
Targeting Orphan G Protein-Coupled Receptor 17 with T0 Ligand Impairs Glioblastoma Growth


 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
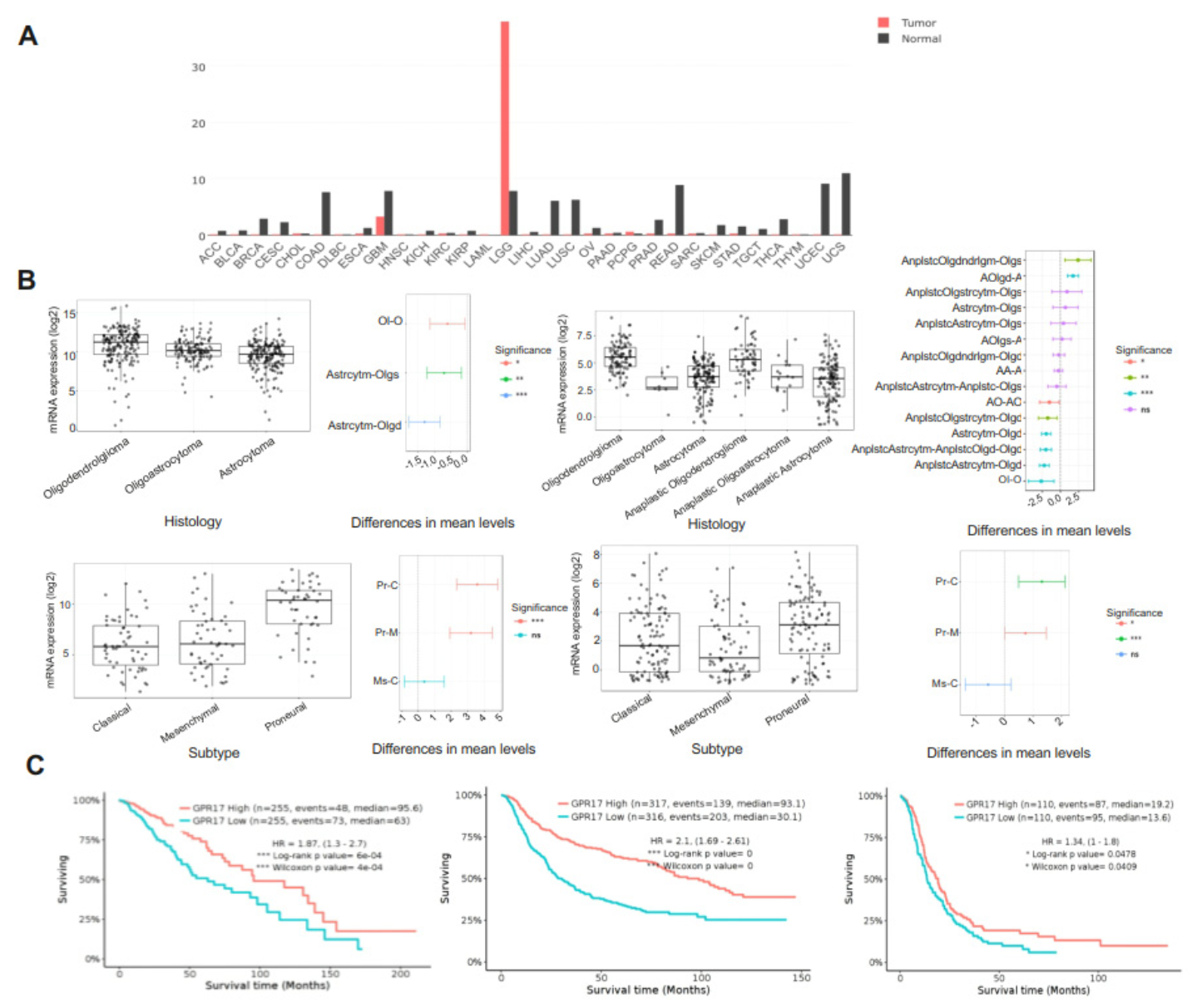
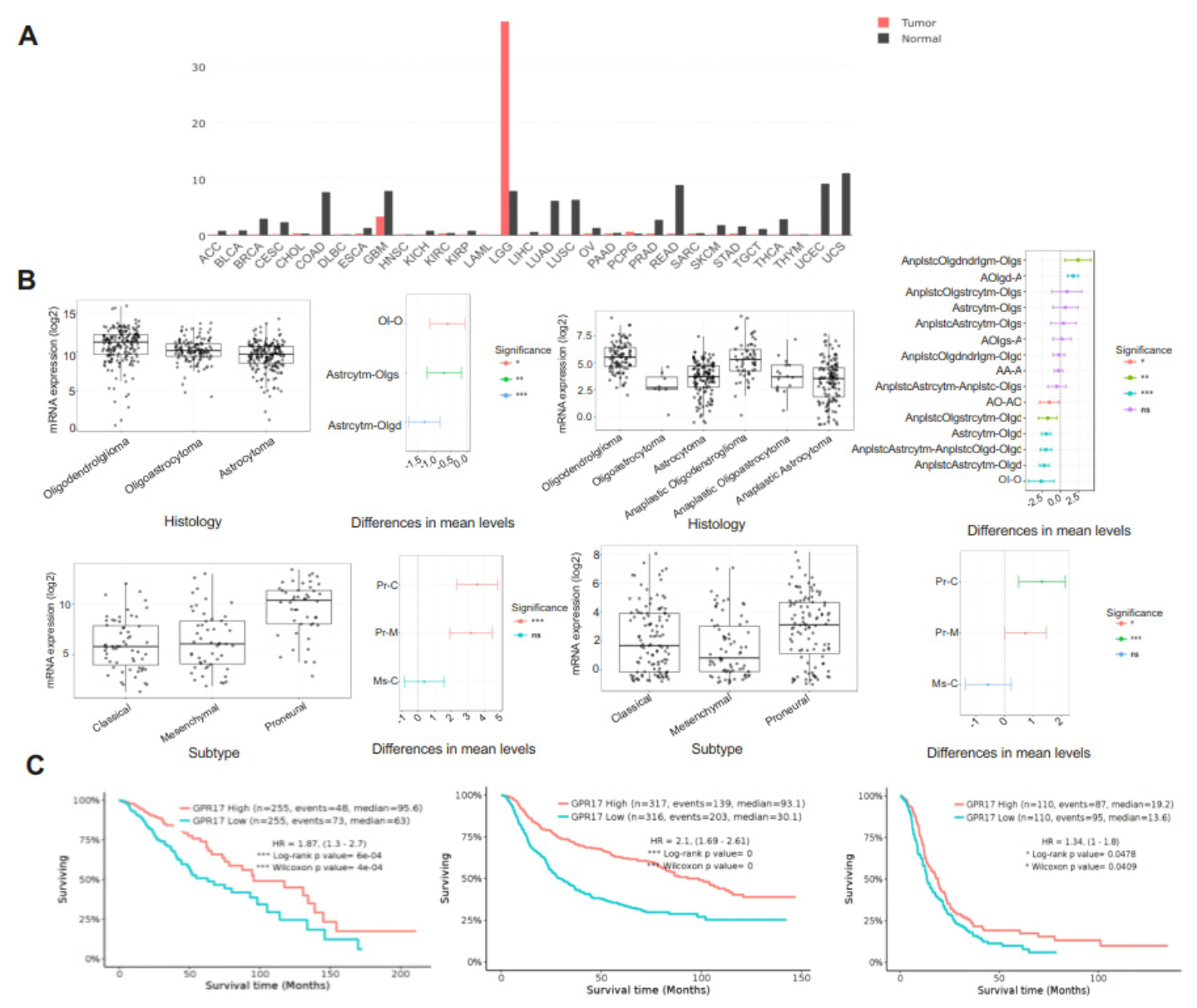
2.1. GPR17 as a Biomarker for LGG and GBM
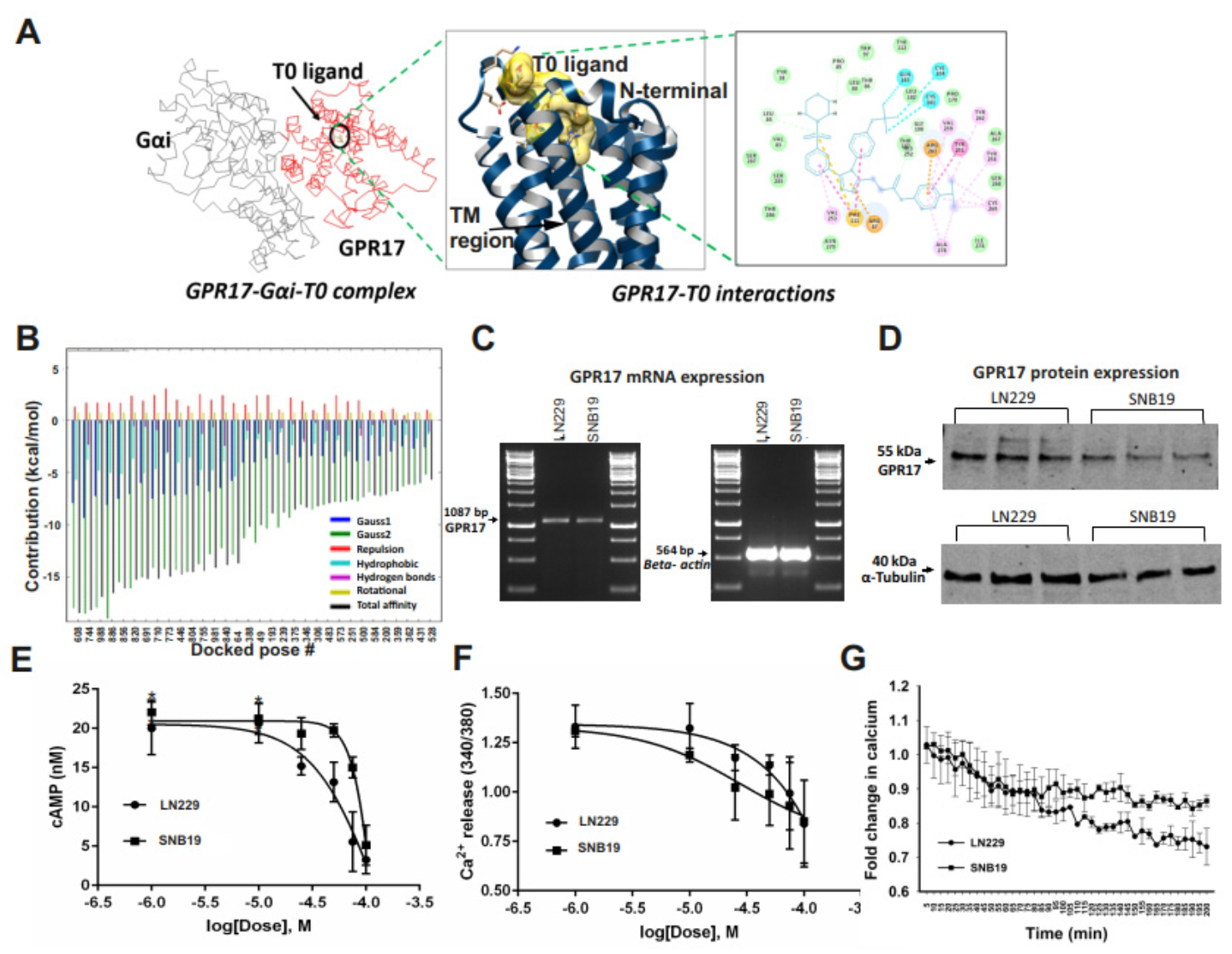
2.2. GA-T0 Activates GPR17 Signaling in GBM Cell Lines
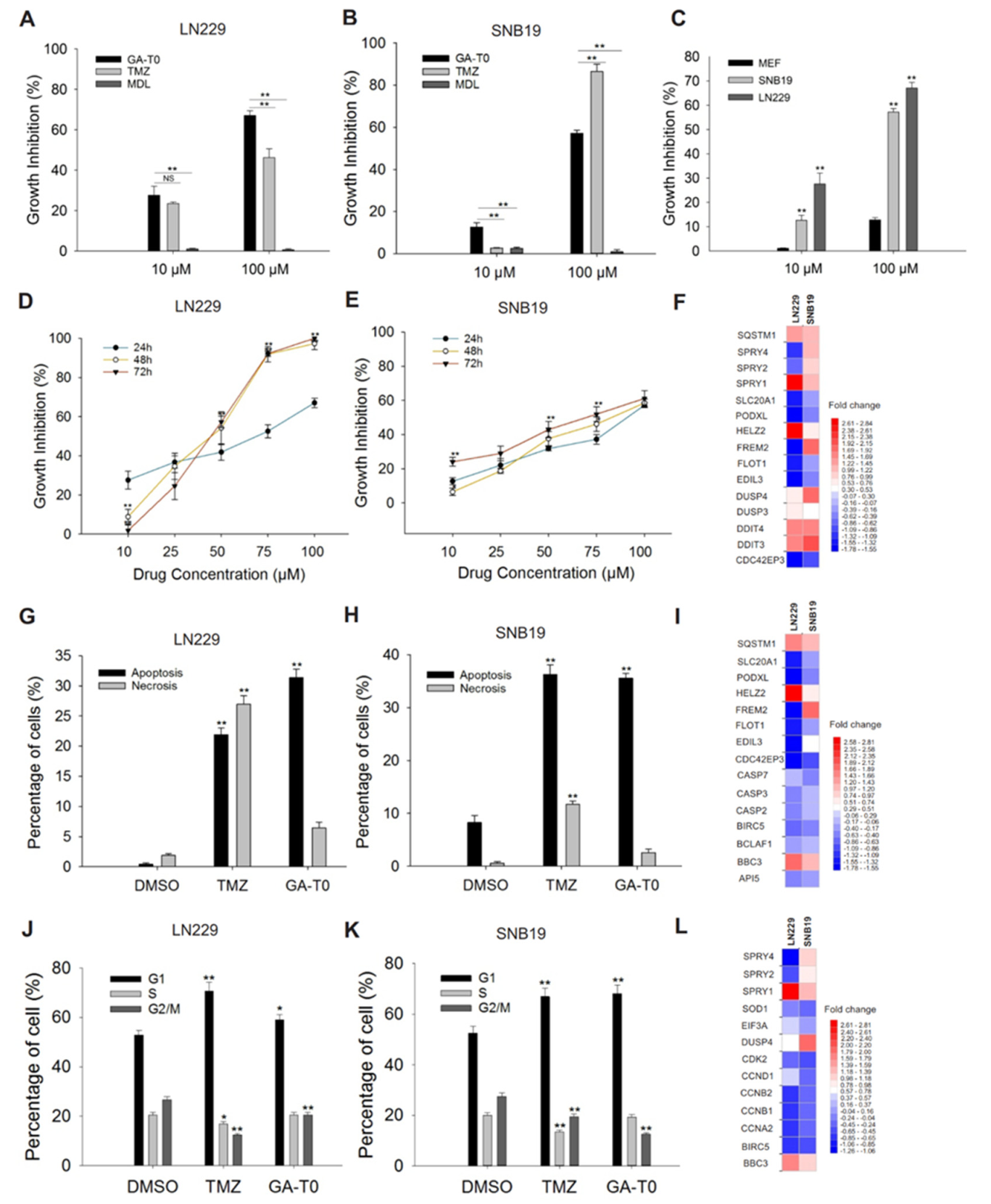
2.3. GPR17 as a Target for Inhibiting GBM Cell Proliferation
2.4. Apoptosis-Mediated Cell Death Induced by GA-T0
2.5. GA-T0 Promoted Cell Cycle Arrest at the G1 Phase
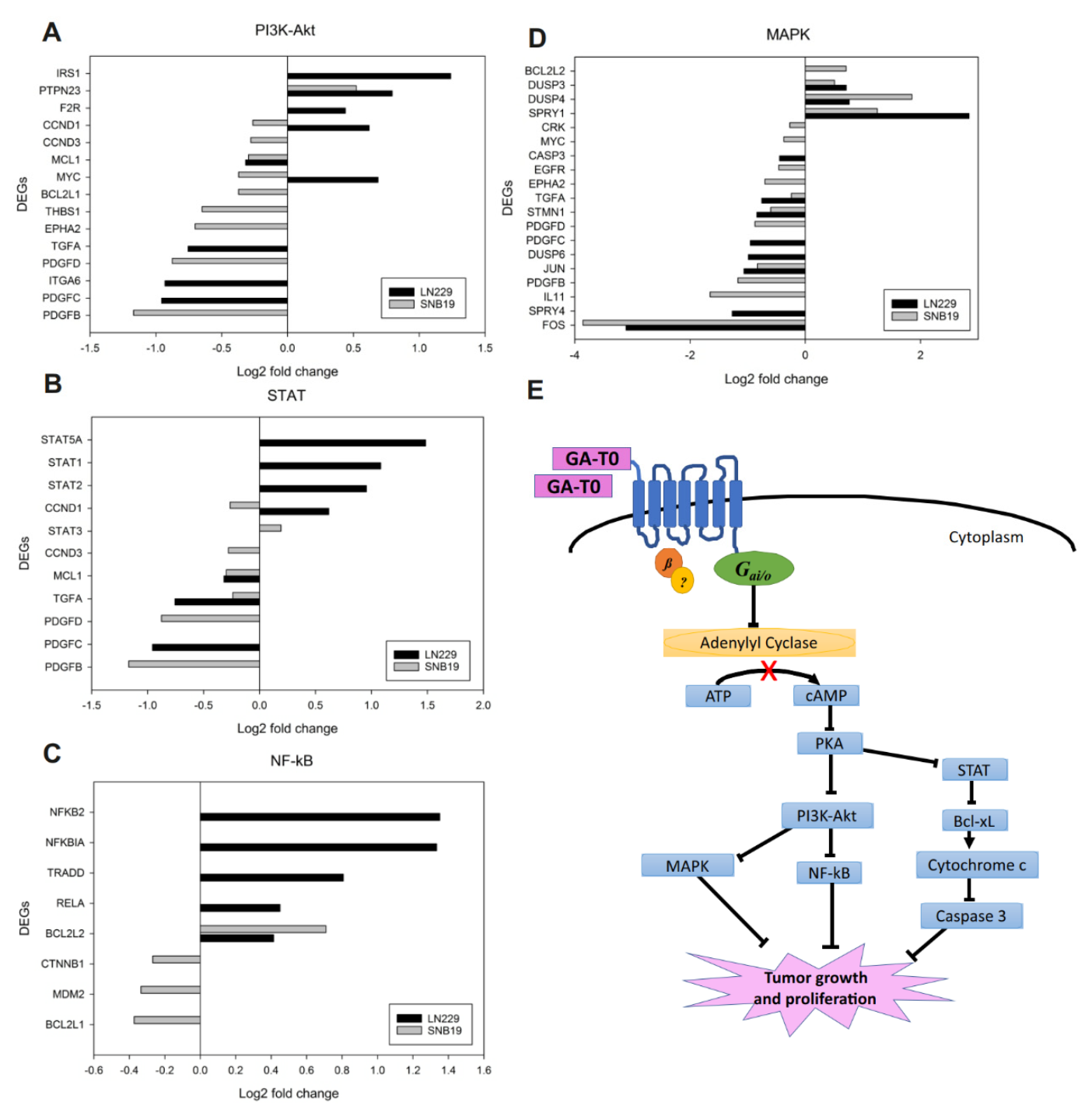
2.6. Effect of GA-T0 Mediated GPR17 Activation on Signal Transduction Pathways
2.6.1. PI3K–Akt Pathway
2.6.2. STAT Pathway
2.6.3. NF-κB Pathway
2.6.4. MAPK Pathway
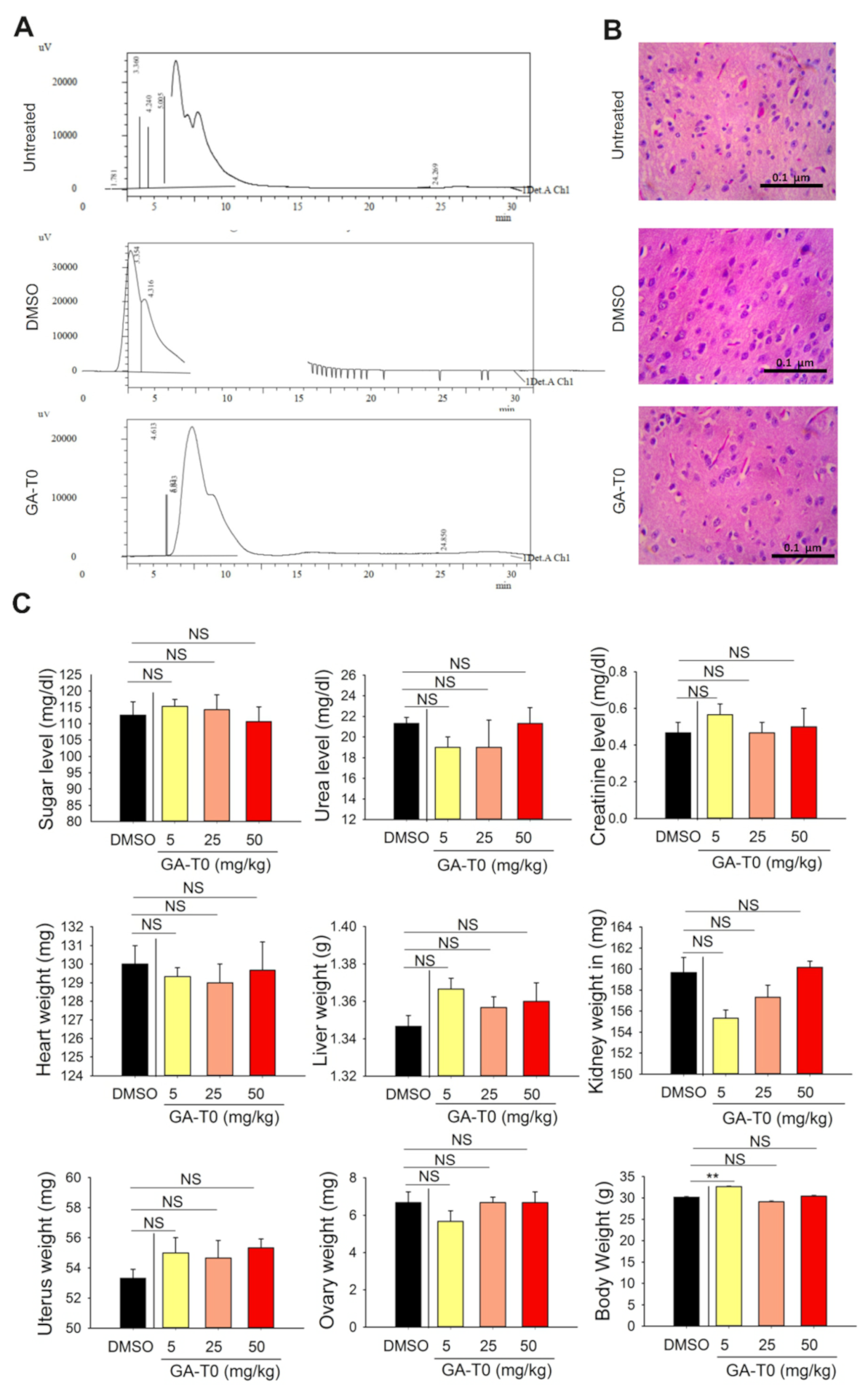
2.7. GA-T0 Crosses the Blood–Brain Barrier
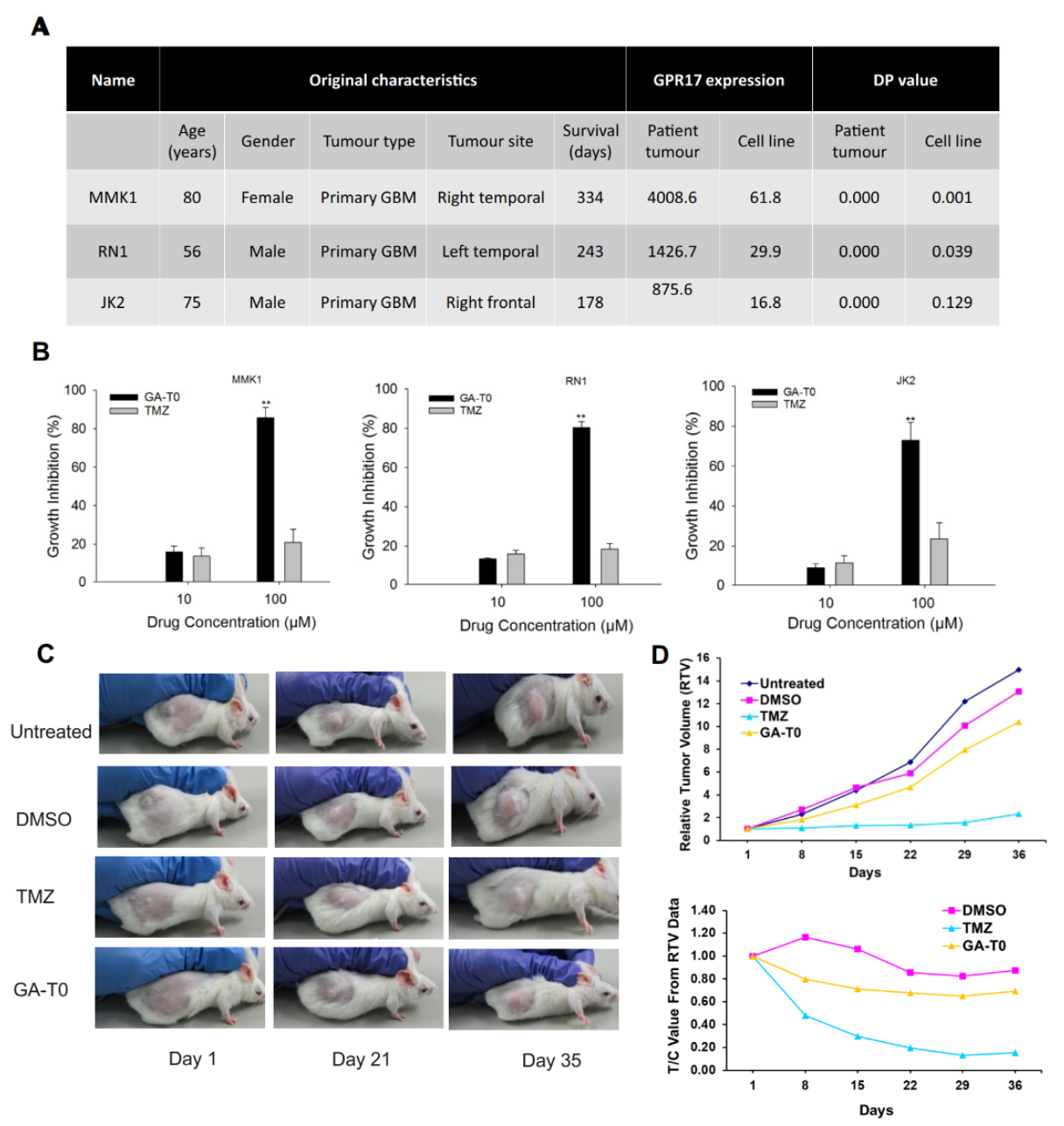
2.8. Preclinical Validation of GA-T0 in Patient-Derived Cell Lines (PDC) and Patient-Derived Xenograft Mouse Models (PDX)
3. Discussion
4. Materials and Methods
4.1. Protein–Protein Docking and Docking Simulations
4.2. Cell Culture
4.3. Expression Analysis of GPR17 at the mRNA and Protein Level in GBM Cells
4.4. cAMP GloTM Assay
4.5. Measurement of the Intracellular Calcium Concentration
4.6. In Vitro Cell Proliferation Assay
4.7. Pharmacodynamics Study
4.8. Apoptosis Annexin V-FITC/PI Apoptotic Assay
4.9. Cell Cycle Analysis by Propidium Iodide (PI)
4.10. Differential Gene Expression Analysis
4.11. Tumor Samples and Cytotoxicity Effect of GA-T0
4.12. In Vivo Experiments
4.12.1. Wild Mice
4.12.2. Histopathology Analysis of the Brain Tissues
4.12.3. Patient-Derived Xenografts (PDXs)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GBM | Glioblastoma multiforme |
| GPCR | G protein-coupled receptor |
| GA-T0 | GPR17 agonist T0510-3657 |
| pDMG | Pediatric diffuse midline glioma |
| CysLT | Cysteinyl-leukotriene |
| TMZ | Temozolomide |
| ROS | Reactive Oxygen Species |
| RNA-seq | RNA-sequencing |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| DNA | Deoxyribonucleic acid |
| FBS | Fetal Bovine Serum |
| DEG | Differentially expressed gene |
References
- Bocangel, D.B.; Finkelstein, S.; Schold, S.C.; Bhakat, K.K.; Mitra, S.; Kokkinakis, D.M. Multifaceted resistance of gliomas to temozolomide. Clin. Cancer Res. 2002, 8, 2725–2734. [Google Scholar]
- Stupp, R.; Mason, W.P.; Bent, M.V.D.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Silbergeld, D.L.; Chicoine, M.R. Isolation and characterization of human malignant glioma cells from histologically normal brain. J. Neurosurg. 1997, 86, 525–531. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nat. Cell Biol. 2012, 488, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nat. Cell Biol. 2013, 501, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-Inducible Factors Regulate Tumorigenic Capacity of Glioma Stem Cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [Green Version]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef]
- Furusawa, C.; Kaneko, K. Chaotic expression dynamics implies pluripotency: When theory and experiment meet. Biol. Direct 2009, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.; Wakimoto, H.; Cahill, D.; Nahed, B.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, A.; Kute, D.; Musa, A.; Mani, S.K.; Sipilä, V.; Emmert-Streib, F.; Zubkov, F.; Gurbanov, A.V.; Yli-Harja, O.; Kandhavelu, M. 2-(2-(2,4-dioxopentan-3-ylidene)hydrazineyl)benzonitrile as novel inhibitor of receptor tyrosine kinase and PI3K/AKT/mTOR signaling pathway in glioblastoma. Eur. J. Med. Chem. 2019, 166, 291–303. [Google Scholar] [CrossRef]
- Doan, P.; Musa, A.; Murugesan, A.; Sipilä, V.; Candeias, N.R.; Emmert-Streib, F.; Ruusuvuori, P.; Granberg, K.; Yli-Harja, O.; Kandhavelu, M. Glioblastoma Multiforme Stem Cell Cycle Arrest by Alkylaminophenol through the Modulation of EGFR and CSC Signaling Pathways. Cells 2020, 9, 681. [Google Scholar] [CrossRef] [Green Version]
- Barros, M.T.; Doan, P.; Kandhavelu, M.; Jennings, B.; Balasubramaniam, S. Engineering calcium signaling of astrocytes for neural–molecular computing logic gates. Sci. Rep. 2021, 11, 595. [Google Scholar] [CrossRef]
- Drews, J. Drug Discovery: A Historical Perspective. Science 2000, 287, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Oprea, T.I.; Bologa, C.; Brunak, S.; Campbell, A.; Gan, G.N.; Gaulton, A.; Gomez, S.M.; Guha, R.; Hersey, A.; Holmes, J.; et al. Unexplored therapeutic opportunities in the human genome. Nat. Rev. Drug Discov. 2018, 17, 317–332. [Google Scholar] [CrossRef]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Ciana, P.; Fumagalli, M.; Trincavelli, M.L.; Verderio, C.; Rosa, P.; Lecca, D.; Ferrario, S.; Parravicini, C.; Capra, V.; Gelosa, P.; et al. The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. EMBO J. 2006, 25, 4615–4627. [Google Scholar] [CrossRef]
- Fratangeli, A.; Parmigiani, E.; Fumagalli, M.; Lecca, D.; Benfante, R.; Passafaro, M.; Buffo, A.; Abbracchio, M.P.; Rosa, P. The Regulated Expression, Intracellular Trafficking, and Membrane Recycling of the P2Y-like Receptor GPR17 in Oli-neu Oligodendroglial Cells. J. Biol. Chem. 2013, 288, 5241–5256. [Google Scholar] [CrossRef] [Green Version]
- Hennen, S.; Wang, H.; Peters, L.; Merten, N.; Simon, K.; Spinrath, A.; Blättermann, S.; Akkari, R.; Schrage, R.; Schröder, R.; et al. Decoding Signaling and Function of the Orphan G Protein-Coupled Receptor GPR17 with a Small-Molecule Agonist. Sci. Signal. 2013, 6, ra93. [Google Scholar] [CrossRef] [Green Version]
- Lecca, D.; Trincavelli, M.L.; Gelosa, P.; Sironi, L.; Ciana, P.; Fumagalli, M.; Villa, G.; Verderio, C.; Grumelli, C.; Guerrini, U.; et al. The Recently Identified P2Y-Like Receptor GPR17 Is a Sensor of Brain Damage and a New Target for Brain Repair. PLoS ONE 2008, 3, e3579. [Google Scholar] [CrossRef] [Green Version]
- Dziedzic, A.; Miller, E.; Saluk-Bijak, J.; Bijak, M. The GPR17 Receptor—A Promising Goal for Therapy and a Potential Marker of the Neurodegenerative Process in Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marucci, G.; Lammi, C.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Amantini, C.; Santoni, G.; Kandhavelu, M.; Abbracchio, M.P.; Lecca, D.; et al. Comparison and optimization of transient transfection methods at human astrocytoma cell line 1321N1. Anal. Biochem. 2011, 414, 300–302. [Google Scholar] [CrossRef]
- Loveson, K.; Lepinay, E.; Robson, S.; Fillmore, H. DIPG-15. The Role of the G-Protein-Coupled Receptor, GPR17 in Paediatric Diffuse Midline Glioma. Neuro. Oncol. 2018, 20, i51. [Google Scholar] [CrossRef]
- Dougherty, J.; Fomchenko, E.I.; Akuffo, A.A.; Schmidt, E.; Helmy, K.Y.; Bazzoli, E.; Brennan, C.; Holland, E.C.; Milosevic, A. Candidate Pathways for Promoting Differentiation or Quiescence of Oligodendrocyte Progenitor-like Cells in Glioma. Cancer Res. 2012, 72, 4856–4868. [Google Scholar] [CrossRef] [Green Version]
- Mutharasu, G.; Murugesan, A.; Mani, S.K.; Yli-Harja, O.; Kandhavelu, M. Transcriptomic analysis of glioblastoma multiforme providing new insights into GPR17 signaling communication. J. Biomol. Struct. Dyn. 2020, 3, 1–14. [Google Scholar] [CrossRef]
- Gnanavel, M.; Yli-Harja, O.; Kandhavelu, M. Protein-Protein Interaction and Coarse Grained Simulation Study of Glioblastoma Multiforme Reveals Novel Pathways of GPR17. TASK Q. 2015, 18, 321–325. [Google Scholar]
- Saravanan, K.M.; Palanivel, S.; Yli-Harja, O.; Kandhavelu, M. Identification of novel GPR17-agonists by structural bioinformatics and signaling activation. Int. J. Biol. Macromol. 2017, 106, 901–907. [Google Scholar] [CrossRef]
- Mantilla, J.G.; Ricciotti, R.W.; Chen, E.Y.; Liu, Y.J.; Hoch, B.L. Amplification of DNA damage-inducible transcript 3 (DDIT3) is associated with myxoid liposarcoma-like morphology and homologous lipoblastic differentiation in dedifferentiated liposarcoma. Mod. Pathol. 2018, 32, 585–592. [Google Scholar] [CrossRef]
- Shoshani, T.; Faerman, A.; Mett, I.; Zelin, E.; Tenne, T.; Gorodin, S.; Moshel, Y.; Elbaz, S.; Budanov, A.; Chajut, A.; et al. Identification of a Novel Hypoxia-Inducible Factor 1-Responsive Gene, RTP801, Involved in Apoptosis. Mol. Cell. Biol. 2002, 22, 2283–2293. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Malone, M.H.; Thomenius, M.J.; Zhong, F.; Xu, F.; Distelhorst, C.W. Dexamethasone-induced Gene 2 (dig2) Is a Novel Pro-survival Stress Gene Induced Rapidly by Diverse Apoptotic Signals. J. Biol. Chem. 2003, 278, 27053–27058. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, G.; Carroll, B.; Sarallah, R.; Correia-Melo, C.; Ogrodnik, M.; Nelson, G.; Otten, E.; Manni, D.; Antrobus, R.; Morgan, B.A.; et al. SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy 2016, 12, 1917–1930. [Google Scholar] [CrossRef]
- Garg, H.; Suri, P.; Gupta, J.C.; Talwar, G.P.; Dubey, S. Survivin: A unique target for tumor therapy. Cancer Cell Int. 2016, 16, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Krejci, P.; Koci, L.; Chlebova, K.; Hýžďalová, M.; Hofmanova, J.; Jira, M.; Kysela, P.; Kozubik, A.; Kala, Z. Apoptosis inhibitor 5 (API-5; AAC-11; FIF) is upregulated in human carcinomas in vivo. Oncol. Lett. 2012, 3, 913–916. [Google Scholar] [CrossRef] [Green Version]
- Han, J.W.; Flemington, C.; Houghton, A.B.; Gu, Z.; Zambetti, G.P.; Lutz, R.J.; Zhu, L.; Chittenden, T. Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc. Natl. Acad. Sci. USA 2001, 98, 11318–11323. [Google Scholar] [CrossRef] [Green Version]
- Kasof, G.M.; Goyal, L.; White, E. Btf, a Novel Death-Promoting Transcriptional Repressor That Interacts with Bcl-2-Related Proteins. Mol. Cell. Biol. 1999, 19, 4390–4404. [Google Scholar] [CrossRef] [Green Version]
- McPherson, J.P.; Sarras, H.; Lemmers, B.; Tamblyn, L.; Migon, E.; Matysiak-Zablocki, E.; Hakem, A.; Azami, S.A.; Cardoso, R.; Fish, J.; et al. Essential role for Bclaf1 in lung development and immune system function. Cell Death Differ. 2009, 16, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Cruzan, M.; Sharma, R.; Tiwari, M.; Karbach, S.; Holstein, D.; Martin, C.R.; Lechleiter, J.D.; Herman, B. Caspase-2 resides in the mitochondria and mediates apoptosis directly from the mitochondrial compartment. Cell Death Discov. 2016, 2, 16005. [Google Scholar] [CrossRef] [Green Version]
- Zhivotovsky, B.; Samali, A.; Gahm, A.; Orrenius, S. Caspases: Their intracellular localization and translocation during apoptosis. Cell Death Differ. 1999, 6, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Chandra, D.; Tang, D. Mitochondrially Localized Active Caspase-9 and Caspase-3 Result Mostly from Translocation from the Cytosol and Partly from Caspase-Mediated Activation in the Organelle. Lack of Evidence for Apaf-1-Mediated Procaspase-9 Activation in the Mitochondria. J. Biol. Chem. 2003, 278, 17408–17420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadesse, S.; Anshabo, A.T.; Portman, N.; Lim, E.; Tilley, W.; Caldon, C.E.; Wang, S. Targeting CDK2 in cancer: Challenges and opportunities for therapy. Drug Discov. Today 2020, 25, 406–413. [Google Scholar] [CrossRef]
- Reyes, J.; Chen, J.Y.; Stewart-Ornstein, J.; Karhohs, K.W.; Mock, C.S.; Lahav, G. Fluctuations in p53 Signaling Allow Escape from Cell-Cycle Arrest. Mol. Cell 2018, 71, 581–591e5. [Google Scholar] [CrossRef] [Green Version]
- Li, R.-Y.; Chen, L.-C.; Zhang, H.-Y.; Du, W.-Z.; Feng, Y.; Wang, H.-B.; Wen, J.-Q.; Liu, X.; Li, X.-F.; Sun, Y.; et al. MiR-139 Inhibits Mcl-1 Expression and Potentiates TMZ-Induced Apoptosis in Glioma. CNS Neurosci. Ther. 2013, 19, 477–483. [Google Scholar] [CrossRef]
- Du, W.; Pang, C.; Xue, Y.; Zhang, Q.; Wei, X. Dihydroartemisinin inhibits the Raf/ERK/MEK and PI3K/AKT pathways in glioma cells. Oncol. Lett. 2015, 21, 3266–3270. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-M.; Chao, J.-R.; Chen, W.; Kuo, M.-L.; Yen, J.J.-Y.; Yang-Yen, H.-F. The Antiapoptotic Gene mcl-1 Is Up-Regulated by the Phosphatidylinositol 3-Kinase/Akt Signaling Pathway through a Transcription Factor Complex Containing CREB. Mol. Cell. Biol. 1999, 19, 6195–6206. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Wu, N.; Li, X.; Guo, C.; Li, C.; Jiang, B.; Wang, H.; Shi, D. Inhibition of PTP1B blocks pancreatic cancer progression by targeting the PKM2/AMPK/mTOC1 pathway. Cell Death Dis. 2019, 10, 874. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, A.; Kortylewski, M.; Kujawski, M.; Zhang, C.; Reckamp, K.; Armstrong, B.; Wang, L.; Kowolik, C.; Deng, J.; Figlin, R.; et al. Targeting Stat3 in the Myeloid Compartment Drastically Improves the In vivo Antitumor Functions of Adoptively Transferred T Cells. Cancer Res. 2010, 70, 7455–7464. [Google Scholar] [CrossRef] [Green Version]
- Kujawski, M.; Kortylewski, M.; Lee, H.; Herrmann, A.; Kay, H.; Yu, H. Stat3 mediates myeloid cell–dependent tumor angiogenesis in mice. J. Clin. Investig. 2008, 118, 3367–3377. [Google Scholar] [CrossRef]
- Akgul, C.; Turner, P.C.; White, M.R.H.; Edwards, S.W. Functional analysis of the human MCL-1 gene. Cell. Mol. Life Sci. 2000, 57, 684–691. [Google Scholar] [CrossRef]
- Nijhawan, D.; Fang, M.; Traer, E.; Zhong, Q.; Gao, W.; Du, F.; Wang, X. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003, 17, 1475–1486. [Google Scholar] [CrossRef] [Green Version]
- Tang, P.; Steck, P.A.; Yung, W.K.A. The autocrine loop of TGF-α/EGFR and brain tumors. J. Neuro. Oncol. 1997, 35, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Cheney, M.D.; McKenzie, P.P.; Volk, E.L.; Fan, L.; Harris, L.C. MDM2 displays differential activities dependent upon the activation status of NFκB. Cancer Biol. Ther. 2008, 7, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashatus, D.; Cogswell, P.; Baldwin, A.S. Expression of the Bcl-3 proto-oncogene suppresses p53 activation. Genes Dev. 2006, 20, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Bredel, M.; Scholtens, D.M.; Yadav, A.K.; Alvarez, A. NFKBIA deletion in glioblastomas. N. Engl. J. Med. 2011, 364, 627–637. [Google Scholar] [CrossRef] [Green Version]
- Spink, C.F.; Gray, L.C.; Davies, F.E.; Morgan, G.J.; Bidwell, J.L. Haplotypic structure across the IκBα gene (NFKBIA) and association with multiple myeloma. Cancer Lett. 2007, 246, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, X.; Zou, J.; Zhang, A.; Wan, Y.; Pu, P.; Song, Z.; Qian, C.; Chen, Y.; Yang, S.; et al. miR-92b controls glioma proliferation and invasion through regulating Wnt/beta-catenin signaling via Nemo-like kinase. Neuro. Oncol. 2013, 15, 578–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai, S.; Yonekawa, A.; Harada, C.; Hamada, M.; Katagiri, W.; Nakazawa, M.; Yura, Y. Involvement of the Wnt-β-catenin pathway in invasion and migration of oral squamous carcinoma cells. Int. J. Oncol. 2010, 37, 1095–1103. [Google Scholar] [CrossRef] [Green Version]
- Celik-Selvi, B.E.; Stütz, A.; Mayer, C.E.; Salhi, J.; Siegwart, G.; Sutterlüty, H. Sprouty3 and Sprouty4, Two Members of a Family Known to Inhibit FGF-Mediated Signaling, Exert Opposing Roles on Proliferation and Migration of Glioblastoma-Derived Cells. Cells 2019, 8, 808. [Google Scholar] [CrossRef] [Green Version]
- Belletti, B.; Baldassarre, G. Stathmin: A protein with many tasks. New biomarker and potential target in cancer. Expert Opin. Ther. Targets 2011, 15, 1249–1266. [Google Scholar] [CrossRef]
- Liu, X.; Dong, B.; Mu, L.; Qin, X.; Qiao, W.; Liu, X.; Yang, L.; Xue, L.; Rainov, N.G. Stathmin expression in glioma-derived microvascular endothelial cells: A novel therapeutic target. Oncol. Rep. 2011, 27, 714–718. [Google Scholar] [CrossRef] [Green Version]
- Weingand, K.; Brown, G.; Hall, R.; Davies, D.; Gossett, K.; Neptun, D.; Waner, T.; Matsuzawa, T.; Salemink, P.; Froelke, W.; et al. Harmonization of animal clinical pathology testing in toxicity and safety studies. Fundam. Appl. Toxicol. 1996, 29, 198–201. [Google Scholar] [CrossRef]
- Stringer, B.W.; Day, B.W.; D’Souza, R.C.J.; Jamieson, P.R.; Ensbey, K.S.; Bruce, Z.C.; Lim, Y.C.; Goasdoué, K.; Offenhäuser, C.; Akgül, S.; et al. A reference collection of patient-derived cell line and xenograft models of proneural, classical and mesenchymal glioblastoma. Sci. Rep. 2019, 9, 4902. [Google Scholar] [CrossRef] [Green Version]
- Berrocal, A.; Segura, P.P.; Gil, M.; Balaña, C.; Lopez, J.G.; Yaya-Tur, R.; Rodríguez, J.; Reynés, G.; Gallego, O.; Iglesias, I.; et al. Extended-schedule dose-dense temozolomide in refractory gliomas. J. Neuro. Oncol. 2010, 96, 417–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, G.; Zhang, N.; Bi, X.; Dou, M. Solid lipid nanoparticles of temozolomide: Potential reduction of cardial and nephric toxicity. Int. J. Pharm. 2008, 355, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-S.; Rait, A.; Kim, E.; DeMarco, J.; Pirollo, K.F.; Chang, E.H. Encapsulation of temozolomide in a tumor-targeting nanocomplex enhances anti-cancer efficacy and reduces toxicity in a mouse model of glioblastoma. Cancer Lett. 2015, 369, 250–258. [Google Scholar] [CrossRef] [Green Version]
- Tentori, L.; Graziani, G. Recent approaches to improve the antitumor efficacy of temozolomide. Curr. Med. Chem. 2009, 16, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, S.; Huang, S. FoxM1: A potential drug target for glioma. Future Oncol. 2012, 8, 223–226. [Google Scholar] [CrossRef] [Green Version]
- Parravicini, C.; Lecca, D.; Marangon, D.; Coppolino, G.T.; Daniele, S.; Bonfanti, E.; Fumagalli, M.; Raveglia, L.; Martini, C.; Gianazza, E.; et al. Development of the first in vivo GPR17 ligand through an iterative drug discovery pipeline: A novel disease-modifying strategy for multiple sclerosis. PLoS ONE 2020, 15, e0231483. [Google Scholar] [CrossRef] [Green Version]
- Mascelli, S.; Barla, A.; Raso, A.; Mosci, S.; Nozza, P.; Biassoni, R.; Morana, G.; Huber, M.; Mircean, C.; Fasulo, D.; et al. Molecular fingerprinting reflects different histotypes and brain region in low grade gliomas. BMC Cancer 2013, 13, 387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durand, B.; Fero, M.; Roberts, J.M.; Raff, M.C. p27Kip1 alters the response of cells to mitogen and is part of a cell-intrinsic timer that arrests the cell cycle and initiates differentiation. Curr. Biol. 1998, 8, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Hindley, C.; Philpott, A. Co-ordination of cell cycle and differentiation in the developing nervous system. Biochem. J. 2012, 444, 375–382. [Google Scholar] [CrossRef] [Green Version]
- Dobashi, Y.; Kudoh, T.; Matsumine, A.; Toyoshima, K.; Akiyama, T. Constitutive Overexpression of CDK2 Inhibits Neuronal Differentiation of Rat Pheochromocytoma PC12 Cells. J. Biol. Chem. 1995, 270, 23031–23037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, C.; Calegari, F. Cdks and cyclins link G1 length and differentiation of embryonic, neural and hematopoietic stem cells. Cell Cycle. 2010, 9, 1893–1900. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Chinot, O.L.; Rouge, T.D.L.M.; Moore, N.; Zeaiter, A.; Das, A.; Phillips, H.; Modrusan, Z.; Cloughesy, T. AVAglio: Phase 3 trial of bevacizumab plus temozolomide and radiotherapy in newly diagnosed glioblastoma multiforme. Adv. Ther. 2011, 28, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Taal, W.; Oosterkamp, H.M.; Walenkamp, A.M.E.; Dubbink, H.J.; Beerepoot, L.V.; Hanse, M.C.J.; Buter, J.; Honkoop, A.H.; Boerman, D.; De Vos, F.; et al. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): A randomised controlled phase 2 trial. Lancet Oncol. 2014, 15, 943–953. [Google Scholar] [CrossRef]
- Eberini, I.; Daniele, S.; Parravicini, C.; Sensi, C.; Trincavelli, M.L.; Martini, C.; Abbracchio, M.P. In silico identification of new ligands for GPR17: A promising therapeutic target for neurodegenerative diseases. J. Comput. Mol. Des. 2011, 25, 743–752. [Google Scholar] [CrossRef]
- Köse, M.; Ritter, K.; Thiemke, K.; Gillard, M.; Kostenis, E.; Müller, C.E. Development of [(3)H]2-Carboxy-4,6-dichloro-1H-indole-3-propionic Acid ([(3)H]PSB-12150): A Useful Tool for Studying GPR17. ACS Med. Chem. Lett. 2014, 5, 326–330. [Google Scholar] [CrossRef] [Green Version]
- Baqi, Y.; Alshaibani, S.; Ritter, K.; Abdelrahman, A.; Spinrath, A.; Kostenis, E.; Müller, C.E. Improved synthesis of 4-/6-substituted 2-carboxy-1H-indole-3-propionic acid derivatives and structure-activity relationships as GPR17 agonists. MedChemComm 2014, 5, 86–92. [Google Scholar] [CrossRef]
- Kimple, R.J.; Kimple, M.E.; Betts, L.; Sondek, J.; Siderovski, D.P. Structural determinants for GoLoco-induced inhibition of nucleotide release by Gα subunits. Nature 2002, 416, 878–881. [Google Scholar] [CrossRef] [PubMed]
- Comeau, S.R.; Gatchell, D.W.; Vajda, S.; Camacho, C.J. ClusPro: A fully automated algorithm for protein-protein docking. Nucleic Acids Res. 2004, 32, W96–W99. [Google Scholar] [CrossRef] [Green Version]
- De Vries, S.J.; van Dijk, M.; Bonvin, A.M. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef] [Green Version]
- Ishak, I.H.; Kamgang, B.; Ibrahim, S.S.; Riveron, J.M.; Irving, H.; Wondji, C.S. Pyrethroid Resistance in Malaysian Populations of Dengue Vector Aedes aegypti Is Mediated by CYP9 Family of Cytochrome P450 Genes. PLoS Negl. Trop. Dis. 2017, 11, e0005302. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Li, L.; Zheng, Z.; Chen, S.; Chen, E.; Hu, Y. Long non-coding RNA linc00261 suppresses gastric cancer progression via promoting Slug degradation. J. Cell. Mol. Med. 2017, 21, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Kumon, R.; Aehle, M.; Sabens, D.; Parikh, P.; Han, Y.; Kourennyi, D.; Deng, C. Spatiotemporal Effects of Sonoporation Measured by Real-Time Calcium Imaging. Ultrasound Med. Biol. 2009, 35, 494–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaiyapuri, P.S.; Ali, A.A.; Mohammad, A.A.; Kandhavelu, J.; Kandhavelu, M. Time lapse microscopy observation of cellular structural changes and image analysis of drug treated cancer cells to characterize the cellular heterogeneity. Environ. Toxicol. 2015, 30, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Day, B.W.; Stringer, B.; Al-Ejeh, F.; Ting, M.J.; Wilson, J.; Ensbey, K.S.; Jamieson, P.R.; Bruce, Z.C.; Lim, Y.C.; Offenhäuser, C.; et al. EphA3 Maintains Tumorigenicity and Is a Therapeutic Target in Glioblastoma Multiforme. Cancer Cell 2013, 23, 238–248. [Google Scholar] [CrossRef] [Green Version]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma Stem Cell Lines Expanded in Adherent Culture Have Tumor-Specific Phenotypes and Are Suitable for Chemical and Genetic Screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doan, P.; Nguyen, P.; Murugesan, A.; Subramanian, K.; Konda Mani, S.; Kalimuthu, V.; Abraham, B.G.; Stringer, B.W.; Balamuthu, K.; Yli-Harja, O.; et al. Targeting Orphan G Protein-Coupled Receptor 17 with T0 Ligand Impairs Glioblastoma Growth. Cancers 2021, 13, 3773. https://doi.org/10.3390/cancers13153773
Doan P, Nguyen P, Murugesan A, Subramanian K, Konda Mani S, Kalimuthu V, Abraham BG, Stringer BW, Balamuthu K, Yli-Harja O, et al. Targeting Orphan G Protein-Coupled Receptor 17 with T0 Ligand Impairs Glioblastoma Growth. Cancers. 2021; 13(15):3773. https://doi.org/10.3390/cancers13153773
Chicago/Turabian StyleDoan, Phuong, Phung Nguyen, Akshaya Murugesan, Kumar Subramanian, Saravanan Konda Mani, Vignesh Kalimuthu, Bobin George Abraham, Brett W. Stringer, Kadalmani Balamuthu, Olli Yli-Harja, and et al. 2021. "Targeting Orphan G Protein-Coupled Receptor 17 with T0 Ligand Impairs Glioblastoma Growth" Cancers 13, no. 15: 3773. https://doi.org/10.3390/cancers13153773
APA StyleDoan, P., Nguyen, P., Murugesan, A., Subramanian, K., Konda Mani, S., Kalimuthu, V., Abraham, B. G., Stringer, B. W., Balamuthu, K., Yli-Harja, O., & Kandhavelu, M. (2021). Targeting Orphan G Protein-Coupled Receptor 17 with T0 Ligand Impairs Glioblastoma Growth. Cancers, 13(15), 3773. https://doi.org/10.3390/cancers13153773