
Emerging Insights into Keratin 16 Expression during Metastatic Progression of Breast Cancer

 , , ,
, , ,  and
and
Abstract
:Simple Summary
Abstract
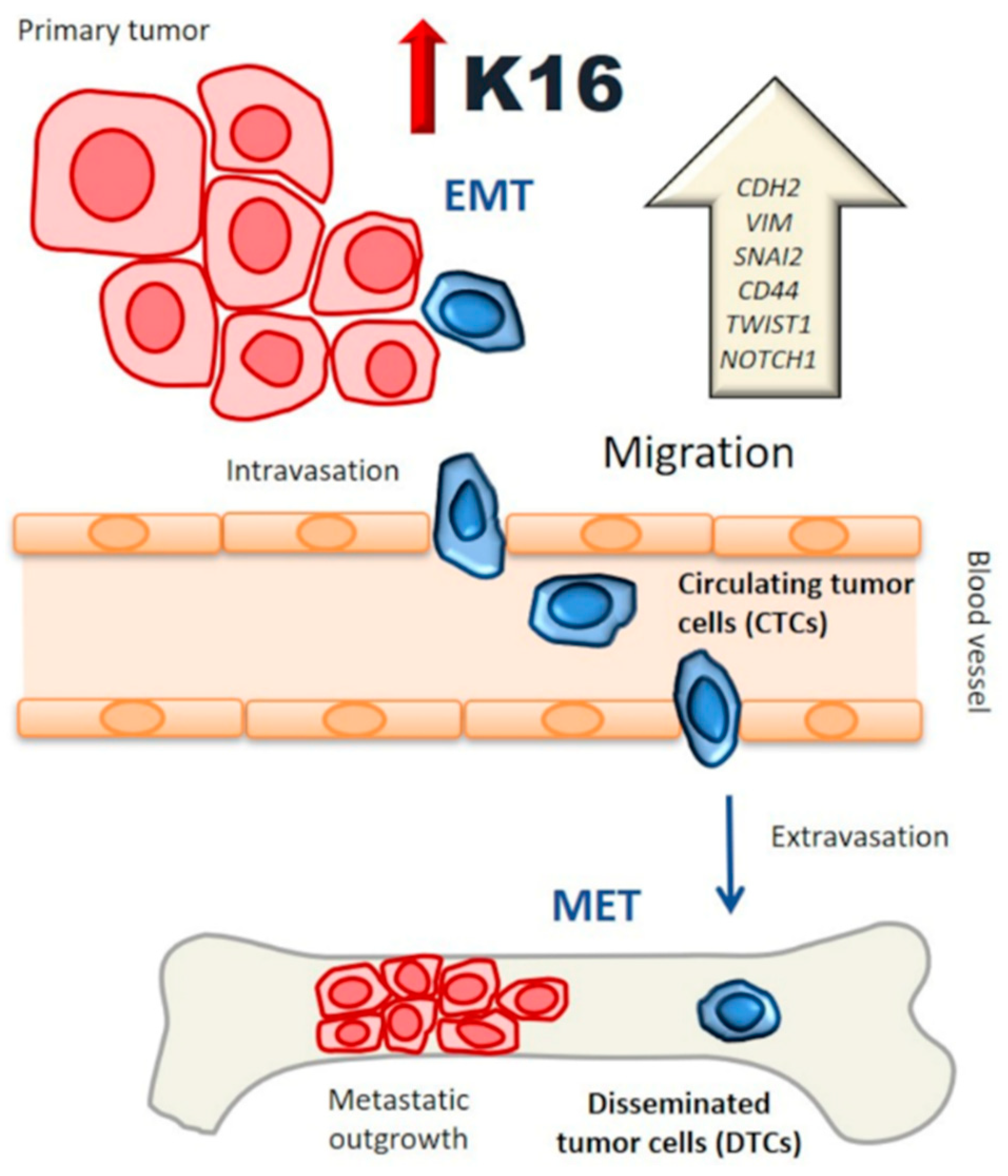
1. Introduction
2. Materials and Methods
2.1. In Silico Analysis
2.2. Cell Culture
2.3. EMT Induction Assay
2.4. KRT16 Overexpression
2.5. KRT16 Knockdown
2.6. Protein Level Assessment
2.7. Quantitative Transcript Analysis
2.8. Migration Assay
2.9. Proliferation Assay
2.10. Immunocytochemical Staining
2.11. Blood Collection and Processing
2.12. Enrichment of CTCs using a Spiral Microfluidic Chip
2.13. CTC Immunophenotyping
2.14. Statistical Analyses
3. Results
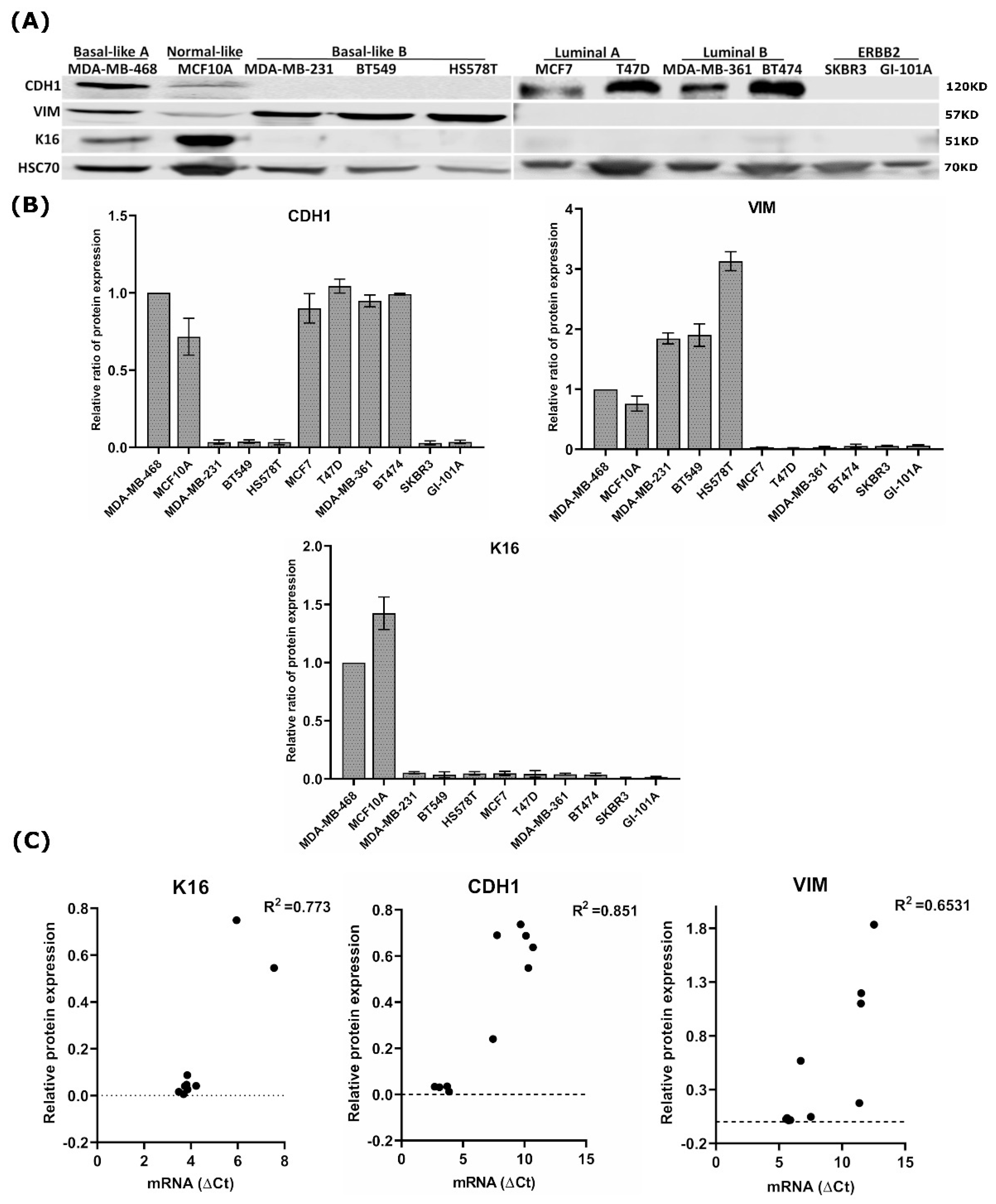
3.1. Keratin 16 Is Overexpressed in the Basal-Like Breast Cancer Subtype
3.2. EMT Induction Leads to Overexpression of K16
3.3. Overexpression of KRT16 Leads to EMT Induction
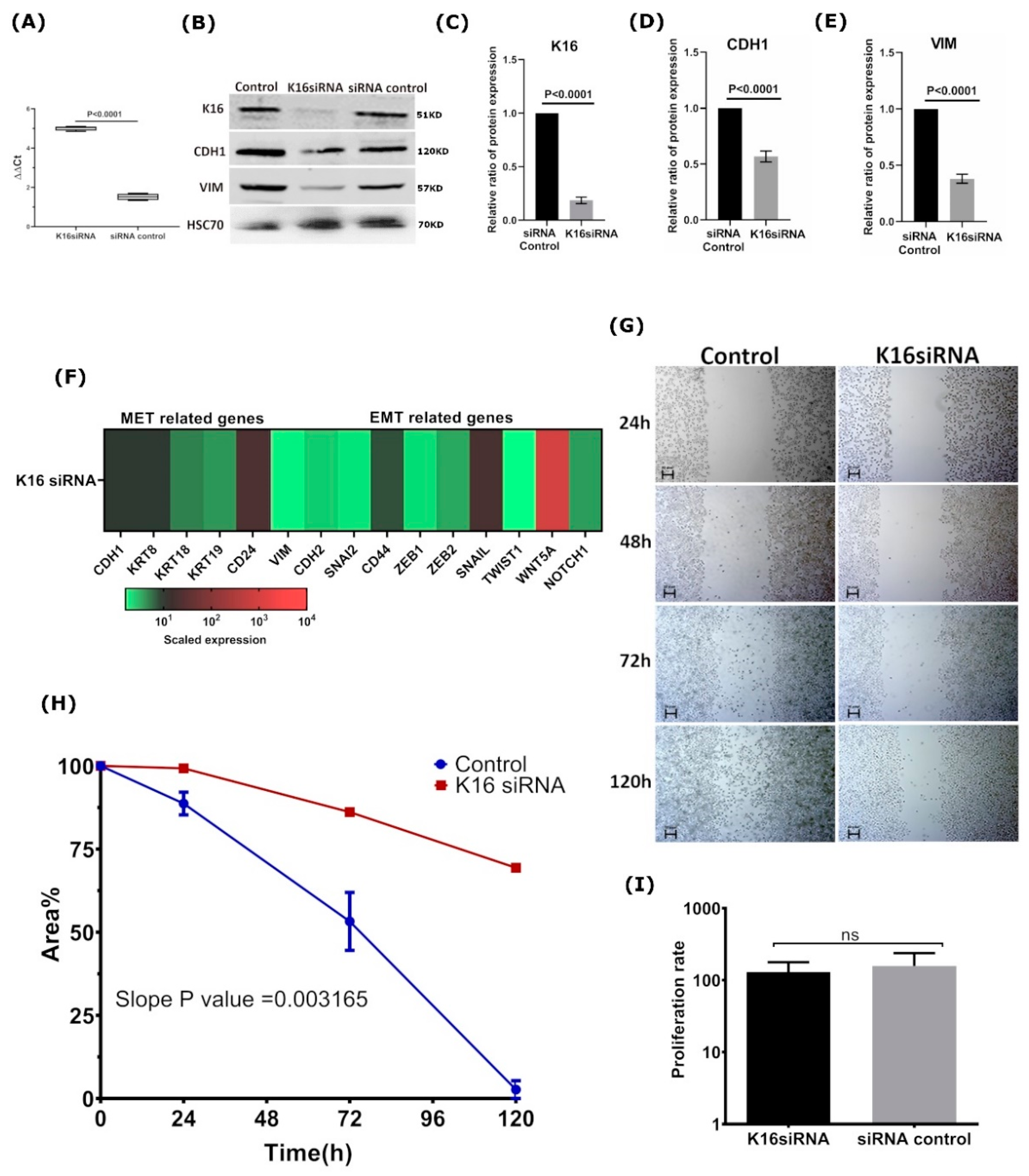
3.4. K16 Knockdown Changes the Mesenchymal Phenotype
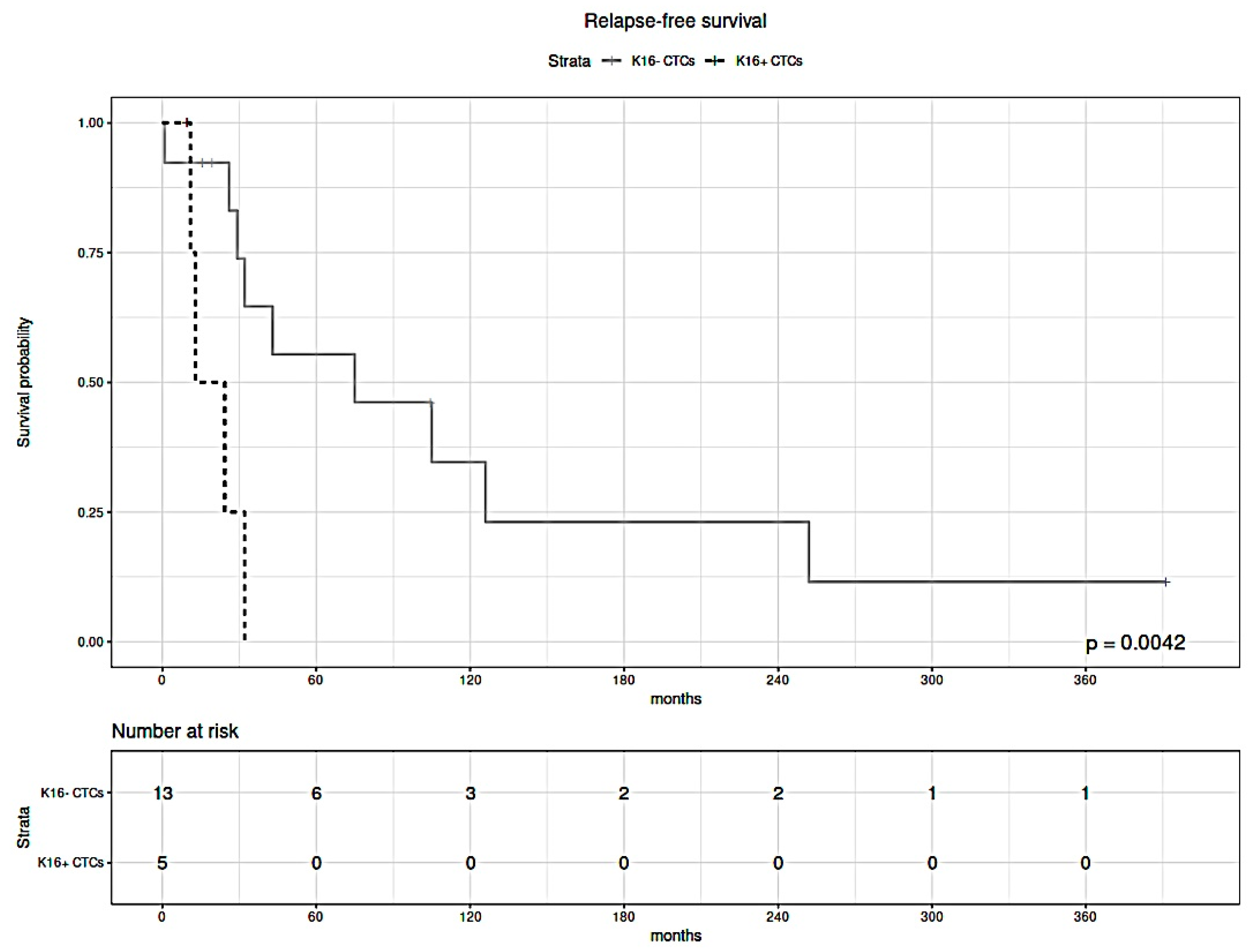
3.5. K16 Expression in CTCs Correlates with a Worse Survival
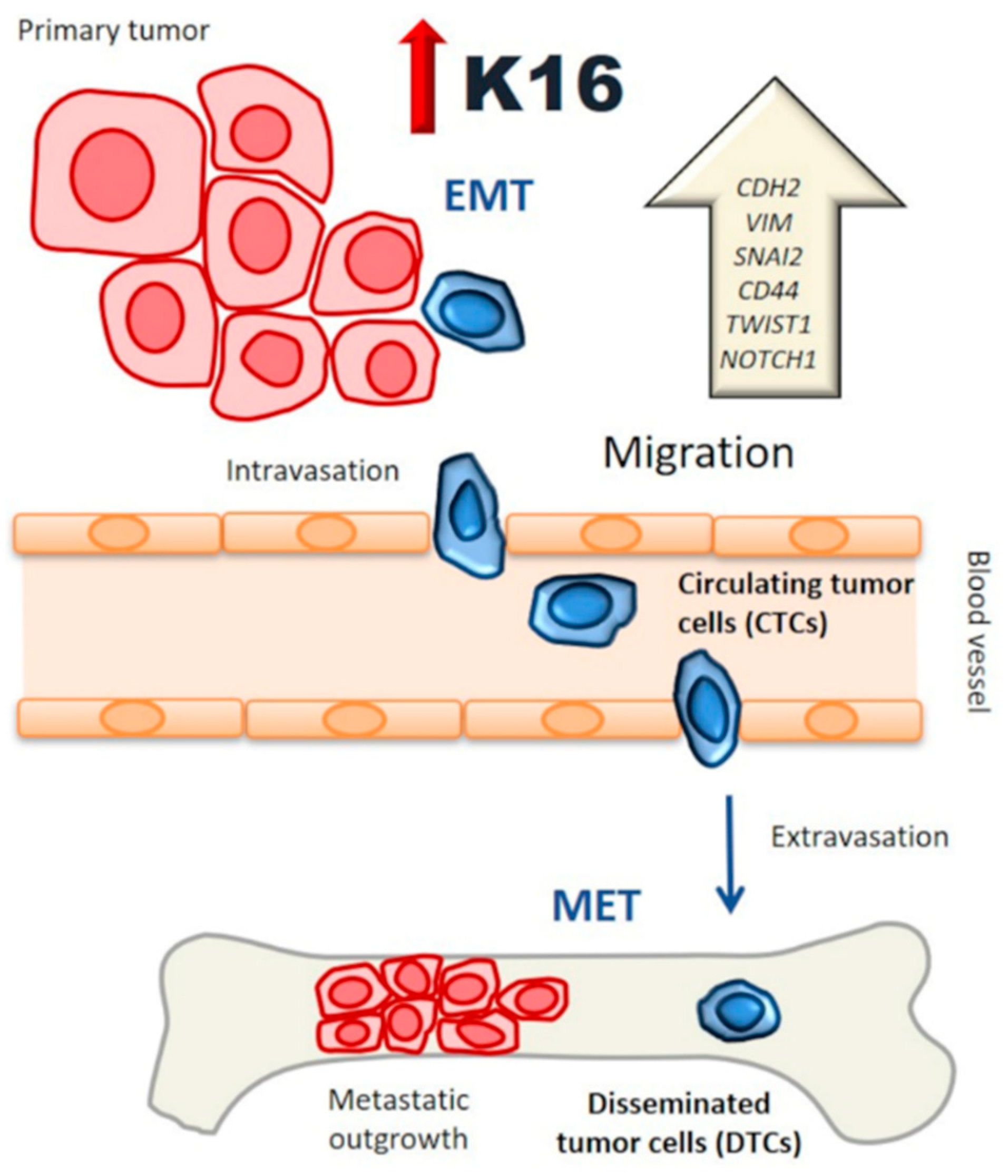
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantel, K.; Hayes, D.F. Disseminated breast tumour cells: Biological and clinical meaning. Nat. Rev. Clin. Oncol. 2017, 15, 129–131. [Google Scholar] [CrossRef]
- Joosse, S.A.; Hannemann, J.; Spötter, J.; Bauche, A.; Andreas, A.; Müller, V.; Pantel, K. Changes in Keratin Expression during Metastatic Progression of Breast Cancer: Impact on the Detection of Circulating Tumor Cells. Clin. Cancer Res. 2012, 18, 993–1003. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.-J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Mader, S.; Pantel, K. Epithelial-mesenchymal plasticity in circulating tumor cells. J. Mol. Med. 2016, 95, 133–142. [Google Scholar] [CrossRef]
- Williams, E.D.; Gao, D.; Redfern, A.; Thompson, E.W. Controversies around epithelial–mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 2019, 19, 716–732. [Google Scholar] [CrossRef] [Green Version]
- Werner, S.; Keller, L.; Pantel, K. Epithelial keratins: Biology and implications as diagnostic markers for liquid biopsies. Mol. Asp. Med. 2020, 72, 100817. [Google Scholar] [CrossRef]
- Moll, R.; Divo, M.; Langbein, L. The human keratins: Biology and pathology. Histochem. Cell Biol. 2008, 129, 705–733. [Google Scholar] [CrossRef] [Green Version]
- Trost, A.; Costa, I.; Jakab, M.; Ritter, M.; Haim, M.; Hintner, H.; Bauer, J.W.; Önder, K. K16 is a further new candidate for homotypic intermediate filament protein interactions. Exp. Dermatol. 2009, 19, e241–e250. [Google Scholar] [CrossRef]
- Wawersik, M.J.; Mazzalupo, S.; Nguyen, D.; Coulombe, P.A. Increased Levels of Keratin 16 Alter Epithelialization Potential of Mouse Skin Keratinocytes In Vivo and Ex Vivo. Mol. Biol. Cell 2001, 12, 3439–3450. [Google Scholar] [CrossRef] [Green Version]
- Rotty, J.D.; Coulombe, P.A. A wound-induced keratin inhibits Src activity during keratinocyte migration and tissue repair. J. Cell Biol. 2012, 197, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Wawersik, M.; Coulombe, P.A. Forced Expression of Keratin 16 Alters the Adhesion, Differentiation, and Migration of Mouse Skin Keratinocytes. Mol. Biol. Cell 2000, 11, 3315–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.-P.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [CrossRef] [Green Version]
- Young, L.; Sung, J.; Stacey, G.; Masters, J.R. Detection of Mycoplasma in cell cultures. Nat. Protoc. 2010, 5, 929–934. [Google Scholar] [CrossRef] [PubMed]
- System, R.D. a biotechne. Brand-manual.pdf. StemXVivo® EMT Inducing Media Supplement (100X). p. Catalog Number: CCM017. Available online: https://resources.rndsystems.com/pdfs/datasheets/ccm017.pdf?v=20210506&_ga=2.157864314.1993146906.1620297005-771850332.1620297004. (accessed on 30 July 2011).
- Tang, Y.; Herr, G.; Johnson, W.; Resnik, E.; Aho, J. Induction and Analysis of Epithelial to Mesenchymal Transition. J. Vis. Exp. 2013, 2013, e50478. [Google Scholar] [CrossRef] [Green Version]
- GenScript. User Manual_GenEZ ORF Clone Products.pdf. Available online: https://www.genscript.com/gsfiles/GenEZ%20ORF%20Clone%20Technical%20Manual.pdf. (accessed on 31 July 2018).
- Li, X.; Yang, H.; Huang, H.; Zhu, T. CELLCOUNTER: Novel Open-Source Software for Counting Cell Migration and Invasion In Vitro. BioMed Res. Int. 2014, 2014, 863564. [Google Scholar] [CrossRef] [PubMed]
- Life technologies, Lipofectamine_RNAiMAX_Reag_protocol.pdf. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/Lipofectamine_RNAiMAX_Reag_protocol.pdf. (accessed on 30 July 2013).
- ThermoScientific, MAN0011430_Pierce_BCA_Protein_Asy_UG.pdf. Available online: https://www.thermofisher.com/document-connect/document-connect.html?url=https%3A%2F%2Fassets.thermofisher.com%2FTFS-Assets%2FLSG%2Fmanuals%2FMAN0011430_Pierce_BCA_Protein_Asy_UG.pdf&title=VXNlciBHdWlkZTogUGllcmNlIEJDQSBQcm90ZWluIEFzc2F5IEtpdA== (accessed on 30 June 2015).
- C-DiGit Blot Scanner--manual.pdf. Available online: https://www.licor.com/documents/uhcazdtkjwc3xax5yqtm6xcaiqtfdqva. (accessed on 25 May 2013).
- NucleoSpin RNA XS Total RNA Isolation User Manual_Rev_09.pdf. Available online: https://www.takarabio.com/documents/User%20Manual/NucleoSpin%20RNA%20XS%20Total%20RNA%20Isolation%20User%20Manual/NucleoSpin%20RNA%20XS%20Total%20RNA%20Isolation%20User%20Manual_Rev_09.pdf. (accessed on 24 July 2015).
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Suarez-Arnedo, A.; Figueroa, F.T.; Clavijo, C.; Arbeláez, P.; Cruz, J.C.; Muñoz-Camargo, C. An image J plugin for the high throughput image analysis of in vitro scratch wound healing assays. PLoS ONE 2020, 15, e0232565. [Google Scholar] [CrossRef] [PubMed]
- LF_biochemlab_BeckmanCoulter_Vi-cell_datasheet_EN.pdf. Available online: http://www.vtpup.cz/common/manual/LF_biochemlab_BeckmanCoulter_Vi-cell_datasheet_EN.pdf. (accessed on 30 May 2010).
- Riebensahm, C.; Joosse, S.A.; Mohme, M.; Hanssen, A.; Matschke, J.; Goy, Y.; Witzel, I.; Lamszus, K.; Kropidlowski, J.; Petersen, C.; et al. Clonality of circulating tumor cells in breast cancer brain metastasis patients. Breast Cancer Res. 2019, 21, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Warkiani, M.E.; Guan, G.; Luan, K.B.; Lee, W.C.; Bhagat, A.A.; Chaudhuri, P.K.; Tan, D.S.-W.; Lim, W.T.; Lee, S.C.; Chen, P.C.Y.; et al. Slanted spiral microfluidics for the ultra-fast, label-free isolation of circulating tumor cells. Lab Chip 2014, 14, 128–137. [Google Scholar] [CrossRef] [Green Version]
- Joosse, S.A. In-Silico Online (Version 2.3.0). 2020. Available online: http://in-silico.online (accessed on 16 June 2020).
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Vasaikar, S.V.; Deshmukh, A.P.; Hollander, P.D.; Addanki, S.; Kuburich, N.A.; Kudaravalli, S.; Joseph, R.; Chang, J.T.; Soundararajan, R.; Mani, S.A. EMTome: A resource for pan-cancer analysis of epithelial-mesenchymal transition genes and signatures. Br. J. Cancer 2021, 124, 259–269. [Google Scholar] [CrossRef]
- Du, W.; Liu, X.; Fan, G.; Zhao, X.; Sun, Y.; Wang, T.; Zhao, R.; Wang, G.; Zhao, C.; Zhu, Y.; et al. From cell membrane to the nucleus: An emerging role of E-cadherin in gene transcriptional regulation. J. Cell. Mol. Med. 2014, 18, 1712–1719. [Google Scholar] [CrossRef]
- Huang, R.Y.-J.; Wong, M.K.; Tan, T.Z.; Kuay, K.T.; Ng, A.H.C.; Chung, V.Y.; Chu, Y.-S.; Matsumura, N.; Lai, H.-C.; Lee, Y.F.; et al. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell Death Dis. 2013, 4, e915. [Google Scholar] [CrossRef] [Green Version]
- Yuanhua, L.; Pudong, Q.; Wei, Z.; Yuan, W.; Delin, L.; Yan, Z.; Geyu, L.; Bo, S. TFAP2A Induced KRT16 as an Oncogene in Lung Adenocarcinoma via EMT. Int. J. Biol. Sci. 2019, 15, 1419–1428. [Google Scholar] [CrossRef]
- Mazzalupo, S.; Wong, P.; Martin, P.; Coulombe, P.A. Role for keratins 6 and 17 during wound closure in embryonic mouse skin. Dev. Dyn. 2003, 226, 356–365. [Google Scholar] [CrossRef]
- Zhang, X.; Yin, M.; Zhang, L.-J. Keratin 6, 16 and 17—Critical Barrier Alarmin Molecules in Skin Wounds and Psoriasis. Cells 2019, 8, 807. [Google Scholar] [CrossRef] [Green Version]
- Tomikawa, K.; Yamamoto, T.; Shiomi, N.; Shimoe, M.; Hongo, S.; Yamashiro, K.; Yamaguchi, T.; Maeda, H.; Takashiba, S. Smad2 Decelerates Re-epithelialization during Gingival Wound Healing. J. Dent. Res. 2012, 91, 764–770. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Liquid biopsy and minimal residual disease—Latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef]
- Keller, L.; Pantel, K. Unravelling tumour heterogeneity by single-cell profiling of circulating tumour cells. Nat. Rev. Cancer 2019, 19, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J.; Obenauf, A. Metastatic colonization by circulating tumour cells. Nat. Cell Biol. 2016, 529, 298–306. [Google Scholar] [CrossRef]
- Pantel, K.; Brakenhoff, R.H. Dissecting the metastatic cascade. Nat. Rev. Cancer 2004, 4, 448–456. [Google Scholar] [CrossRef]
- Agnoletto, C.; Corrà, F.; Minotti, L.; Baldassari, F.; Crudele, F.; Cook, W.J.J.; Di Leva, G.; Pio d’Adamo, A.; Gasparini, P. Heterogeneity in Circulating Tumor Cells: The Relevance of the Stem-Cell Subset. Cancers 2019, 11, 483. [Google Scholar] [CrossRef] [Green Version]
- Lecharpentier, A.; Vielh, P.; Perez-Moreno, P.; Planchard, D.; Soria, J.C.; Farace, F. Detection of circulating tumour cells with a hybrid (epithelial/mesenchymal) phenotype in patients with metastatic non-small cell lung cancer. Br. J. Cancer 2011, 105, 1338–1341. [Google Scholar] [CrossRef] [Green Version]
- Bednarz-Knoll, N.; Alix-Panabières, C.; Pantel, K. Plasticity of disseminating cancer cells in patients with epithelial malignancies. Cancer Metastasis Rev. 2012, 31, 673–687. [Google Scholar] [CrossRef]
- Nel, I.; Baba, H.A.; Ertle, J.; Weber, F.; Sitek, B.; Eisenacher, M.; Meyer, H.E.; Schlaak, J.F.; Hoffmann, A.-C. Individual Profiling of Circulating Tumor Cell Composition and Therapeutic Outcome in Patients with Hepatocellular Carcinoma. Transl. Oncol. 2013, 6, 420–428. [Google Scholar] [CrossRef]
- Satelli, A.; Mitra, A.; Brownlee, Z.; Xia, X.; Bellister, S.; Overman, M.J.; Kopetz, S.; Ellis, L.M.; Meng, Q.H.; Li, S. Epithelial–Mesenchymal Transitioned Circulating Tumor Cells Capture for Detecting Tumor Progression. Clin. Cancer Res. 2015, 21, 899–906. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, A.J.; Marengo, M.S.; Oltean, S.; Kemeny, G.; Bitting, R.; Turnbull, J.D.; Herold, C.I.; Marcom, P.K.; George, D.J.; Garcia-Blanco, M.A. Circulating Tumor Cells from Patients with Advanced Prostate and Breast Cancer Display Both Epithelial and Mesenchymal Markers. Mol. Cancer Res. 2011, 9, 997–1007. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5’-3′) | Reverse Primer (5’-3′) | Product Length |
|---|---|---|---|
| E-cadherin (CDH1) | CGAGAGCTACACGTTCACGG | GGGTGTCGAGGGAAAAATAGG | 119 bp |
| N-cadherin (CDH2) | TGCGGTACAGTGTAACTGGG | GAAACCGGGCTATCTGCTCG | 123 bp |
| Vimentin (VIM) | GACGCCATCAACACCGAGTT | CTTTGTCGTTGGTTAGCTGGT | 238 bp |
| SNAI2 (SLUG) | TGTGACAAGGAATATGTGAGCC | TGAGCCCTCAGATTTGACCTG | 203 bp |
| SNAI1 | ACTGCAACAAGGAATACCTCAG | GCACTGGTACTTCTTGACATCTG | 242 bp |
| ZEB1 | GATGATGAATGCGAGTCAGATGC | ACAGCAGTGTCTTGTTGTTGT | 86 bp |
| ZEB2 | GGAGACGAGTCCAGCTAGTGT | CCACTCCACCCTCCCTTATTTC | 107 bp |
| TWIST1 | AAGGCATCACTATGGACTTTCTCT | GCCAGTTTGATCCCAGTATTTT | 96 bp |
| WNT5A | ATTCTTGGTGGTCGCTAGGTA | CGCCTTCTCCGATGTACTGC | 159 bp |
| NOTCH1 | GAGGCGTGGCAGACTATGC | CTTGTACTCCGTCAGCGTGA | 140 bp |
| KRT8 | CAGAAGTCCTACAAGGTGTCCA | CTCTGGTTGACCGTAACTGCG | 194 bp |
| KRT18 | GCTCAGATCTTCGCAAATACTGT | CTTCCTCTTCGTGGTTCTTCTTC | 250 bp |
| KRT19 | ACCAAGTTTGAGACGGAACAG | CCCTCAGCGTACTGATTTCCT | 181 bp |
| KRT16 | GACCGGCGGAGATGTGAAC | CTGCTCGTACTGGTCACGC | 91 bp |
| CD24 | CTCCTACCCACGCAGATTTATTC | AGAGTGAGACCACGAAGAGAC | 166 bp |
| CD44 | CTGCCGCTTTGCAGGTGTA | CATTGTGGGCAAGGTGCTATT | 109 bp |
| GAPDH | GGAGCGAGATCCCTCCAAAAT | GGCTGTTGTCATACTTCTCATGG | 197 bp |
| Univariable Analysis | Multivariable Analysis | |||||||
|---|---|---|---|---|---|---|---|---|
| Covariate | Coefficient (bi) | HR [exp(bi)] | HR 95% CI | p-Value | Coefficient (bi) | HR [exp(bi)] | HR 95% CI | p-Value |
| K16+ CTCs | 2.061 | 7.856 | (1.677, 36.81) | 0.0089 | 4.864 | 129.6 | (1.6, 1.120) | 0.0287 |
| Age | −0.072 | 0.930 | (0.867, 0.998) | 0.0432 | −0.217 | 0.804 | (0.627, 1.032) | 0.0869 |
| ER+ | −0.638 | 0.528 | (0.166, 1.680) | 0.280 | −5.909 | 0.003 | (0.0, 0.951) | 0.0481 |
| PR+ | −0.150 | 0.861 | (0.275, 2.693) | 0.797 | 3.857 | 47.3 | (0.108, 2.670) | 0.2136 |
| ERBB2+ | 0.078 | 1.081 | (0.283, 4.123) | 0.910 | −0.435 | 0.647 | (0.030, 14.1) | 0.7820 |
| T3-4 | 0.272 | 1.312 | (0.286, 6.420) | 0.737 | 0.117 | 1.352 | (0.080, 15.9) | 0.9308 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elazezy, M.; Schwentesius, S.; Stegat, L.; Wikman, H.; Werner, S.; Mansour, W.Y.; Failla, A.V.; Peine, S.; Müller, V.; Thiery, J.P.; et al. Emerging Insights into Keratin 16 Expression during Metastatic Progression of Breast Cancer. Cancers 2021, 13, 3869. https://doi.org/10.3390/cancers13153869
Elazezy M, Schwentesius S, Stegat L, Wikman H, Werner S, Mansour WY, Failla AV, Peine S, Müller V, Thiery JP, et al. Emerging Insights into Keratin 16 Expression during Metastatic Progression of Breast Cancer. Cancers. 2021; 13(15):3869. https://doi.org/10.3390/cancers13153869
Chicago/Turabian StyleElazezy, Maha, Sandra Schwentesius, Luisa Stegat, Harriet Wikman, Stefan Werner, Wael Y. Mansour, Antonio Virgilio Failla, Sven Peine, Volkmar Müller, Jean Paul Thiery, and et al. 2021. "Emerging Insights into Keratin 16 Expression during Metastatic Progression of Breast Cancer" Cancers 13, no. 15: 3869. https://doi.org/10.3390/cancers13153869
APA StyleElazezy, M., Schwentesius, S., Stegat, L., Wikman, H., Werner, S., Mansour, W. Y., Failla, A. V., Peine, S., Müller, V., Thiery, J. P., Ebrahimi Warkiani, M., Pantel, K., & Joosse, S. A. (2021). Emerging Insights into Keratin 16 Expression during Metastatic Progression of Breast Cancer. Cancers, 13(15), 3869. https://doi.org/10.3390/cancers13153869