
Expression of CCRL2 Inhibits Tumor Growth by Concentrating Chemerin and Inhibiting Neoangiogenesis

 ,
,  , , and
, , and 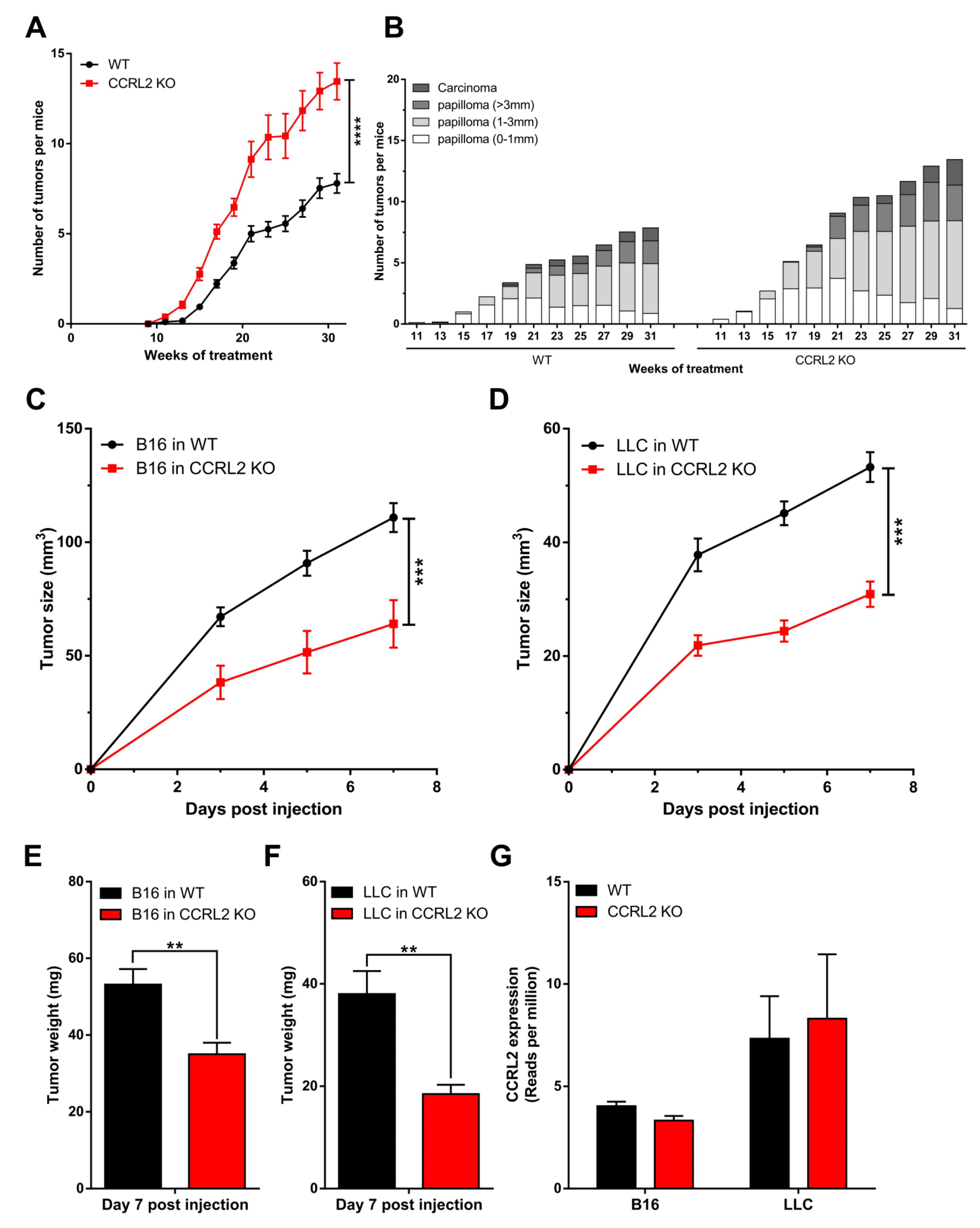{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Cell Lines
2.3. Knockout of Ccrl2 in Tumoral Cell Lines
2.4. Overexpression of CCRL2 in Tumoral Cell Lines
2.5. Proliferation Assays
2.6. Chemerin Presentation Assay
2.7. GFP-Chemerin Binding Assay
2.8. Tumor Models
2.9. Flow Cytometry Analysis
2.10. Histological Procedures
2.11. RT-qPCR
2.12. RNA Sequencing and Transcriptome Analysis
2.13. Statistical Analyses
3. Results
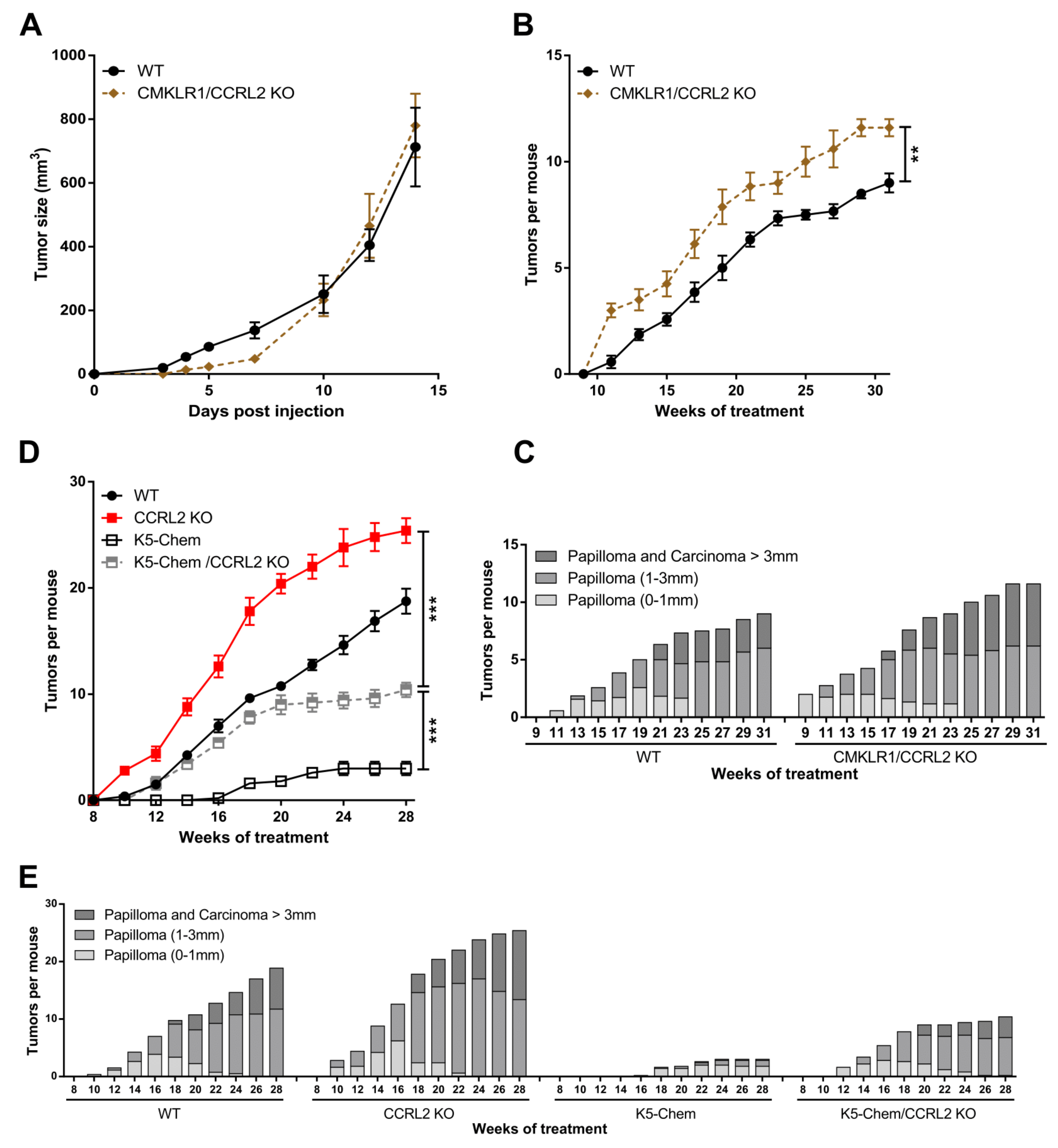
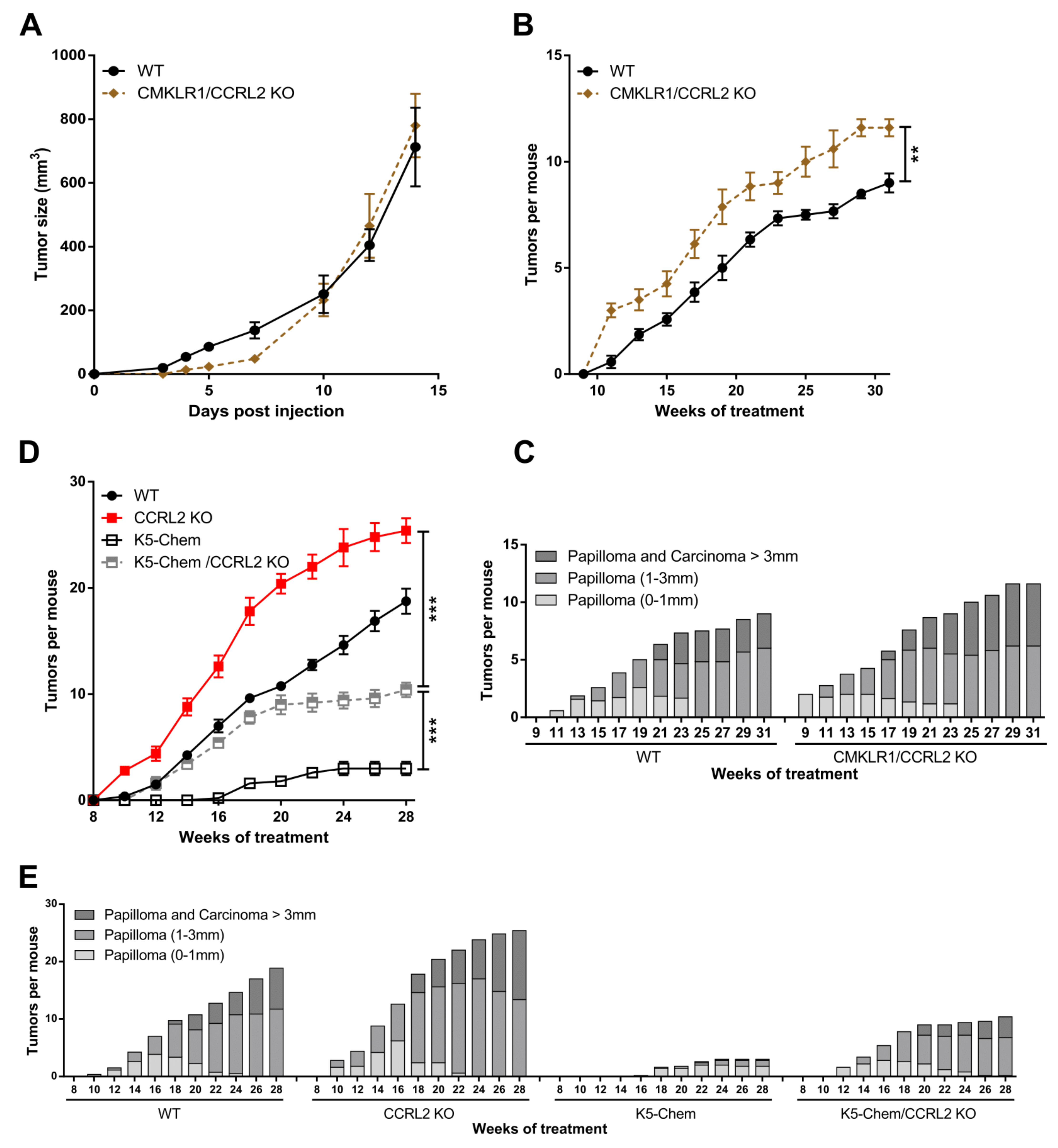
3.1. CCRL2 Deficiency Accelerates Tumor Progression in a Chemical Model of Skin Carcinogenesis and Delays Tumor Growth in Graft Models
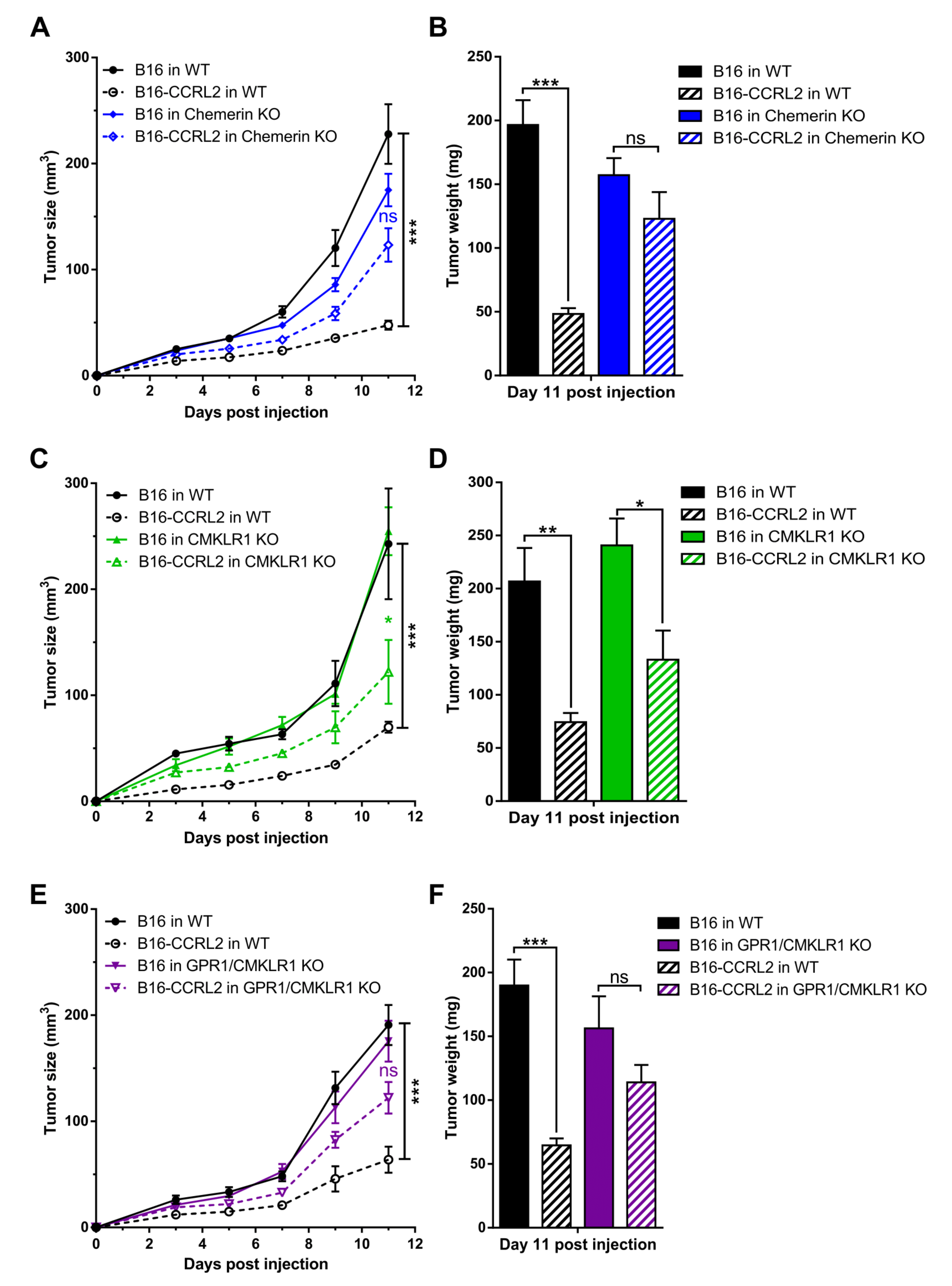
3.2. The Consequences of Ccrl2 Loss of Function Are Linked to the Chemerin/CMKLR1 System
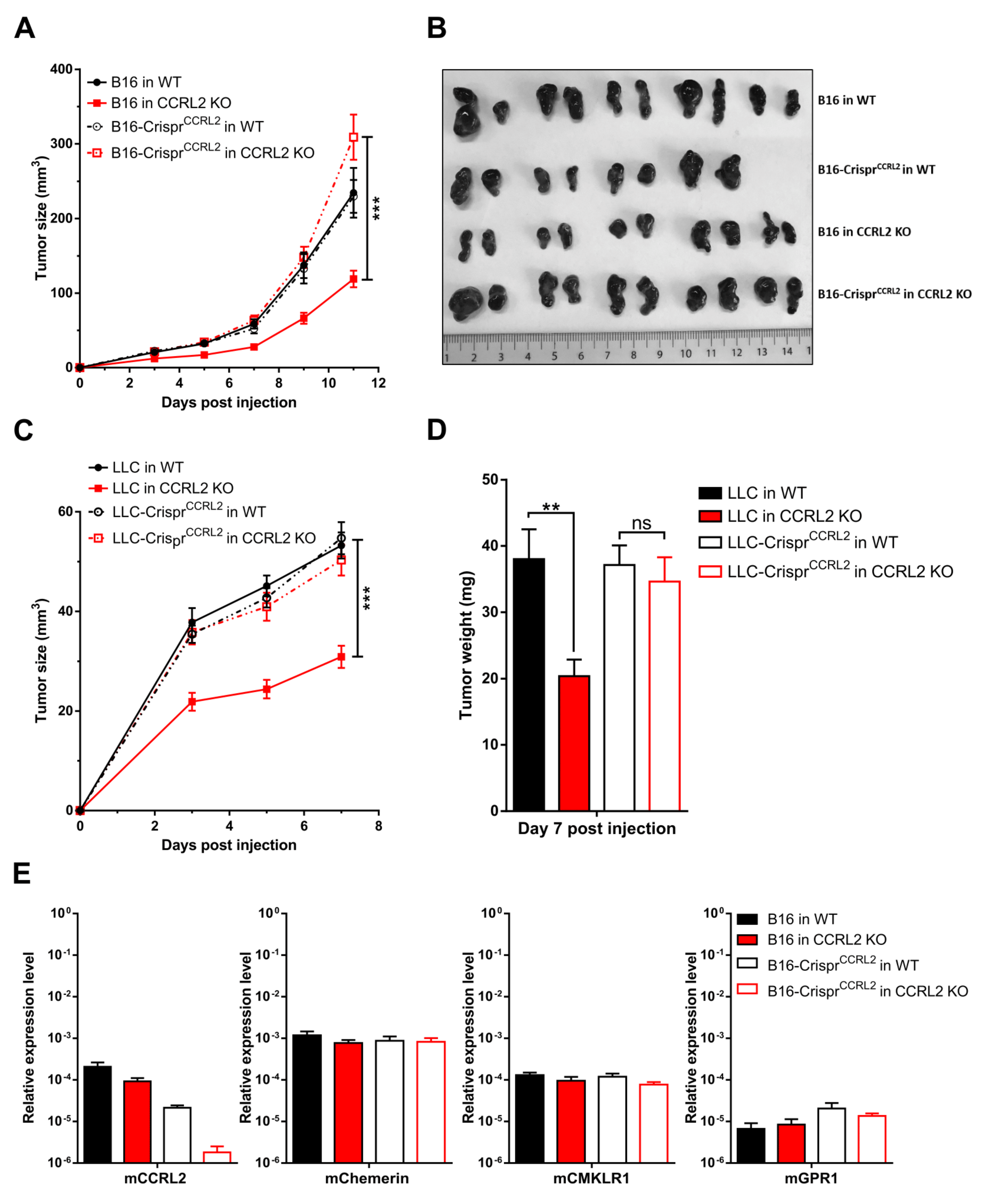
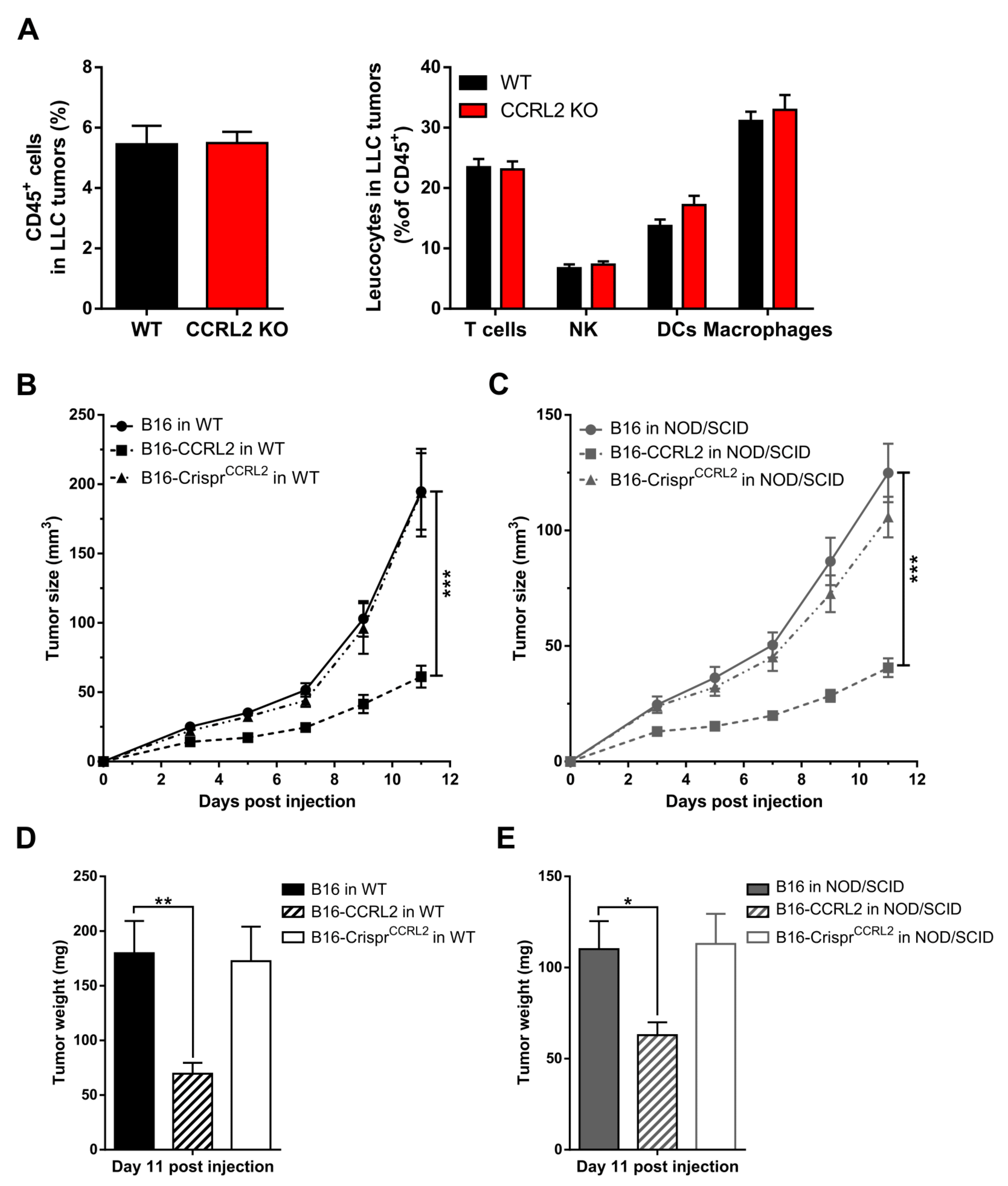
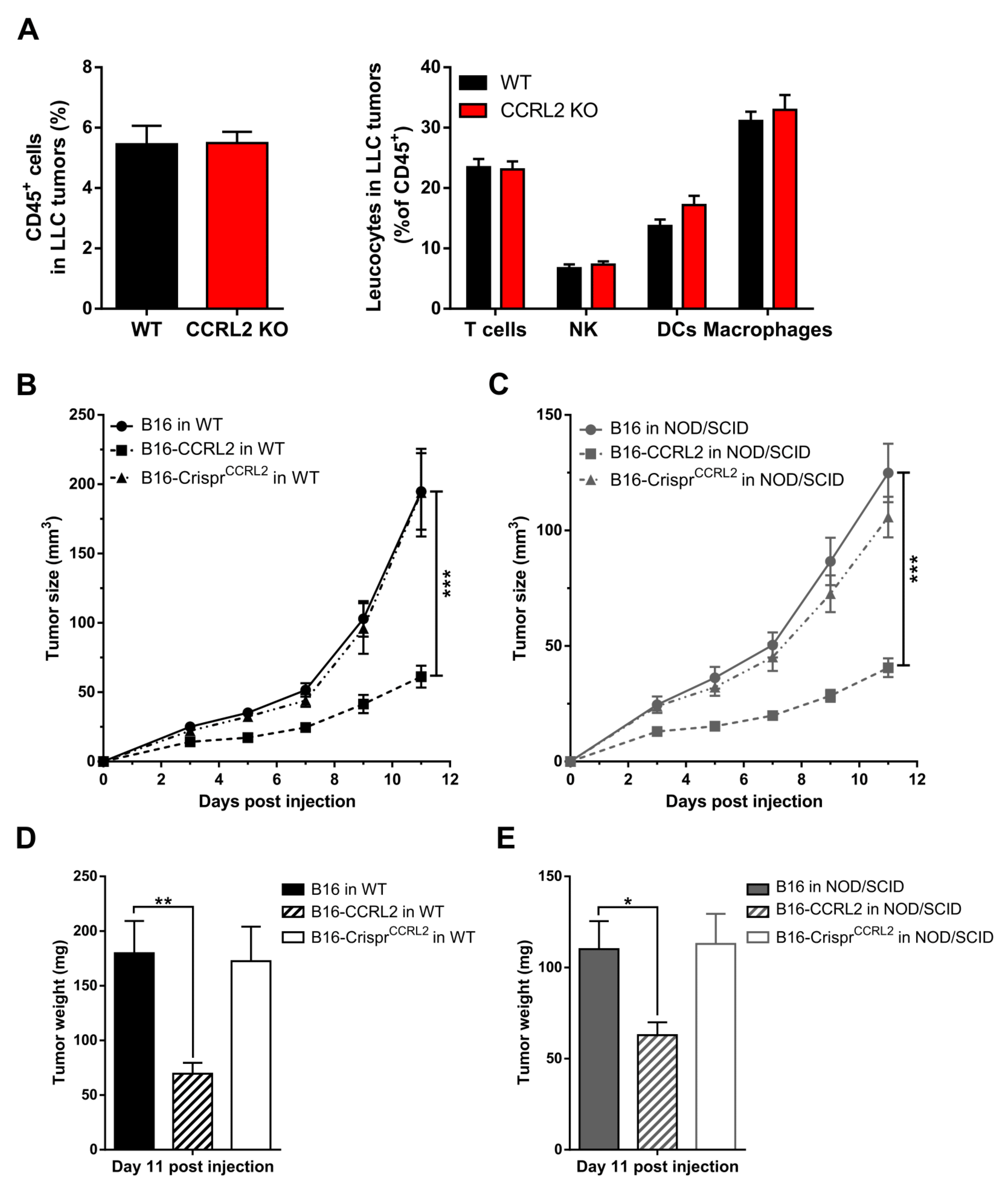
3.3. Knocking out Ccrl2 in Tumor Cells Restores the Growth of Tumor Grafts in Ccrl2 KO Mice
3.4. CCRL2 Overexpression by Tumor Cells Delays Tumor Growth
3.5. Overexpression or Knockout of Ccrl2 Do Not Affect Proliferation of B16 and LLC Cells Ex Vivo
3.6. The Recruitment of Immune Cells to Tumors Is Not Affected in Ccrl2 KO Mice
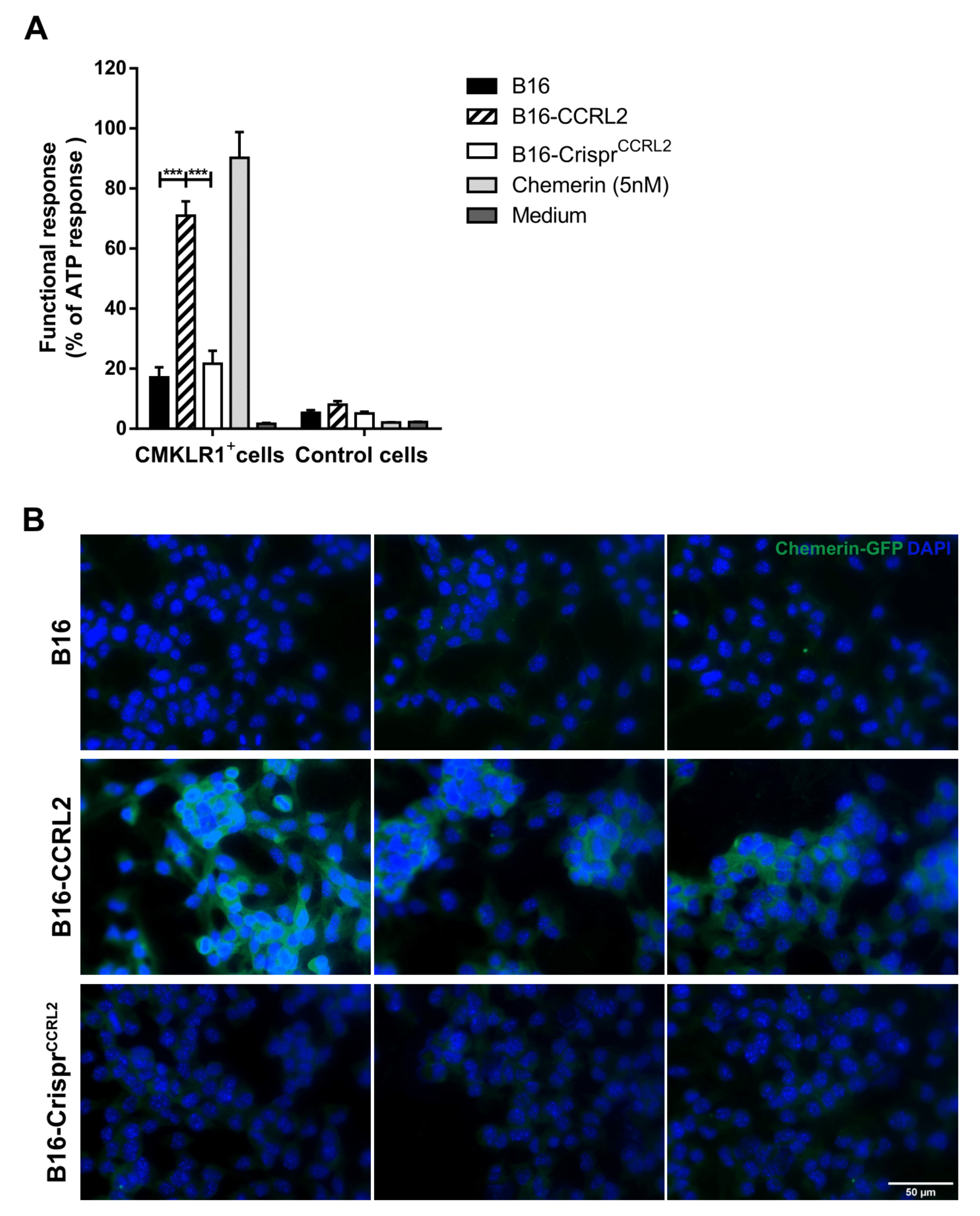
3.7. CCRL2-Expressing Cells Concentrate Chemerin and Activate CMKLR1+ Cells
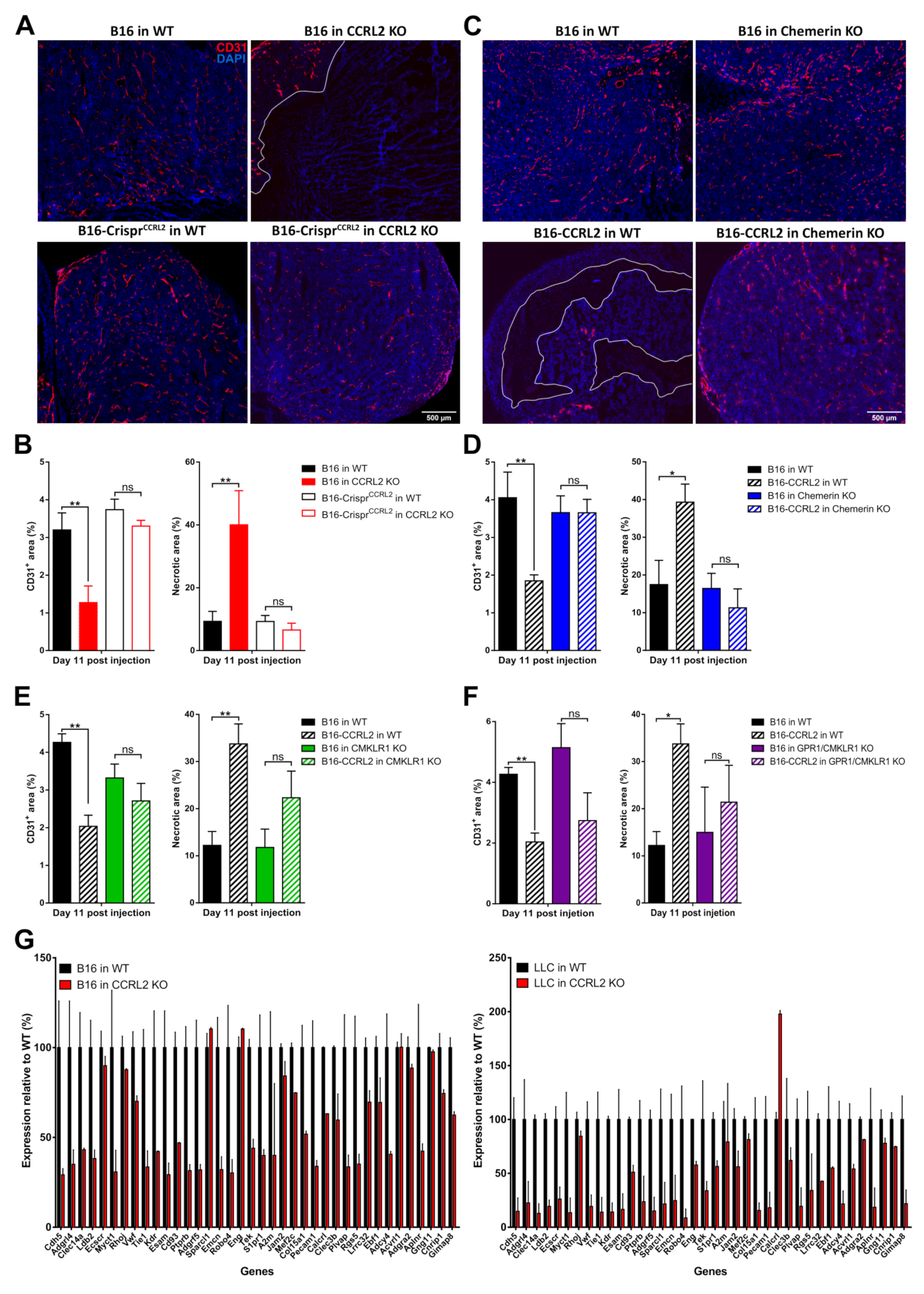
3.8. CCRL2 Expression by Tumor Cells Impairs Tumor Vascularization
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A. The chemokine system: Redundancy for robust outputs. Immunol. Today 1999, 20, 254–257. [Google Scholar] [CrossRef]
- Bondue, B.; Wittamer, V.; Parmentier, M. Chemerin and its receptors in leukocyte trafficking, inflammation and metabolism. Cytokine Growth Factor Rev. 2011, 22, 331–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, A.; Davenport, A.P. International Union of Basic and Clinical Pharmacology CIII: Chemerin Receptors CMKLR1 (Chemerin1) and GPR1 (Chemerin2) Nomenclature, Pharmacology, and Function. Pharmacol. Rev. 2017, 70, 174–196. [Google Scholar] [CrossRef] [Green Version]
- Goralski, K.B.; Jackson, A.E.; McKeown, B.T.; Sinal, C.J. More Than an Adipokine: The Complex Roles of Chemerin Signaling in Cancer. Int. J. Mol. Sci. 2019, 20, 4778. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Luo, S.; Wang, G.; Peng, Z.; Zeng, W.; Tan, S.; Xi, Y.; Fan, J. Downregulation of tazarotene induced gene-2 (TIG2) in skin squamous cell carcinoma. Eur. J. Dermatol. 2008, 18, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Li, C.; Ye, Y.-B.; Peng, F.; Chen, Q. Expression of Chemerin Correlates with a Favorable Prognosis in Patients with Non-Small Cell Lung Cancer. Lab. Med. 2011, 42, 553–557. [Google Scholar] [CrossRef]
- Pachynski, R.; Zabel, B.A.; Kohrt, H.E.; Tejeda, N.M.; Monnier, J.; Swanson, C.D.; Holzer, A.K.; Gentles, A.J.; Sperinde, G.V.; Edalati, A.; et al. The chemoattractant chemerin suppresses melanoma by recruiting natural killer cell antitumor defenses. J. Exp. Med. 2012, 209, 1427–1435. [Google Scholar] [CrossRef]
- Treeck, O.; Buechler, C.; Ortmann, O. Chemerin and Cancer. Int. J. Mol. Sci. 2019, 20, 3750. [Google Scholar] [CrossRef] [Green Version]
- Dubois-Vedrenne, I.; Al Delbany, D.; De Henau, O.; Robert, V.; Vernimmen, M.; Langa, F.; Lefort, A.; Libert, F.; Wittamer, V.; Parmentier, M. The antitumoral effects of chemerin are independent from leukocyte recruitment and mediated by inhibition of neoangiogenesis. Oncotarget 2021, 12, 1903–1919. [Google Scholar] [CrossRef] [PubMed]
- Bonecchi, R.; Graham, G.J. Atypical Chemokine Receptors and Their Roles in the Resolution of the Inflammatory Response. Front. Immunol. 2016, 7, 224. [Google Scholar] [CrossRef] [Green Version]
- Massara, M.; Bonavita, O.; Mantovani, A.; Locati, M.; Bonecchi, R. Atypical chemokine receptors in cancer: Friends or foes? J. Leukoc. Biol. 2016, 99, 927–933. [Google Scholar] [CrossRef] [Green Version]
- Sjöberg, E.; Meyrath, M.; Chevigné, A.; Östman, A.; Augsten, M.; Szpakowska, M. The diverse and complex roles of atypical chemokine receptors in cancer: From molecular biology to clinical relevance and therapy. Adv. Cancer Res. 2020, 145, 99–138. [Google Scholar] [CrossRef]
- Zabel, B.A.; Nakae, S.; Zúñiga, L.; Kim, J.-Y.; Ohyama, T.; Alt, C.; Pan, J.; Suto, H.; Soler, D.; Allen, S.J.; et al. Mast cell–expressed orphan receptor CCRL2 binds chemerin and is required for optimal induction of IgE-mediated passive cutaneous anaphylaxis. J. Exp. Med. 2008, 205, 2207–2220. [Google Scholar] [CrossRef]
- Monnier, J.; Lewén, S.; O’Hara, E.; Huang, K.; Tu, H.; Butcher, E.C.; Zabel, B.A. Expression, Regulation, and Function of Atypical Chemerin Receptor CCRL2 on Endothelial Cells. J. Immunol. 2012, 189, 956–967. [Google Scholar] [CrossRef] [Green Version]
- Schioppa, T.; Sozio, F.; Barbazza, I.; Scutera, S.; Bosisio, D.; Sozzani, S.; Del Prete, A. Molecular Basis for CCRL2 Regulation of Leukocyte Migration. Front. Cell Dev. Biol. 2020, 8, 615031. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Xu, Z.; Wang, Z.; Yao, H.; Shen, Z.; Yu, F.; Tang, Y.; Fu, D.; Lin, S.; Lu, G.; et al. Elevated chemokine CC-motif receptor-like 2 (CCRL2) promotes cell migration and invasion in glioblastoma. Biochem. Biophys. Res. Commun. 2012, 429, 168–172. [Google Scholar] [CrossRef]
- Wang, L.-P.; Cao, J.; Zhang, J.; Wang, B.-Y.; Hu, X.-C.; Shao, Z.-M.; Wang, Z.-H.; Ou, Z.-L. The human chemokine receptor CCRL2 suppresses chemotaxis and invasion by blocking CCL2-induced phosphorylation of p38 MAPK in human breast cancer cells. Med. Oncol. 2015, 32, 254. [Google Scholar] [CrossRef] [PubMed]
- Akram, I.G.; Georges, R.; Hielscher, T.; Adwan, H.; Berger, M.R. The chemokines CCR1 and CCRL2 have a role in colorectal cancer liver metastasis. Tumor Biol. 2015, 37, 2461–2471. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, A.; Sozio, F.; Schioppa, T.; Ponzetta, A.; Vermi, W.; Calza, S.; Bugatti, M.; Salvi, V.; Bernardini, G.; Benvenuti, F.; et al. The Atypical Receptor CCRL2 Is Essential for Lung Cancer Immune Surveillance. Cancer Immunol. Res. 2019, 7, 1775–1788. [Google Scholar] [CrossRef]
- Otero, K.; Vecchi, A.; Hirsch, E.; Kearley, J.; Vermi, W.; Prete, A.D.; Gonzalvo-Feo, S.; Garlanda, C.; Azzolino, O.; Salogni, L.; et al. Europe PMC Funders Group Non-redundant role of CCRL2 in lung dendritic cell trafficking. Blood 2012, 116, 2942–2949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luangsay, S.; Wittamer, V.; Bondue, B.; De Henau, O.; Rouger, L.; Brait, M.; Franssen, J.-D.; De Nadai, P.; Huaux, F.; Parmentier, M. Mouse ChemR23 Is Expressed in Dendritic Cell Subsets and Macrophages, and Mediates an Anti-Inflammatory Activity of Chemerin in a Lung Disease Model. J. Immunol. 2009, 183, 6489–6499. [Google Scholar] [CrossRef]
- Dubois-Vedrenne, I.; De Henau, O.; Robert, V.; Langa, F.; Javary, J.; Al Delbany, D.; Vosters, O.; Canals, E.A.; Vernimmen, M.; Luangsay, S.; et al. Expression of Bioactive Chemerin by Keratinocytes Inhibits Late Stages of Tumor Development in a Chemical Model of Skin Carcinogenesis. Front. Oncol. 2019, 9, 1253. [Google Scholar] [CrossRef] [PubMed]
- Danciu, C.; Falamas, A.; Dehelean, C.; Soica, C.; Radeke, H.; Barbu-Tudoran, L.; Bojin, F.; Pînzaru, S.C.; Munteanu, M.F. A characterization of four B16 murine melanoma cell sublines molecular fingerprint and proliferation behavior. Cancer Cell Int. 2013, 13, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittamer, V.; Franssen, J.-D.; Vulcano, M.; Mirjolet, J.-F.; Le Poul, E.; Migeotte, I.; Brézillon, S.; Tyldesley, R.; Blanpain, C.; Detheux, M.; et al. Specific Recruitment of Antigen-presenting Cells by Chemerin, a Novel Processed Ligand from Human Inflammatory Fluids. J. Exp. Med. 2003, 198, 977–985. [Google Scholar] [CrossRef]
- Bondue, B.; De Henau, O.; Luangsay, S.; Devosse, T.; de Nadai, P.; Springael, J.-Y.; Parmentier, M.; Vosters, O. The Chemerin/ChemR23 System Does Not Affect the Pro-Inflammatory Response of Mouse and Human Macrophages Ex Vivo. PLoS ONE 2012, 7, e40043. [Google Scholar] [CrossRef] [Green Version]
- Peyrassol, X.; Laeremans, T.; Gouwy, M.; Lahura, V.; Debulpaep, M.; Van Damme, J.; Steyaert, J.; Parmentier, M.; Langer, I. Development by Genetic Immunization of Monovalent Antibodies (Nanobodies) Behaving as Antagonists of the Human ChemR23 Receptor. J. Immunol. 2016, 196, 2893–2901. [Google Scholar] [CrossRef] [Green Version]
- Abel, E.; Angel, J.M.; Kiguchi, K.; DiGiovanni, J. Multi-stage chemical carcinogenesis in mouse skin: Fundamentals and applications. Nat. Protoc. 2009, 4, 1350–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Chow, M.T.; Luster, A.D. Chemokines in Cancer. Cancer Immunol. Res. 2014, 2, 1125–1131. [Google Scholar] [CrossRef] [Green Version]
- Marcuzzi, E.; Angioni, R.; Molon, B.; Calì, B. Chemokines and Chemokine Receptors: Orchestrating Tumor Metastasization. Int. J. Mol. Sci. 2018, 20, 96. [Google Scholar] [CrossRef] [Green Version]
- Del Prete, A.; Bonecchi, R.; Vecchi, A.; Mantovani, A.; Sozzani, S. CCRL2, a fringe member of the atypical chemoattractant receptor family. Eur. J. Immunol. 2013, 43, 1418–1422. [Google Scholar] [CrossRef]
- Shimada, T.; Matsumoto, M.; Tatsumi, Y.; Kanamaru, A.; Akira, S. A novel lipopolysaccharide inducible C-C chemokine receptor related gene in murine macrophages. FEBS Lett. 1998, 425, 490–494. [Google Scholar] [CrossRef] [Green Version]
- Del Prete, A.; Locati, M.; Otero, K.; Riboldi, E.; Mantovani, A.; Vecchi, A.; Sozzani, S. Migration of dendritic cells across blood and lymphatic endothelial barriers. Thromb. Haemost. 2006, 95, 22–28. [Google Scholar] [CrossRef] [Green Version]
- Gonzalvo-Feo, S.; Del Prete, A.; Pruenster, M.; Salvi, V.; Wang, L.; Sironi, M.; Bierschenk, S.; Sperandio, M.; Vecchi, A.; Sozzani, S. Endothelial Cell–Derived Chemerin Promotes Dendritic Cell Transmigration. J. Immunol. 2014, 192, 2366–2373. [Google Scholar] [CrossRef] [Green Version]
- Del Prete, A.; Muñoz, L.M.; Mazzon, C.; Toffali, L.; Sozio, F.; Za, L.; Bosisio, D.; Gazzurelli, L.; Salvi, V.; Tiberio, L.; et al. The atypical receptor CCRL2 is required for CXCR2-dependent neutrophil recruitment and tissue damage. Blood 2017, 130, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Mazzotti, C.; Gagliostro, V.; Bosisio, D.; Del Prete, A.; Tiberio, L.; Thelen, M.; Sozzani, S. The Atypical Receptor CCRL2 (C-C Chemokine Receptor-Like 2) Does Not Act as a Decoy Receptor in Endothelial Cells. Front. Immunol. 2017, 8, 1233. [Google Scholar] [CrossRef] [Green Version]
- De Henau, O.; DeGroot, G.-N.; Imbault, V.; Robert, V.; De Poorter, C.; Mcheik, S.; Gales, C.; Parmentier, M.; Springael, J.-Y. Signaling Properties of Chemerin Receptors CMKLR1, GPR1 and CCRL2. PLoS ONE 2016, 11, e0164179. [Google Scholar] [CrossRef] [PubMed]
- Masiero, M.; Simões, F.; Han, H.D.; Snell, C.; Peterkin, T.; Bridges, E.; Mangala, L.S.; Wu, S.; Pradeep, S.; Li, D.; et al. A Core Human Primary Tumor Angiogenesis Signature Identifies the Endothelial Orphan Receptor ELTD1 as a Key Regulator of Angiogenesis. Cancer Cell 2013, 24, 229–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzon, C.; Zanotti, L.; Wang, L.; Del Prete, A.; Fontana, E.; Salvi, V.; Poliani, P.L.; Sozzani, S. CCRL2 regulates M1/M2 polarization during EAE recovery phase. J. Leukoc. Biol. 2016, 99, 1027–1033. [Google Scholar] [CrossRef] [Green Version]
- Reyes, N.; Benedetti, I.; Rebollo, J.; Correa, O.; Geliebter, J. Atypical chemokine receptor CCRL2 is overexpressed in prostate cancer cells. J. Biomed. Res. 2018, 33, 17–23. [Google Scholar] [CrossRef]
- Ben Dhaou, C.; Mandi, K.; Frye, M.; Acheampong, A.; Radi, A.; De Becker, B.; Antoine, M.; Baeyens, N.; Wittamer, V.; Parmentier, M. Chemerin regulates normal angiogenesis and hypoxia-driven neovascularization. Angiogenesis 2021, 1–21. [Google Scholar] [CrossRef]
- Bozaoglu, K.; Curran, J.E.; Stocker, C.J.; Zaibi, M.S.; Segal, D.; Konstantopoulos, N.; Morrison, S.; Carless, M.; Dyer, T.D.; Cole, S.A.; et al. Chemerin, a Novel Adipokine in the Regulation of Angiogenesis. J. Clin. Endocrinol. Metab. 2010, 95, 2476–2485. [Google Scholar] [CrossRef] [Green Version]
- Kaur, J.; Adya, R.; Tan, B.K.; Chen, J.; Randeva, H.S. Identification of chemerin receptor (ChemR23) in human endothelial cells: Chemerin-induced endothelial angiogenesis. Biochem. Biophys. Res. Commun. 2010, 391, 1762–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, N.; Naruse, K.; Kobayashi, Y.; Miyabe, M.; Saiki, T.; Enomoto, A.; Takahashi, M.; Matsubara, T. Chemerin promotes angiogenesis in vivo. Physiol. Rep. 2018, 6, e13962. [Google Scholar] [CrossRef] [PubMed]
- Wittamer, V.; Bondue, B.; Guillabert, A.; Vassart, G.; Parmentier, M.; Communi, D. Neutrophil-Mediated Maturation of Chemerin: A Link between Innate and Adaptive Immunity. J. Immunol. 2005, 175, 487–493. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Delbany, D.; Robert, V.; Dubois-Vedrenne, I.; Del Prete, A.; Vernimmen, M.; Radi, A.; Lefort, A.; Libert, F.; Wittamer, V.; Sozzani, S.; et al. Expression of CCRL2 Inhibits Tumor Growth by Concentrating Chemerin and Inhibiting Neoangiogenesis. Cancers 2021, 13, 5000. https://doi.org/10.3390/cancers13195000
Al Delbany D, Robert V, Dubois-Vedrenne I, Del Prete A, Vernimmen M, Radi A, Lefort A, Libert F, Wittamer V, Sozzani S, et al. Expression of CCRL2 Inhibits Tumor Growth by Concentrating Chemerin and Inhibiting Neoangiogenesis. Cancers. 2021; 13(19):5000. https://doi.org/10.3390/cancers13195000
Chicago/Turabian StyleAl Delbany, Diana, Virginie Robert, Ingrid Dubois-Vedrenne, Annalisa Del Prete, Maxime Vernimmen, Ayoub Radi, Anne Lefort, Frédérick Libert, Valérie Wittamer, Silvano Sozzani, and et al. 2021. "Expression of CCRL2 Inhibits Tumor Growth by Concentrating Chemerin and Inhibiting Neoangiogenesis" Cancers 13, no. 19: 5000. https://doi.org/10.3390/cancers13195000
APA StyleAl Delbany, D., Robert, V., Dubois-Vedrenne, I., Del Prete, A., Vernimmen, M., Radi, A., Lefort, A., Libert, F., Wittamer, V., Sozzani, S., & Parmentier, M. (2021). Expression of CCRL2 Inhibits Tumor Growth by Concentrating Chemerin and Inhibiting Neoangiogenesis. Cancers, 13(19), 5000. https://doi.org/10.3390/cancers13195000