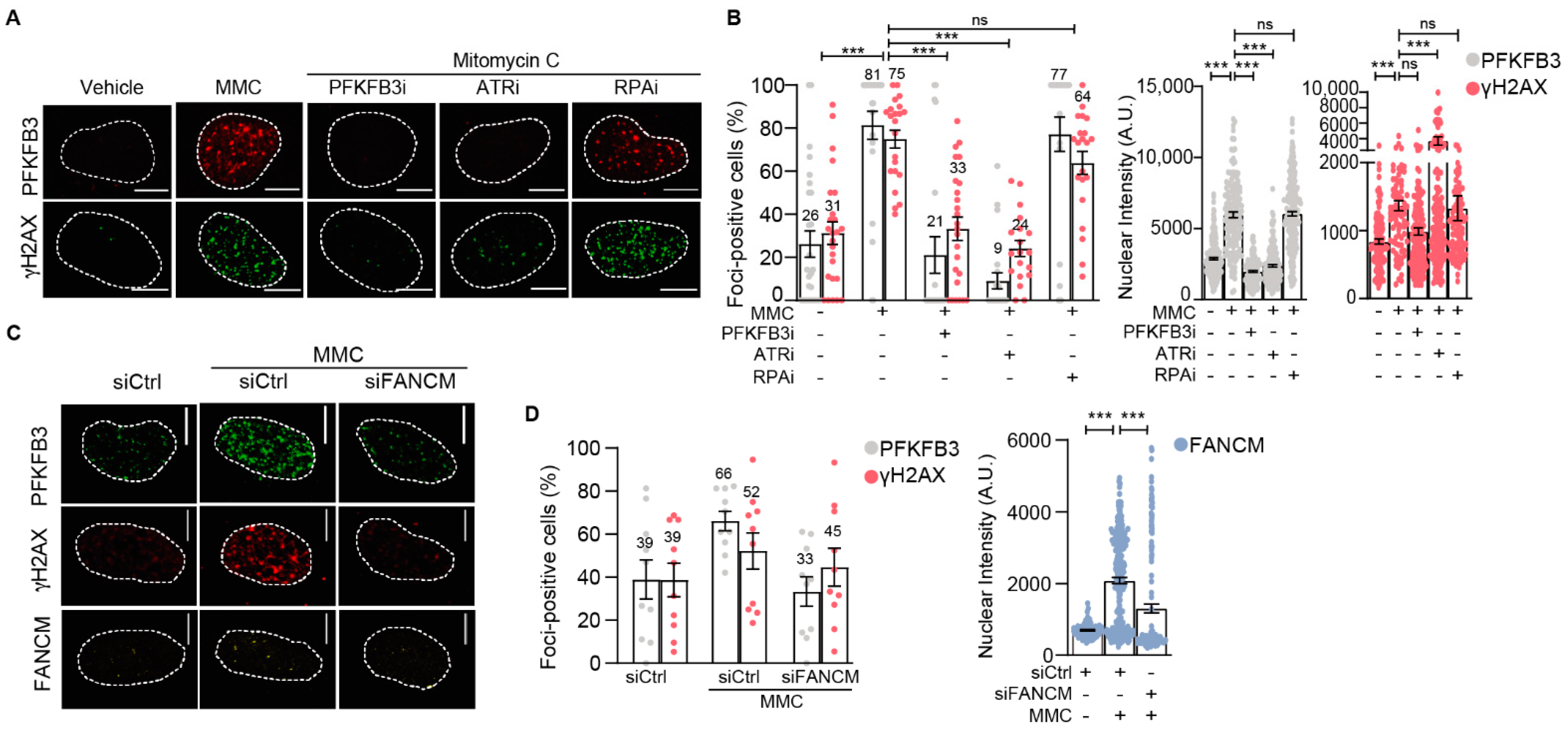
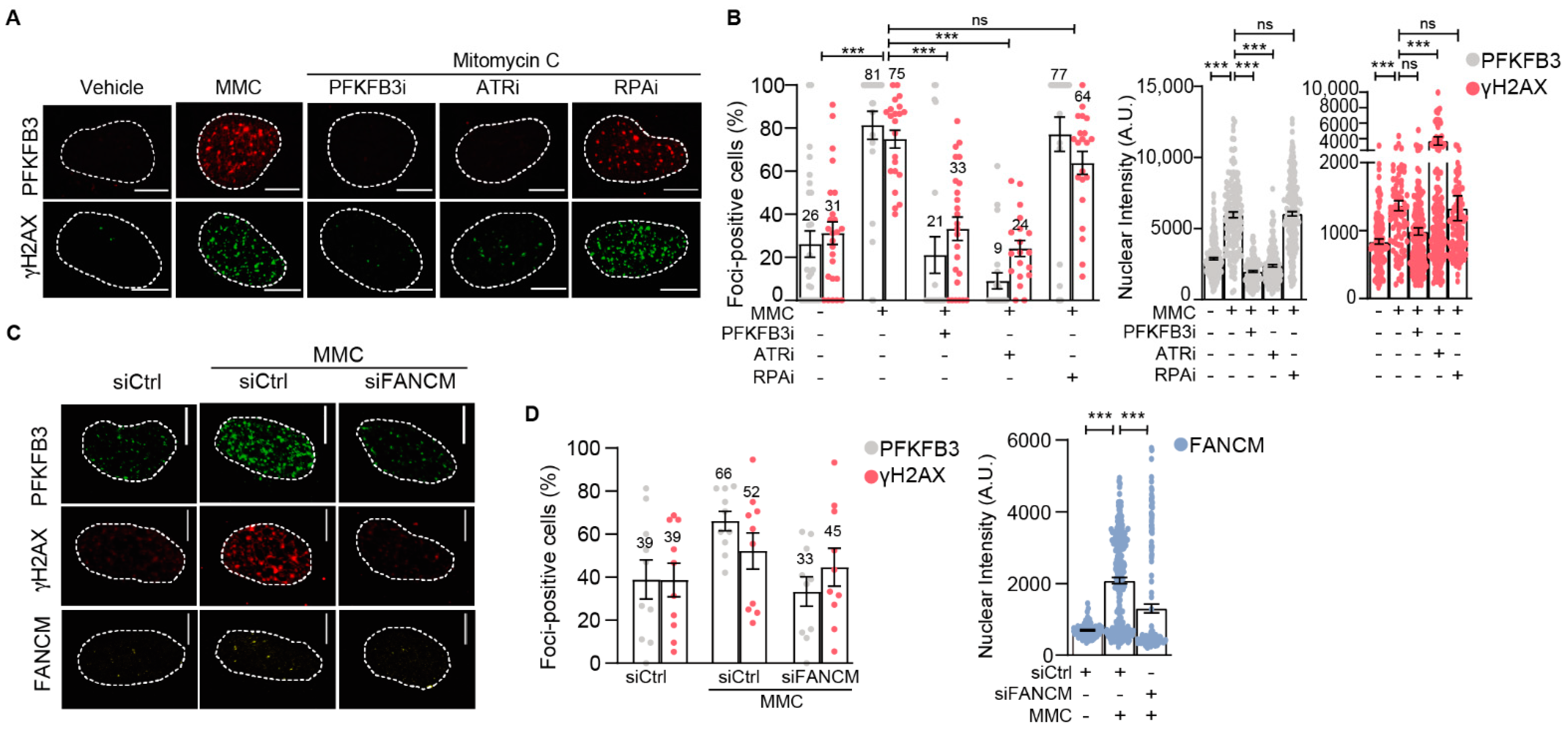
3.4. PFKFB3 Is Recruited into Foci upon Initiation of Replicative S-Phase FA Repair by FANCM and ATR
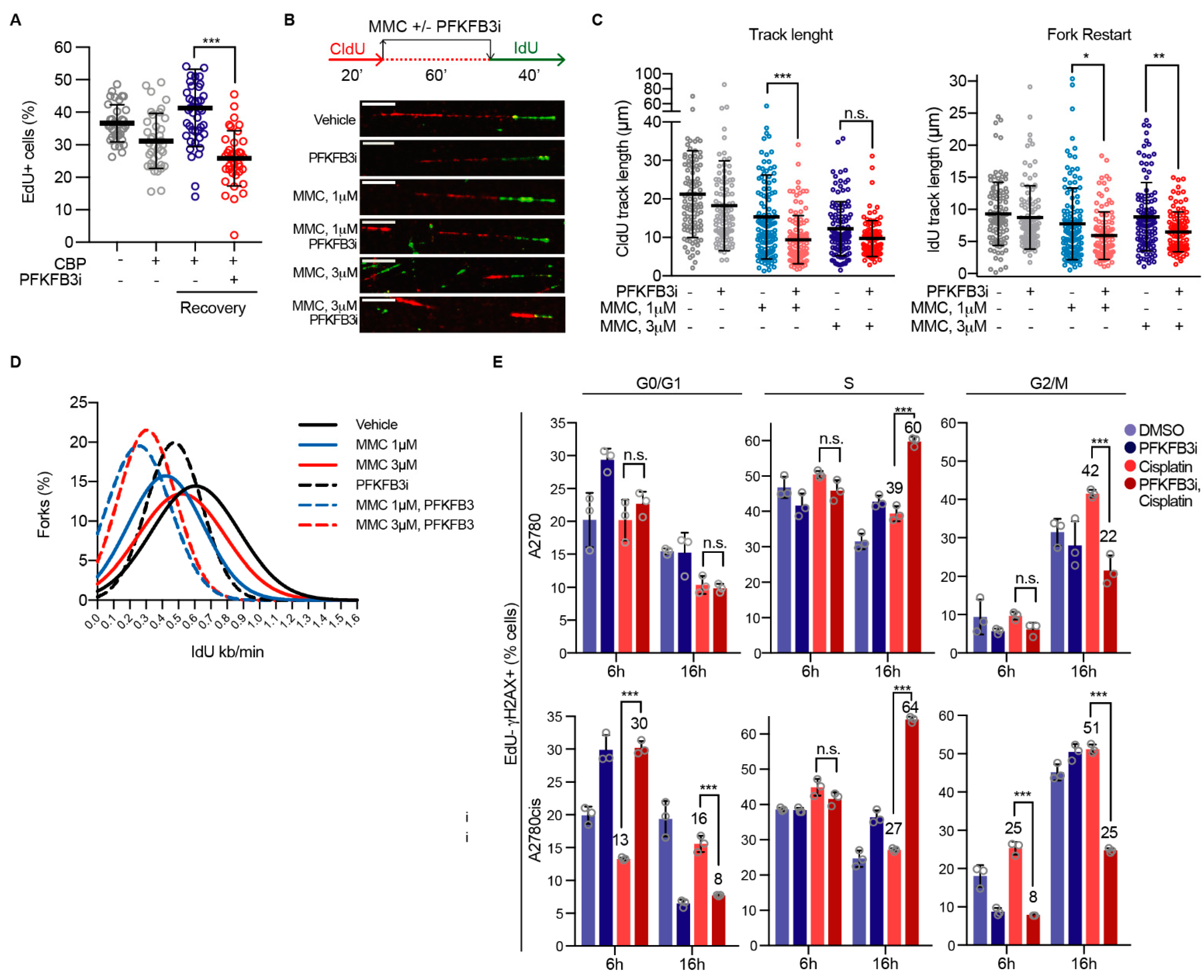
3.5. PFKFB3 Kinase Activity Promotes Assembly of FA Factors into Foci at the Chromatin
3.6. PFKFB3 Inhibition Impairs Replication Fork Restart upon ICL-Induction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Niraj, J.; Farkkila, A.; D’Andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, A.D. Fanconi anemia diagnosis and the diepoxybutane (DEB) test. Exp. Hematol. 1993, 21, 731–733. [Google Scholar] [PubMed]
- Gari, K.; Decaillet, C.; Stasiak, A.Z.; Stasiak, A.; Constantinou, A. The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol. Cell 2008, 29, 141–148. [Google Scholar] [CrossRef]
- Ciccia, A.; Ling, C.; Coulthard, R.; Yan, Z.; Xue, Y.; Meetei, A.R.; Laghmani el, H.; Joenje, H.; McDonald, N.; de Winter, J.P.; et al. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol. Cell 2007, 25, 331–343. [Google Scholar] [CrossRef]
- Ling, C.; Huang, J.; Yan, Z.; Li, Y.; Ohzeki, M.; Ishiai, M.; Xu, D.; Takata, M.; Seidman, M.; Wang, W. Bloom syndrome complex promotes FANCM recruitment to stalled replication forks and facilitates both repair and traverse of DNA interstrand crosslinks. Cell Discov. 2016, 2, 16047. [Google Scholar] [CrossRef]
- Deans, A.J.; West, S.C. FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia. Mol. Cell 2009, 36, 943–953. [Google Scholar] [CrossRef]
- Meetei, A.R.; Sechi, S.; Wallisch, M.; Yang, D.; Young, M.K.; Joenje, H.; Hoatlin, M.E.; Wang, W. A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol. Cell Biol. 2003, 23, 3417–3426. [Google Scholar] [CrossRef] [Green Version]
- Singh, T.R.; Ali, A.M.; Paramasivam, M.; Pradhan, A.; Wahengbam, K.; Seidman, M.M.; Meetei, A.R. ATR-dependent phosphorylation of FANCM at serine 1045 is essential for FANCM functions. Cancer Res. 2013, 73, 4300–4310. [Google Scholar] [CrossRef] [Green Version]
- Collis, S.J.; Ciccia, A.; Deans, A.J.; Horejsi, Z.; Martin, J.S.; Maslen, S.L.; Skehel, J.M.; Elledge, S.J.; West, S.C.; Boulton, S.J. FANCM and FAAP24 function in ATR-mediated checkpoint signaling independently of the Fanconi anemia core complex. Mol. Cell 2008, 32, 313–324. [Google Scholar] [CrossRef]
- Schwab, R.A.; Blackford, A.N.; Niedzwiedz, W. ATR activation and replication fork restart are defective in FANCM-deficient cells. EMBO J. 2010, 29, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Kim, J.M.; Shiotani, B.; Yang, K.; Zou, L.; D’Andrea, A.D. The FANCM/FAAP24 complex is required for the DNA interstrand crosslink-induced checkpoint response. Mol. Cell 2010, 39, 259–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.C.; Li, Z.; Lopez-Martinez, D.; Nicholson, W.V.; Venien-Bryan, C.; Cohn, M.A. The FANCD2-FANCI complex is recruited to DNA interstrand crosslinks before monoubiquitination of FANCD2. Nat. Commun. 2016, 7, 12124. [Google Scholar] [CrossRef]
- Garcia-Higuera, I.; Taniguchi, T.; Ganesan, S.; Meyn, M.S.; Timmers, C.; Hejna, J.; Grompe, M.; D’Andrea, A.D. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell 2001, 7, 249–262. [Google Scholar] [CrossRef]
- Sims, A.E.; Spiteri, E.; Sims, R.J., III; Arita, A.G.; Lach, F.P.; Landers, T.; Wurm, M.; Freund, M.; Neveling, K.; Hanenberg, H.; et al. FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat. Struct. Mol. Biol. 2007, 14, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; van Twest, S.; Leis, A.; Bythell-Douglas, R.; Murphy, V.J.; Sharp, M.; Parker, M.W.; Crismani, W.; Deans, A.J. Monoubiquitination by the human Fanconi anemia core complex clamps FANCI:FANCD2 on DNA in filamentous arrays. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Smogorzewska, A.; Matsuoka, S.; Vinciguerra, P.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Ballif, B.A.; Gygi, S.P.; Hofmann, K.; D’Andrea, A.D.; et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 2007, 129, 289–301. [Google Scholar] [CrossRef] [Green Version]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Knipscheer, P.; Raschle, M.; Smogorzewska, A.; Enoiu, M.; Ho, T.V.; Scharer, O.D.; Elledge, S.J.; Walter, J.C. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 2009, 326, 1698–1701. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Pan, H.; Liu, Z.; Xie, J.; Han, W. Roles of PFKFB3 in cancer. Signal Transduct. Target. Ther. 2017, 2, 17044. [Google Scholar] [CrossRef]
- Zhou, Q.-Q.; Chen, S.-S.; Zhang, Q.-Q.; Liu, P.-F.; Fang, H.-Z.; Yang, Y.; Zhang, L.-C. Cerebrospinal fluid-contacting nucleus mediates nociception via release of fractalkine. Braz. J. Med. Biol. Res. 2017, 50, e6275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atsumi, T.; Chesney, J.; Metz, C.; Leng, L.; Donnelly, S.; Makita, Z.; Mitchell, R.; Bucala, R. High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res. 2002, 62, 5881–5887. [Google Scholar] [PubMed]
- Li, F.-L.; Liu, J.-P.; Bao, R.-X.; Yan, G.; Feng, X.; Xu, Y.-P.; Sun, Y.-P.; Yan, W.; Ling, Z.-Q.; Xiong, Y.; et al. Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin-induced apoptosis. Nat. Commun. 2018, 9, 508. [Google Scholar] [CrossRef] [Green Version]
- Franklin, D.A.; He, Y.; Leslie, P.L.; Tikunov, A.P.; Fenger, N.; Macdonald, J.M.; Zhang, Y. p53 coordinates DNA repair with nucleotide synthesis by suppressing PFKFB3 expression and promoting the pentose phosphate pathway. Sci. Rep. 2016, 6, 38067. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, N.M.S.; Farnegardh, K.; Bonagas, N.; Ninou, A.H.; Groth, P.; Wiita, E.; Jonsson, M.; Hallberg, K.; Lehto, J.; Pennisi, R.; et al. Targeting PFKFB3 radiosensitizes cancer cells and suppresses homologous recombination. Nat. Commun. 2018, 9, 3872. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Jin, L.; Deng, C.; Guan, Y.; Kalogera, E.; Ray, U.; Thirusangu, P.; Staub, J.; Sarkar Bhattacharya, S.; Xu, H.; et al. Inhibition of PFKFB3 induces cell death and synergistically enhances chemosensitivity in endometrial cancer. Oncogene 2021, 40, 1409–1424. [Google Scholar] [CrossRef] [PubMed]
- Hahn, W.C.; Counter, C.M.; Lundberg, A.S.; Beijersbergen, R.L.; Brooks, M.W.; Weinberg, R.A. Creation of human tumour cells with defined genetic elements. Nature 1999, 400, 464–468. [Google Scholar] [CrossRef]
- Lu, R.; O’Rourke, J.J.; Sobinoff, A.P.; Allen, J.A.M.; Nelson, C.B.; Tomlinson, C.G.; Lee, M.; Reddel, R.R.; Deans, A.J.; Pickett, H.A. The FANCM-BLM-TOP3A-RMI complex suppresses alternative lengthening of telomeres (ALT). Nat. Commun. 2019, 10, 2252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yalcin, A.; Clem, B.F.; Simmons, A.; Lane, A.; Nelson, K.; Clem, A.L.; Brock, E.; Siow, D.; Wattenberg, B.; Telang, S.; et al. Nuclear targeting of 6-phosphofructo-2-kinase (PFKFB3) increases proliferation via cyclin-dependent kinases. J. Biol. Chem. 2009, 284, 24223–24232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehto, J.; Huguet Ninou, A.; Chioureas, D.; Jonkers, J.; Gustafsson, N.M.S. Targeting CX3CR1 Suppresses the Fanconi Anemia DNA Repair Pathway and Synergizes with Platinum. Cancers 2021, 13, 1442. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Prevo, R.; Fokas, E.; Reaper, P.M.; Charlton, P.A.; Pollard, J.R.; McKenna, W.G.; Muschel, R.J.; Brunner, T.B. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol. Ther. 2012, 13, 1072–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuck, S.C.; Turchi, J.J. Targeted inhibition of Replication Protein A reveals cytotoxic activity, synergy with chemotherapeutic DNA-damaging agents, and insight into cellular function. Cancer Res. 2010, 70, 3189–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson Sheppard, N.; Heldring, N.; Dahlman-Wright, K. Estrogen receptor-alpha, RBCK1, and protein kinase C beta 1 cooperate to regulate estrogen receptor-alpha gene expression. J. Mol. Endocrinol. 2012, 49, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- McQuin, C.; Goodman, A.; Chernyshev, V.; Kamentsky, L.; Cimini, B.A.; Karhohs, K.W.; Doan, M.; Ding, L.; Rafelski, S.M.; Thirstrup, D.; et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 2018, 16, e2005970. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, T.; Goldhardt, T.; Edelmann, M.; Rogge, T.; Rauch, K.; Kyuchukov, N.D.; Menck, K.; Bleckman, A.; Kalucka, J.; Khan, S.; et al. Effects of the Novel PFKFB3 Inhibitor KAN0438757 on Colorectal Cancer Cells and Its Systemic Toxicity Evaluation In Vivo. Cancers 2021, 13, 1011. [Google Scholar] [CrossRef]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Castella, M.; Jacquemont, C.; Thompson, E.L.; Yeo, J.E.; Cheung, R.S.; Huang, J.W.; Sobeck, A.; Hendrickson, E.A.; Taniguchi, T. FANCI Regulates Recruitment of the FA Core Complex at Sites of DNA Damage Independently of FANCD2. PLoS Genet. 2015, 11, e1005563. [Google Scholar] [CrossRef]
- Chirnomas, D.; Taniguchi, T.; de la Vega, M.; Vaidya, A.P.; Vasserman, M.; Hartman, A.R.; Kennedy, R.; Foster, R.; Mahoney, J.; Seiden, M.V.; et al. Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway. Mol. Cancer Ther. 2006, 5, 952–961. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, T.; Tischkowitz, M.; Ameziane, N.; Hodgson, S.V.; Mathew, C.G.; Joenje, H.; Mok, S.C.; D’Andrea, A.D. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat. Med. 2003, 9, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Li, J.; Chen, Y.-C.; Qian, H.; Chen, Y.-J.; Su, J.-Y.; Wu, M.; Lan, T. The functional status of DNA repair pathways determines the sensitization effect to cisplatin in non-small cell lung cancer cells. Cell. Oncol. 2016, 39, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef]
- Wang, Y.; Leung, J.W.; Jiang, Y.; Lowery, M.G.; Do, H.; Vasquez, K.M.; Chen, J.; Wang, W.; Li, L. FANCM and FAAP24 maintain genome stability via cooperative as well as unique functions. Mol. Cell 2013, 49, 997–1009. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.L.; North, P.S.; Hickson, I.D. Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat. Struct. Mol. Biol. 2007, 14, 677–679. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Martinez, D.; Liang, C.-C.; Cohn, M.A. Cellular response to DNA interstrand crosslinks: The Fanconi anemia pathway. Cell Mol. Life Sci. 2016, 73, 3097–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, W.-P.; Ho, Y.-C.; Wu, C.-K.; Hsu, S.-H.; Shiu, J.-L.; Huang, J.-C.; Chang, S.-B.; Chiu, W.-T.; Hung, J.-J.; Liu, T.-L.; et al. Chronic treatment with cisplatin induces chemoresistance through the TIP60-mediated Fanconi anemia and homologous recombination repair pathways. Sci. Rep. 2017, 7, 3879. [Google Scholar] [CrossRef] [PubMed]
- Ralf, C.; Hickson, I.D.; Wu, L. The Bloom’s syndrome helicase can promote the regression of a model replication fork. J. Biol. Chem. 2006, 281, 22839–22846. [Google Scholar] [CrossRef] [Green Version]
- Lachaud, C.; Moreno, A.; Marchesi, F.; Toth, R.; Blow, J.J.; Rouse, J. Ubiquitinated Fancd2 recruits Fan1 to stalled replication forks to prevent genome instability. Science 2016, 351, 846–849. [Google Scholar] [CrossRef] [Green Version]
- Michl, J.; Zimmer, J.; Buffa, F.M.; McDermott, U.; Tarsounas, M. FANCD2 limits replication stress and genome instability in cells lacking BRCA2. Nat. Struct. Mol. Biol. 2016, 23, 755–757. [Google Scholar] [CrossRef]
- Akkari, Y.M.; Bateman, R.L.; Reifsteck, C.A.; D’Andrea, A.D.; Olson, S.B.; Grompe, M. The 4N cell cycle delay in Fanconi anemia reflects growth arrest in late S phase. Mol. Genet. Metab. 2001, 74, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, P.R.; D’Andrea, A.D.; Taniguchi, T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004, 18, 1958–1963. [Google Scholar] [CrossRef] [Green Version]
- Mutreja, K.; Krietsch, J.; Hess, J.; Ursich, S.; Berti, M.; Roessler, F.K.; Zellweger, R.; Patra, M.; Gasser, G.; Lopes, M. ATR-Mediated Global Fork Slowing and Reversal Assist Fork Traverse and Prevent Chromosomal Breakage at DNA Interstrand Cross-Links. Cell Rep. 2018, 24, 2629–2642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Zhang, J.; Bellani, M.A.; Pokharel, D.; Gichimu, J.; James, R.C.; Gali, H.; Ling, C.; Yan, Z.; Xu, D.; et al. Remodeling of Interstrand Crosslink Proximal Replisomes Is Dependent on ATR, FANCM, and FANCD2. Cell Rep. 2019, 27, 1794–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichierri, P.; Franchitto, A.; Rosselli, F. BLM and the FANC proteins collaborate in a common pathway in response to stalled replication forks. EMBO J. 2004, 23, 3154–3163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damia, G.; Broggini, M. Platinum Resistance in Ovarian Cancer: Role of DNA Repair. Cancers 2019, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Dobbelstein, M.; Sorensen, C.S. Exploiting replicative stress to treat cancer. Nat. Rev. Drug Discov. 2015, 14, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.A.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Shimomuki, M.; Katsuki, Y.; Takahashi, D.; Kobayashi, W.; Ishiai, M.; Miyoshi, H.; Takata, M.; Kurumizaka, H. FANCI-FANCD2 stabilizes the RAD51-DNA complex by binding RAD51 and protects the 5’-DNA end. Nucleic Acids Res. 2016, 44, 10758–10771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaufort, C.M.; Helmijr, J.C.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van IJcken, W.F.; Heine, A.A.; Smid, M.; et al. Ovarian cancer cell line panel (OCCP): Clinical importance of in vitro morphological subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.E. Control of glycaemia. Bailliere’s Clin. Endocrinol. Metab. 1993, 7, 551–586. [Google Scholar] [CrossRef]
- Alabert, C.; Bukowski-Wills, J.C.; Lee, S.B.; Kustatscher, G.; Nakamura, K.; de Lima Alves, F.; Menard, P.; Mejlvang, J.; Rappsilber, J.; Groth, A. Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat. Cell Biol. 2014, 16, 281–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ninou, A.H.; Lehto, J.; Chioureas, D.; Stigsdotter, H.; Schelzig, K.; Åkerlund, E.; Gudoityte, G.; Joneborg, U.; Carlson, J.; Jonkers, J.; et al. PFKFB3 Inhibition Sensitizes DNA Crosslinking Chemotherapies by Suppressing Fanconi Anemia Repair. Cancers 2021, 13, 3604. https://doi.org/10.3390/cancers13143604
Ninou AH, Lehto J, Chioureas D, Stigsdotter H, Schelzig K, Åkerlund E, Gudoityte G, Joneborg U, Carlson J, Jonkers J, et al. PFKFB3 Inhibition Sensitizes DNA Crosslinking Chemotherapies by Suppressing Fanconi Anemia Repair. Cancers. 2021; 13(14):3604. https://doi.org/10.3390/cancers13143604
Chicago/Turabian StyleNinou, Anna Huguet, Jemina Lehto, Dimitrios Chioureas, Hannah Stigsdotter, Korbinian Schelzig, Emma Åkerlund, Greta Gudoityte, Ulrika Joneborg, Joseph Carlson, Jos Jonkers, and et al. 2021. "PFKFB3 Inhibition Sensitizes DNA Crosslinking Chemotherapies by Suppressing Fanconi Anemia Repair" Cancers 13, no. 14: 3604. https://doi.org/10.3390/cancers13143604
APA StyleNinou, A. H., Lehto, J., Chioureas, D., Stigsdotter, H., Schelzig, K., Åkerlund, E., Gudoityte, G., Joneborg, U., Carlson, J., Jonkers, J., Seashore-Ludlow, B., & Gustafsson, N. M. S. (2021). PFKFB3 Inhibition Sensitizes DNA Crosslinking Chemotherapies by Suppressing Fanconi Anemia Repair. Cancers, 13(14), 3604. https://doi.org/10.3390/cancers13143604