
Cyclic Derivatives of the Chemerin C-Terminus as Metabolically Stable Agonists at the Chemokine-like Receptor 1 for Cancer Treatment

 ,
,  ,
,
Abstract
:Simple Summary
Abstract
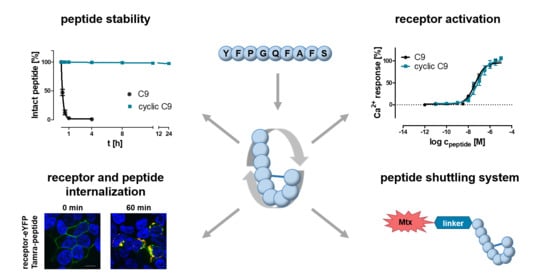
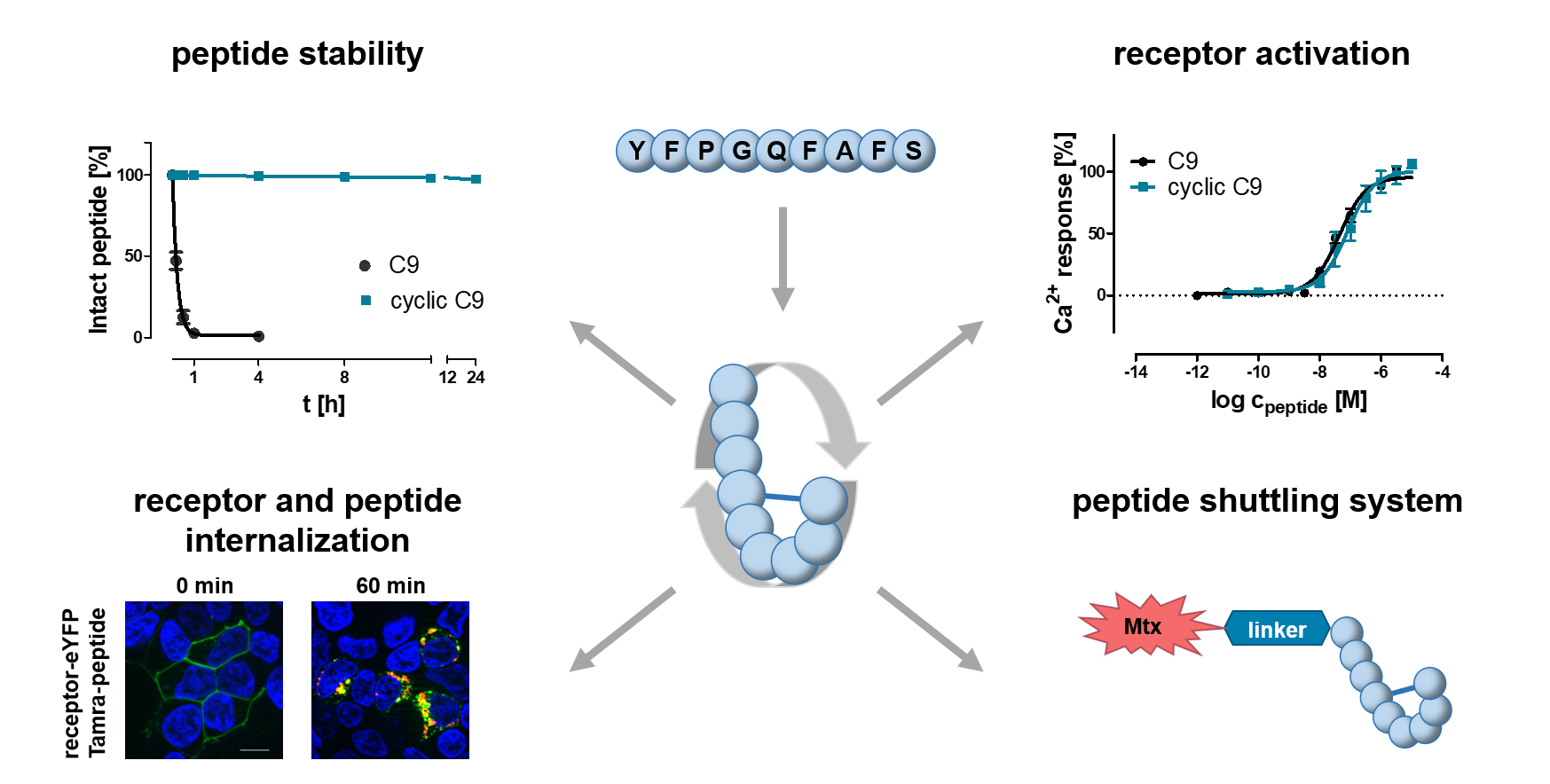
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Peptide Synthesis
2.3. Investigation of Plasma Stability
2.4. Cell Culture
2.5. Calcium Flux Assay
2.6. Bioluminescence Resonance Energy Transfer (BRET)
2.7. Statistical Analysis
2.8. Fluorescence Microscopy
2.9. Quantification of Peptide Uptake Using a High Content Imager
2.10. Resazurin Assay
3. Results
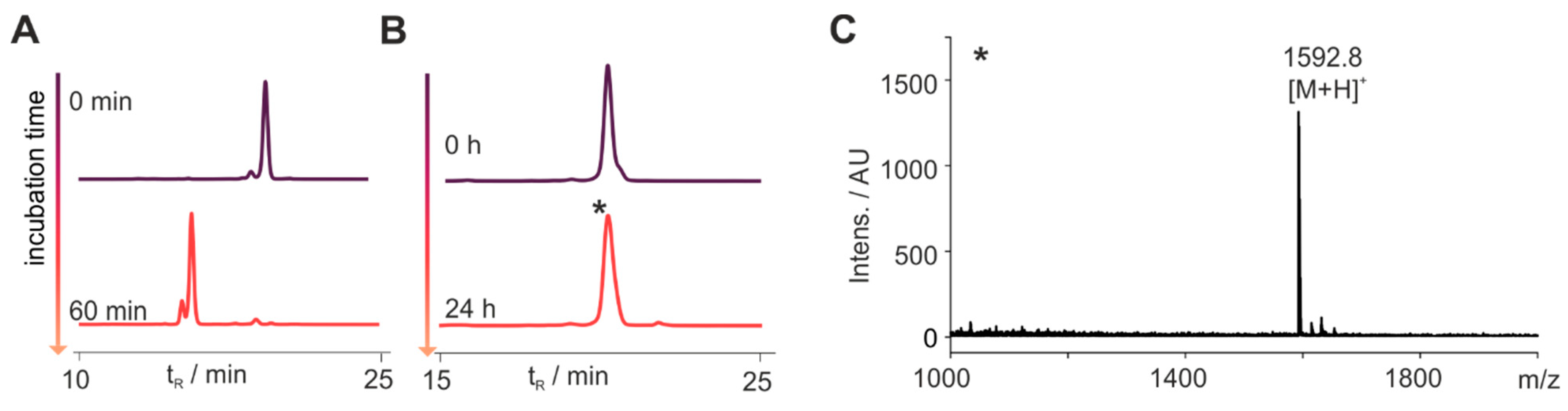
3.1. Set-Up to Investigate Stabilized Chemerin Analogues
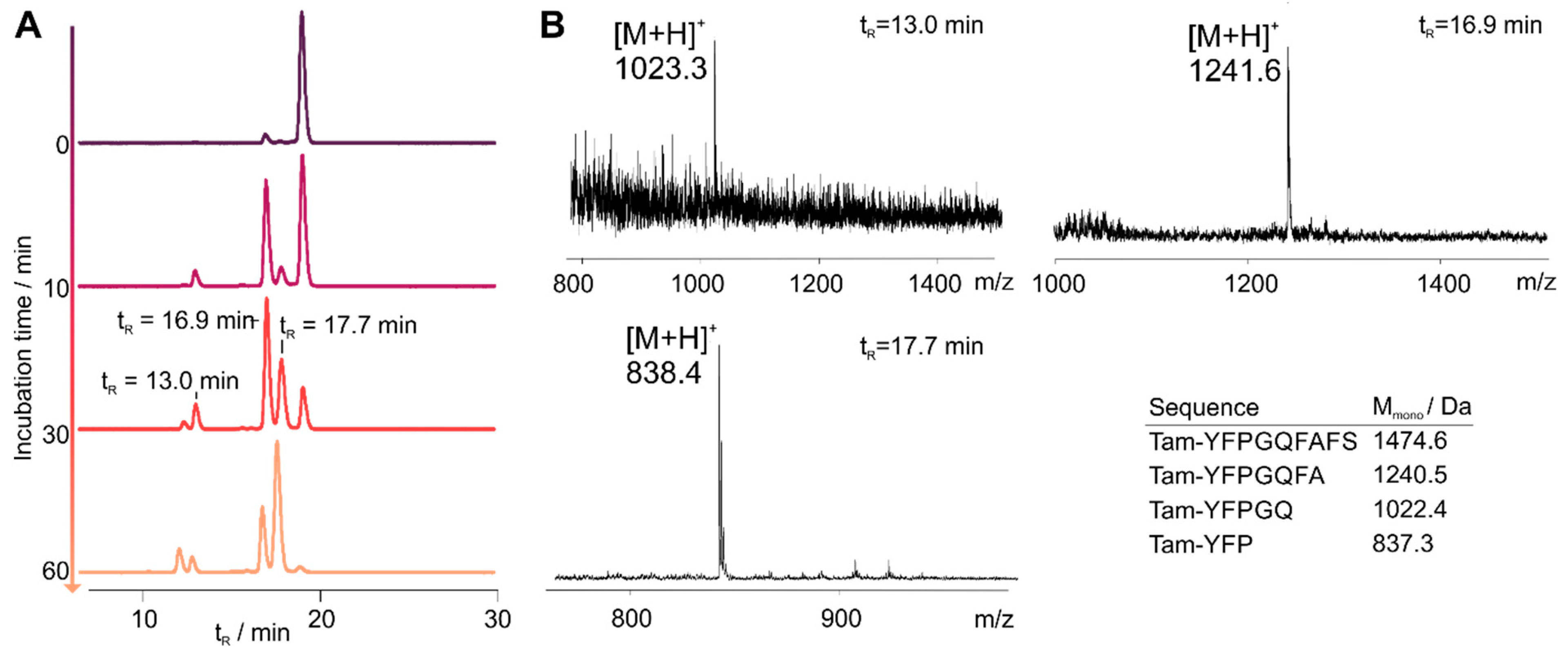
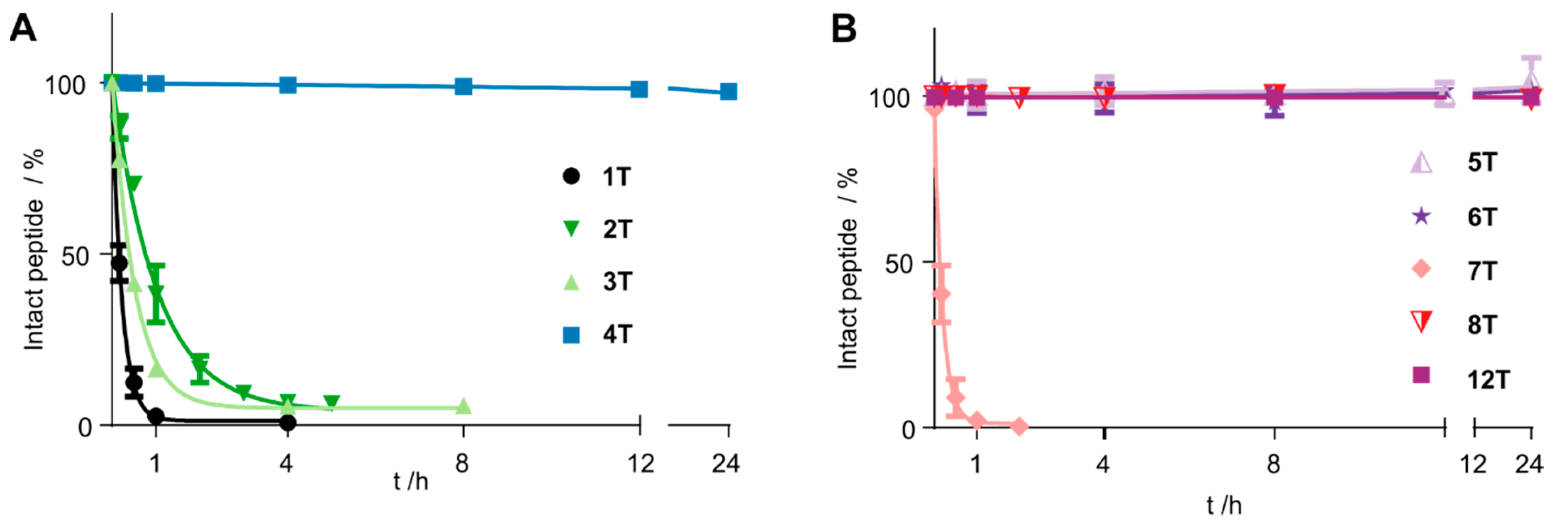
3.2. Cyclization Prevents C-Terminal Degradation
3.3. N-Terminal Modification Yields Metabolically Stable Peptides
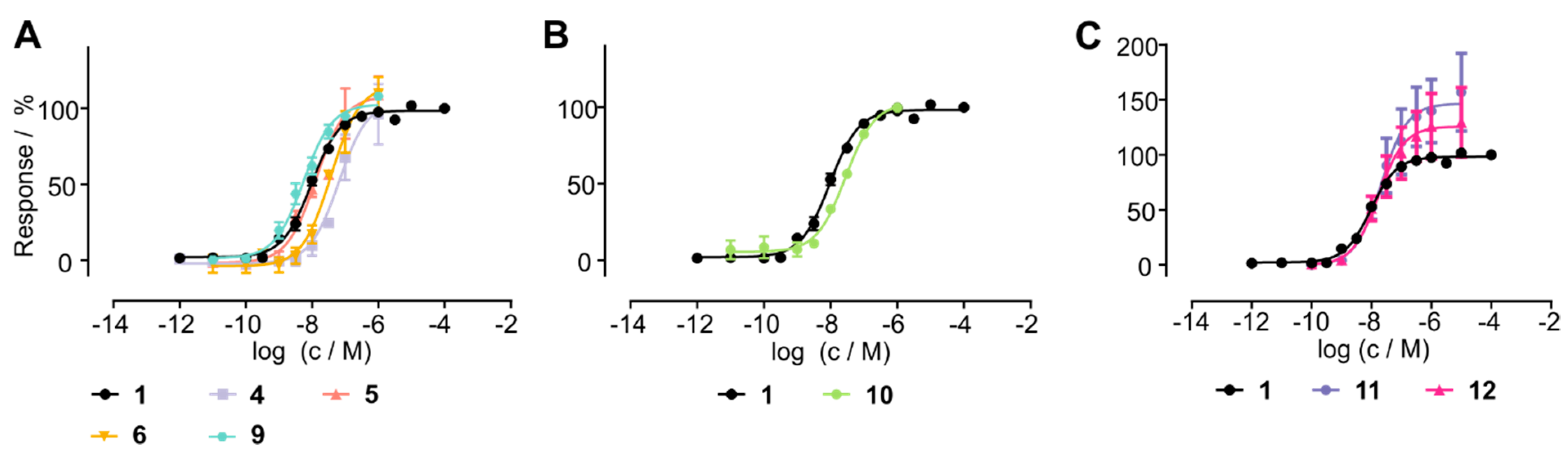
3.4. Stabilized Chemerin-9 Analogs Activate CMKLR1
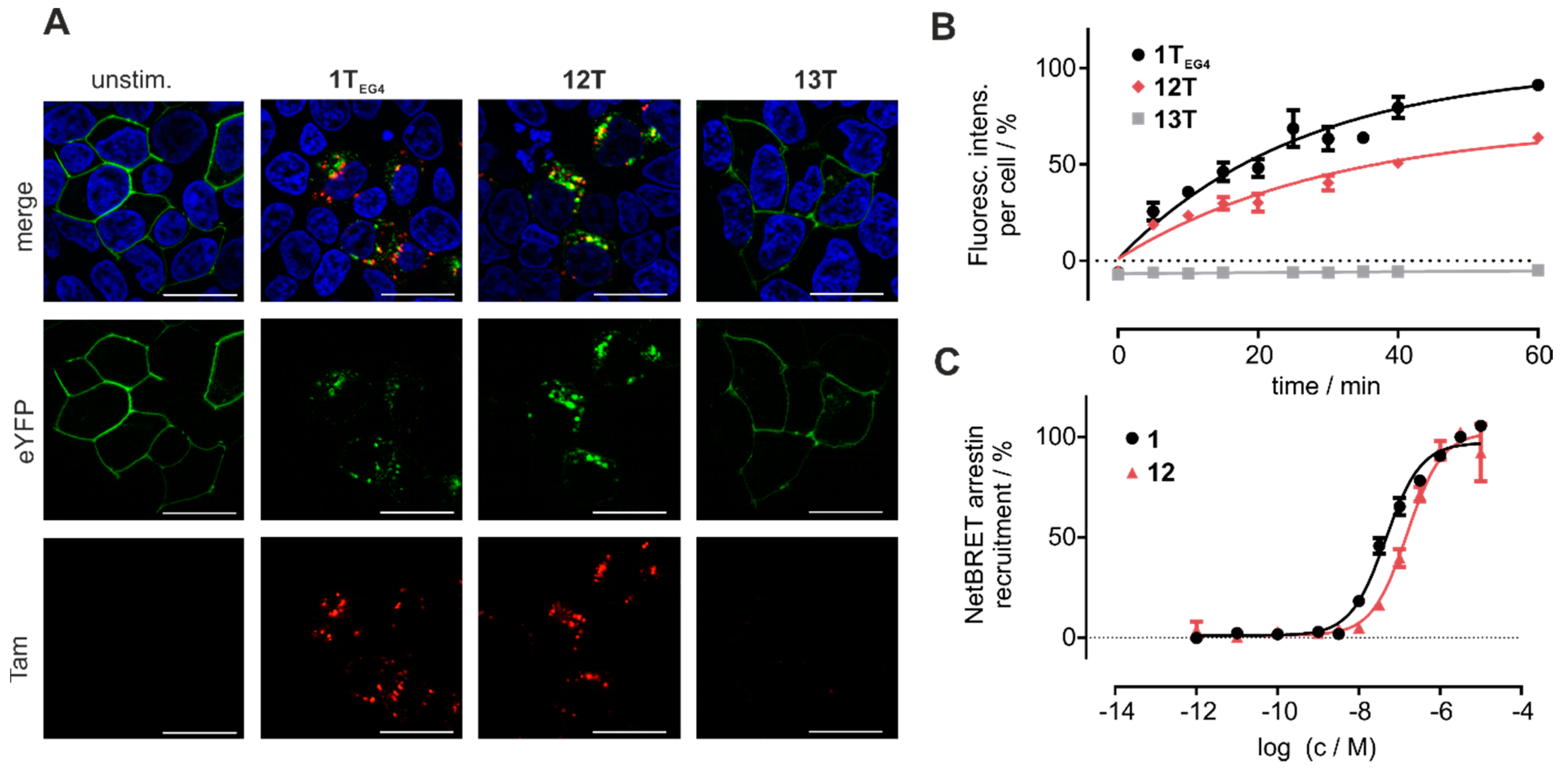
3.5. Cyclic Chemerin-9 Derivatives Are Suitable for Intracellular Delivery
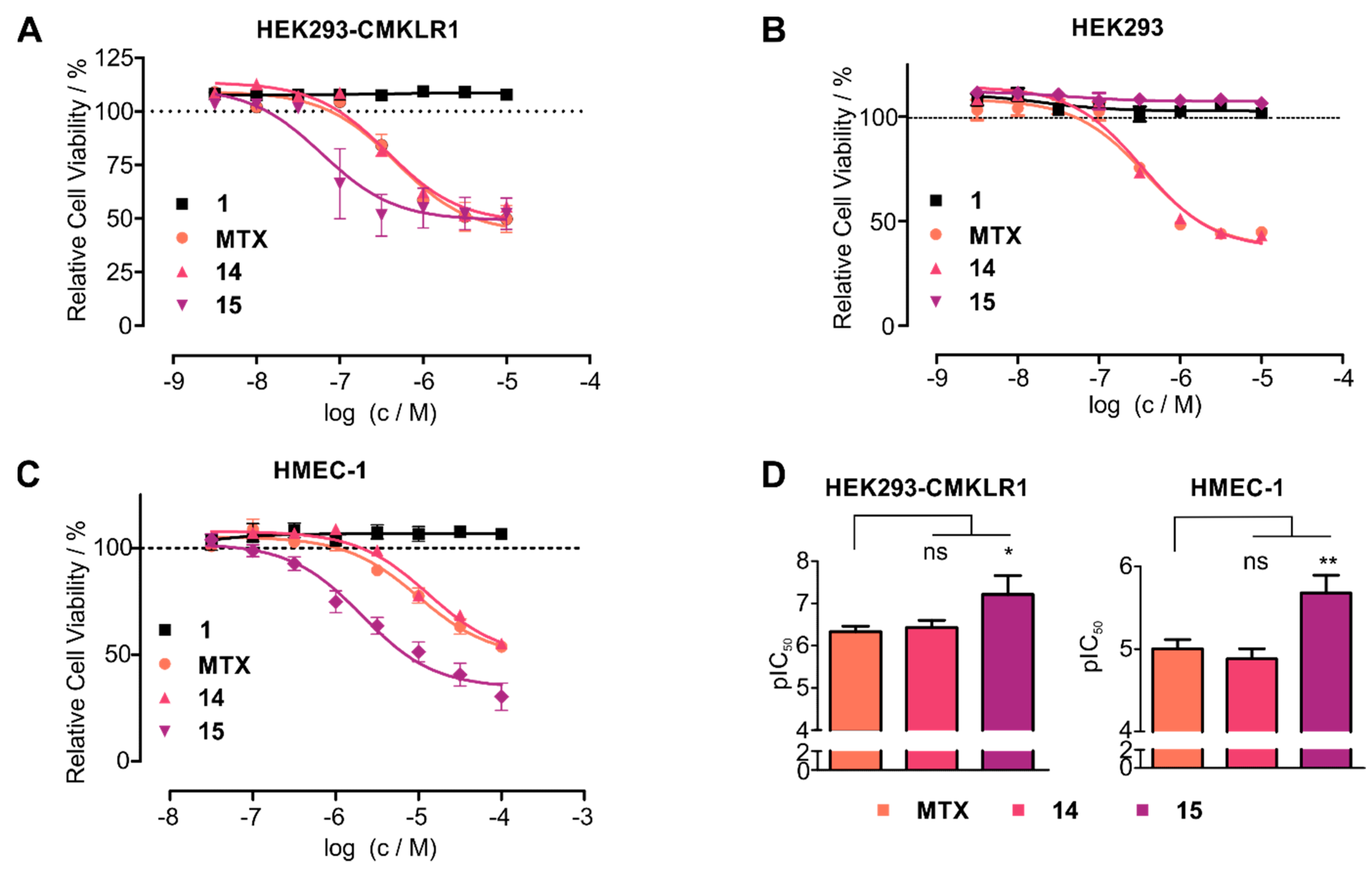
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. (Eds.) Innate Immunity. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Wittamer, V.; Franssen, J.-D.; Vulcano, M.; Mirjolet, J.-F.; Le Poul, E.; Migeotte, I.; Brezillon, S.; Tyldesley, R.; Blanpain, C.; Detheux, M.; et al. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J. Exp. Med. 2003, 198, 977–985. [Google Scholar] [CrossRef]
- Roh, S.-G.; Song, S.-H.; Choi, K.-C.; Katoh, K.; Wittamer, V.; Parmentier, M.; Sasaki, S.-I. Chemerin—A new adipokine that modulates adipogenesis via its own receptor. Biochem. Biophys. Res. Commun. 2007, 362, 1013–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banas, M.; Zabieglo, K.; Kasetty, G.; Kapinska-Mrowiecka, M.; Borowczyk, J.; Drukala, J.; Murzyn, K.; Zabel, B.A.; Butcher, E.C.; Schroeder, J.M.; et al. Chemerin is an antimicrobial agent in human epidermis. PLoS ONE 2013, 8, e58709. [Google Scholar] [CrossRef]
- Zabel, B.A.; Allen, S.J.; Kulig, P.; Allen, J.A.; Cichy, J.; Handel, T.M.; Butcher, E.C. Chemerin activation by serine proteases of the coagulation, fibrinolytic, and inflammatory cascades. J. Biol. Chem. 2005, 280, 34661–34666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, S.; Saalbach, A.; Heiker, J.T.; Meier, R.; Zellmann, T.; Simon, J.C.; Beck-Sickinger, A.G. Proteolytic activation of prochemerin by kallikrein 7 breaks an ionic linkage and results in C-terminal rearrangement. Biochem. J. 2013, 452, 271–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.-Y.; Zabel, B.A.; Myles, T.; Allen, S.J.; Handel, T.M.; Lee, P.P.; Butcher, E.C.; Leung, L.L. Regulation of chemerin bioactivity by plasma carboxypeptidase N, carboxypeptidase B (activated thrombin-activable fibrinolysis inhibitor), and platelets. J. Biol. Chem. 2009, 284, 751–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, Y.; Du, X.-Y.; Zhao, L.; Morser, J.; Leung, L.L.K. Proteolytic cleavage of chemerin protein is necessary for activation to the active form, Chem157S, which functions as a signaling molecule in glioblastoma. J. Biol. Chem. 2011, 286, 39510–39519. [Google Scholar] [CrossRef] [Green Version]
- Wittamer, V.; Gregoire, F.; Robberecht, P.; Vassart, G.; Communi, D.; Parmentier, M. The C-terminal nonapeptide of mature chemerin activates the chemerin receptor with low nanomolar potency. J. Biol. Chem. 2004, 279, 9956–9962. [Google Scholar] [CrossRef] [Green Version]
- Fischer, T.F.; Schoeder, C.T.; Zellmann, T.; Stichel, J.; Meiler, J.; Beck-Sickinger, A.G. Cyclic analogues of the chemerin C-terminus mimic a loop conformation essential for activating the chemokine-like receptor 1. J. Med. Chem. 2021, 64, 3048–3058. [Google Scholar] [CrossRef]
- Luangsay, S.; Wittamer, V.; Bondue, B.; de Henau, O.; Rouger, L.; Brait, M.; Franssen, J.-D.; de Nadai, P.; Huaux, F.; Parmentier, M. Mouse ChemR23 is expressed in dendritic cell subsets and macrophages, and mediates an anti-inflammatory activity of chemerin in a lung disease model. J. Immunol. 2009, 183, 6489–6499. [Google Scholar] [CrossRef]
- Parolini, S.; Santoro, A.; Marcenaro, E.; Luini, W.; Massardi, L.; Facchetti, F.; Communi, D.; Parmentier, M.; Majorana, A.; Sironi, M.; et al. The role of chemerin in the colocalization of NK and dendritic cell subsets into inflamed tissues. Blood 2007, 109, 3625–3632. [Google Scholar] [CrossRef]
- Berg, V.; Sveinbjörnsson, B.; Bendiksen, S.; Brox, J.; Meknas, K.; Figenschau, Y. Human articular chondrocytes express ChemR23 and chemerin; ChemR23 promotes inflammatory signalling upon binding the ligand chemerin 21-157. Arthritis Res. Ther. 2010, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kumar, J.D.; Aolymat, I.; Tiszlavicz, L.; Reisz, Z.; Garalla, H.M.; Beynon, R.; Simpson, D.; Dockray, G.J.; Varro, A. Chemerin acts via CMKLR1 and GPR1 to stimulate migration and invasion of gastric cancer cells: Putative role of decreased TIMP-1 and TIMP-2. Oncotarget 2019, 10, 98–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, J.D.; Kandola, S.; Tiszlavicz, L.; Reisz, Z.; Dockray, G.J.; Varro, A. The role of chemerin and ChemR23 in stimulating the invasion of squamous oesophageal cancer cells. Br. J. Cancer 2016, 114, 1152–1159. [Google Scholar] [CrossRef] [Green Version]
- Zabel, B.A.; Nakae, S.; Zúñiga, L.; Kim, J.-Y.; Ohyama, T.; Alt, C.; Pan, J.; Suto, H.; Soler, D.; Allen, S.J.; et al. Mast cell-expressed orphan receptor CCRL2 binds chemerin and is required for optimal induction of IgE-mediated passive cutaneous anaphylaxis. J. Exp. Med. 2008, 205, 2207–2220. [Google Scholar] [CrossRef]
- De Henau, O.; Degroot, G.-N.; Imbault, V.; Robert, V.; de Poorter, C.; Mcheik, S.; Galés, C.; Parmentier, M.; Springael, J.-Y. Signaling properties of chemerin receptors CMKLR1, GPR1 and CCRL2. PLoS ONE 2016, 11, e0164179. [Google Scholar] [CrossRef]
- Barnea, G.; Strapps, W.; Herrada, G.; Berman, Y.; Ong, J.; Kloss, B.; Axel, R.; Lee, K.J. The genetic design of signaling cascades to record receptor activation. Proc. Natl. Acad. Sci. USA 2008, 105, 64–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goralski, K.B.; Jackson, A.E.; McKeown, B.T.; Sinal, C.J. More than an adipokine: The complex roles of chemerin signaling in cancer. Int. J. Mol. Sci. 2019, 20, 4778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rennier, K.; Shin, W.J.; Krug, E.; Virdi, G.; Pachynski, R.K. Chemerin Reactivates PTEN and Suppresses PD-L1 in Tumor Cells via Modulation of a Novel CMKLR1-mediated Signaling Cascade. Clin. Cancer Res. 2020, 26, 5019–5035. [Google Scholar] [CrossRef] [PubMed]
- Kourra, C.M.B.K.; Cramer, N. Converting disulfide bridges in native peptides to stable methylene thioacetals. Chem. Sci. 2016, 7, 7007–7012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppenz, P.; Els-Heindl, S.; Beck-Sickinger, A.G. Identification and stabilization of a highly selective gastrin-releasing peptide receptor agonist. J. Pept. Sci. 2019, 25, e3224. [Google Scholar] [CrossRef]
- Hoppenz, P.; Els-Heindl, S.; Kellert, M.; Kuhnert, R.; Saretz, S.; Lerchen, H.-G.; Köbberling, J.; Riedl, B.; Hey-Hawkins, E.; Beck-Sickinger, A.G. A Selective Carborane-Functionalized Gastrin-Releasing Peptide Receptor Agonist as Boron Delivery Agent for Boron Neutron Capture Therapy. J. Org. Chem. 2020, 85, 1446–1457. [Google Scholar] [CrossRef]
- Bandholtz, S.; Wichard, J.; Kühne, R.; Grötzinger, C. Molecular evolution of a peptide GPCR ligand driven by artificial neural networks. PLoS ONE 2012, 7, e36948. [Google Scholar] [CrossRef]
- Kaur, J.; Adya, R.; Tan, B.K.; Chen, J.; Randeva, H.S. Identification of chemerin receptor (ChemR23) in human endothelial cells: Chemerin-induced endothelial angiogenesis. Biochem. Biophys. Res. Commun. 2010, 4, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.J.; Zabel, B.A.; Pachynski, R.K. Mechanisms and functions of chemerin in cancer: Potential roles in therapeutic intervention. Front. Immunol. 2018, 9, 2772. [Google Scholar] [CrossRef]
- Shimamura, K.; Matsuda, M.; Miyamoto, Y.; Yoshimoto, R.; Seo, T.; Tokita, S. Identification of a stable chemerin analog with potent activity toward ChemR23. Peptides 2009, 30, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Böhme, D.; Beck-Sickinger, A.G. Controlling toxicity of Peptide-drug conjugates by different chemical linker structures. ChemMedChem 2015, 10, 804–814. [Google Scholar] [CrossRef]
- Pachynski, R.K.; Wang, P.; Salazar, N.; Zheng, Y.; Nease, L.; Rosalez, J.; Leong, W.-I.; Virdi, G.; Rennier, K.; Shin, W.J.; et al. Chemerin suppresses breast cancer growth by recruiting immune effector cells into the tumor microenvironment. Front. Immunol. 2019, 10, 983. [Google Scholar] [CrossRef] [Green Version]
- Pachynski, R.K.; Zabel, B.A.; Kohrt, H.E.; Tejeda, N.M.; Monnier, J.; Swanson, C.D.; Holzer, A.K.; Gentles, A.J.; Sperinde, G.V.; Edalati, A.; et al. The chemoattractant chemerin suppresses melanoma by recruiting natural killer cell antitumor defenses. J. Exp. Med. 2012, 209, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-J.; Yin, H.-K.; Guan, D.-X.; Zhao, J.-S.; Feng, Y.-X.; Deng, Y.-Z.; Wang, X.; Li, N.; Wang, X.-F.; Cheng, S.-Q.; et al. Chemerin suppresses hepatocellular carcinoma metastasis through CMKLR1-PTEN-Akt axis. Br. J. Cancer 2018, 118, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Haberl, E.M.; Pohl, R.; Rein-Fischboeck, L.; Feder, S.; Sinal, C.J.; Bruckmann, A.; Hoering, M.; Krautbauer, S.; Liebisch, G.; Buechler, C. Overexpression of hepatocyte chemerin-156 lowers tumor burden in a murine model of diethylnitrosamine-induced hepatocellular carcinoma. Int. J. Mol. Sci. 2019, 21, 252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillerey, C.; Huntington, N.D.; Smyth, M.J. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 2016, 17, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Tümmler, C.; Snapkov, I.; Wickström, M.; Moens, U.; Ljungblad, L.; Maria Elfman, L.H.; Winberg, J.-O.; Kogner, P.; Johnsen, J.I.; Sveinbjørnsson, B. Inhibition of chemerin/CMKLR1 axis in neuroblastoma cells reduces clonogenicity and cell viability in vitro and impairs tumor growth in vivo. Oncotarget 2017, 8, 95135–95151. [Google Scholar] [CrossRef] [Green Version]
- Kiczmer, P.; Seńkowska, A.P.; Kula, A.; Dawidowicz, M.; Strzelczyk, J.K.; Zajdel, E.N.; Walkiewicz, K.; Waniczek, D.; Ostrowska, Z.; Świętochowska, E. Assessment of CMKLR1 level in colorectal cancer and its correlation with angiogenic markers. Exp. Mol. Pathol. 2020, 113, 104377. [Google Scholar] [CrossRef]
- Erdmann, S.; Niederstadt, L.; Koziolek, E.J.; Gómez, J.D.C.; Prasad, S.; Wagener, A.; von Hacht, J.L.; Reinicke, S.; Exner, S.; Bandholtz, S.; et al. CMKLR1-targeting peptide tracers for PET/MR imaging of breast cancer. Theranostics 2019, 9, 6719–6733. [Google Scholar] [CrossRef]
- Worm, D.J.; Els-Heindl, S.; Beck-Sickinger, A.G. Targeting of peptide-binding receptors on cancer cells with peptide-drug conjugates. Pept. Sci. 2020, 112, e24171. [Google Scholar] [CrossRef]
- Böhme, D.; Krieghoff, J.; Beck-Sickinger, A.G. Double methotrexate-modified neuropeptide Y analogues express increased toxicity and overcome drug resistance in breast cancer cells. J. Med. Chem. 2016, 59, 3409–3417. [Google Scholar] [CrossRef] [PubMed]
- Worm, D.J.; Hoppenz, P.; Els-Heindl, S.; Kellert, M.; Kuhnert, R.; Saretz, S.; Köbberling, J.; Riedl, B.; Hey-Hawkins, E.; Beck-Sickinger, A.G. Selective neuropeptide Y conjugates with maximized carborane loading as promising boron delivery agents for boron neutron capture therapy. J. Med. Chem. 2020, 63, 2358–2371. [Google Scholar] [CrossRef]
- Wittrisch, S.; Klöting, N.; Mörl, K.; Chakaroun, R.; Blüher, M.; Beck-Sickinger, A.G. NPY1R-targeted peptide-mediated delivery of a dual PPARα/γ agonist to adipocytes enhances adipogenesis and prevents diabetes progression. Mol. Metab. 2020, 31, 163–180. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fluorophore | Used for Labeling | Excitation (nm) | Emission (nm) |
|---|---|---|---|
| Hoechst33342 | cell nucleus | 352 | 455 |
| Tam | peptide | 549 | 577 |
| eYFP | CMKLR1 | 514 | 526 |
| Compound Number | Sequence | t1/2/h |
|---|---|---|
| 1T | Tam-YFPGQFAFS | <0.2 |
| 2T | Tam-YFP(cQFC)FS | 0.75 |
| 3T | Tam- YFP(c(CH2)QFC)FS | 0.3 |
| 4T | Tam-YFP(cQFAFC) | >24 |
| 5T | Tam-YFP(c(CH2)QFAFC) | >24 |
| 6T | Tam-YFP(c(CH2)QFAFX) | >24 |
| 7T | YFP(cQFK(Tam)FC) | <0.2 |
| 8T | yFP(cQFK(Tam)FC) | >24 |
| 12T | Tam-EG(4)-YFP(xQFAFC) | >48 h |
| Compound Number | Sequence | Mmono | Mobs | tR/%B | tR/%B | Purity |
|---|---|---|---|---|---|---|
| 1 | YFPGQFAFS | 1062.5 | 1063.5 | 41.7 a | 30.2 b | >95 |
| 1T | Tam-YFPGQFAFS | 1474.6 | 1475.6 | 50.5 a | 43.8 c | >95 |
| 1TEG4 | Tam-EG(4)-YFPGQFAFS | 1721.7 | 1722.7 | 51.0 a | 40.5 b | >95 |
| 2T | Tam-YFP(cQFCFS) | 1550.6 | 1551.6 | 54.2 a | 48.4 c | >95 |
| 3T | Tam-YFP(c(CH2)QFCFS) | 1564.6 | 1565.5 | 53.8 a | 48.5 c | >95 |
| 4 | YFP(cQFAFC) | 1122.4 | 1123.4 | 46.5 a | 34.2 b | >95 |
| 4T | Tam-YFP(cQFAFC) | 1534.6 | 1535.6 | 54.4 a | 47.9 c | >95 |
| 5 | YFP(c(CH2)QFAFC) | 1136.5 | 1137.5 | 45.5 a | 36.0 c | >95 |
| 5T | Tam-YFP(c(CH2)QFAFC) | 1548.6 | 1549.6 | 53.6 a | 48.2 c | >95 |
| 6 | YFP(c(CH2)QFAFX) | 1150.5 | 1151.5 | 46.1 a | 37.4 b | >95 |
| 6T | Tam-YFP(c(CH2)QFAFX) | 1562.6 | 1563.6 | 55.0 a | 43.9 b | >95 |
| 7T | YFP(cQFK(Tam)FC) | 1591.6 | 1592.6 | 49.1 a | 42.6 c | >95 |
| 8T | yFP(cQFK(Tam)FC) | 1591.6 | 1592.7 | 50.1 a | 15.9 b | >95 |
| 9 | YFP(xQFAFC) | 1136.4 | 1137.5 | 45.6 a | 34.3 b | >95 |
| 10 | yFP(c(CH2)QFAFX) | 1150.5 | 1151.4 | 47.9 a | 36.1 b | >95 |
| 11 | GFLGYFP(xQFAFC) | 1552.6 | 1553.6 | 48.1 a | 37.6 b | >95 |
| 12 | EG(4)-YFP(xQFAFC) | 1383.6 | 1384.6 | 47.4 a | 37.4 b | >95 |
| 12T | Tam-EG(4)-YFP(xQFAFC) | 1795.7 | 1796.7 | 48.6 a | 38.6 b | >95 |
| 13T | Tam-EG(4)-YFPGQFAAS | 1233.6 | 1234.6 | 47.5 a | 36.8 b | >95 |
| 14 | Mtx-GFLGK(Ac)YFPGQFAFS | 2042.9 | 2043.9 | 42.8/43.1 a | 38.3/38.6 b | >95 |
| 15 | Mtx-GFLGK(Ac)YFP(xQFAFC) | 2116.9 | 2117.9 | 39.4/40 a | 35/35.6 b | >95 |
| Compound Number | Sequence | EC50/nM | pEC50 ± SEM |
|---|---|---|---|
| Ca2+ Flux | |||
| 1 | YFPGQFAFS | 10 | 8.021 ± 0.04 |
| 4 | YFP(cQFAFC) | 64 | 7.192 ± 0.15 |
| 5 | YFP(c(CH2)QFAFC) | 13 | 7.888 ± 0.13 |
| 6 | YFP(c(CH2)QFAFX) | 37 | 7.429 ± 0.11 |
| 9 | YFP(xQFAFC) | 5 | 8.259 ± 0.07 |
| 10 | yFP(c(CH2)QFAFX) | 28 | 7.553 ± 0.07 |
| 11 | GFLGYFP(xQFAFC) | 21 | 7.676 ± 0.22 |
| 12 | EG(4)-YFP(xQFAFC) | 17 | 7.766 ± 0.23 |
| Arrestin3 Recruitment | |||
| 1 | YFPGQFAFS | 46 | 7.338 ± 0.04 |
| 12 | EG(4)-YFP(xQFAFC) | 154 | 6.813 ± 0.06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischer, T.F.; Czerniak, A.S.; Weiß, T.; Zellmann, T.; Zielke, L.; Els-Heindl, S.; Beck-Sickinger, A.G. Cyclic Derivatives of the Chemerin C-Terminus as Metabolically Stable Agonists at the Chemokine-like Receptor 1 for Cancer Treatment. Cancers 2021, 13, 3788. https://doi.org/10.3390/cancers13153788
Fischer TF, Czerniak AS, Weiß T, Zellmann T, Zielke L, Els-Heindl S, Beck-Sickinger AG. Cyclic Derivatives of the Chemerin C-Terminus as Metabolically Stable Agonists at the Chemokine-like Receptor 1 for Cancer Treatment. Cancers. 2021; 13(15):3788. https://doi.org/10.3390/cancers13153788
Chicago/Turabian StyleFischer, Tobias F., Anne S. Czerniak, Tina Weiß, Tristan Zellmann, Lina Zielke, Sylvia Els-Heindl, and Annette G. Beck-Sickinger. 2021. "Cyclic Derivatives of the Chemerin C-Terminus as Metabolically Stable Agonists at the Chemokine-like Receptor 1 for Cancer Treatment" Cancers 13, no. 15: 3788. https://doi.org/10.3390/cancers13153788
APA StyleFischer, T. F., Czerniak, A. S., Weiß, T., Zellmann, T., Zielke, L., Els-Heindl, S., & Beck-Sickinger, A. G. (2021). Cyclic Derivatives of the Chemerin C-Terminus as Metabolically Stable Agonists at the Chemokine-like Receptor 1 for Cancer Treatment. Cancers, 13(15), 3788. https://doi.org/10.3390/cancers13153788