Simple Summary
Although cisplatin is very effective as a treatment strategy in triple-negative breast cancer (TNBC), it has unwarranted outcomes owing to recurrence, chemoresistance and neurotoxicity. In the current studies we determined a novel therapeutic option that enhances the efficacy of cisplatin and at the same time protects neuronal cells from cisplatin induced toxicity.
Abstract
Although cisplatin is very effective as a treatment strategy in triple-negative breast cancer (TNBC), it has unwarranted outcomes owing to recurrence, chemoresistance and neurotoxicity. There is critically important to find new, effective and safe therapeutics for TNBC. We determined if SP-receptor antagonism in combination with cisplatin may serve as a novel, more efficacious and safer therapeutic option than existing therapies for TNBC. We used a neuronal cell line (PC12) and two TNBC cell lines (Sum 185 and Sum 159) for these studies. We determined that the levels of cells expressing the high-affinity SP-receptor (neurokinin 1 receptor (NK1R)), as determined by flow-cytometry was significantly elevated in response to cisplatin in all three cells. We determined that treatment with aprepitant, an SP-receptor antagonist decreased cisplatin-induced, loss of viability (studied by MTT assay), production of reactive oxygen species (by DCFDA assay) and apoptosis (by flow-cytometry) in PC12 cells while it was increased in the two TNBC cells. Furthermore, we demonstrated that important genes associated with metastases, inflammation, chemoresistance and cell cycle progression are attenuated by SP-receptor antagonism in the TNBC cell line, Sum 185. These studies implicate that SP-receptor antagonism in combination with cisplatin may possibly serve as a novel, more efficacious and safer therapeutic option than existing therapies for TNBC.
1. Introduction
Breast cancer is amongst the foremost cause of cancer-related mortality in women [1]. Triple negative breast cancer (TNBC) is more prevalent in young women and is characterized by aggressive clinical characteristics, the early peak of distant recurrences at 3 years after diagnosis, poor prognosis and high mortality rate within the first 5 years [2,3]. Cisplatin forms the basis of chemotherapy regimens for many malignancies, including TNBC, ovarian and cervical cancers, prostate and testicular cancers, bladder cancer, head and neck cancer, lung cancer and non-Hodgkin’s lymphoma [4,5,6,7]. It mediates its effects by crosslinking with the purine bases on the DNA, thus leading to interference with DNA repair leading to DNA damage and ensuing apoptosis of tumor cells. Though cisplatin is highly effective as first-line therapy in TNBC, it can lead to chemoresistance and unwanted dire manifestations such as peripheral neuropathy, inhibited immune responses to infections, severe kidney damage, hearing loss, allergic reactions, hemorrhage, and gastrointestinal disorders [8,9]. Novel therapeutic combinations are needed to increase the efficacy of cisplatin and at the same time ablate or reduce cisplatin-induced toxicity and chemoresistance.
Substance P (SP), a neuropeptide and pain transmitter acting via its high-affinity receptor, neurokinin1 receptor (NK-1R), has shown to be mitogenic for human cancer cells in vitro [10,11,12,13]. NK1R is overexpressed in TNBC cells [14] and many other cancers, such as head and neck cancer, glioma, astrocytoma, retinoblastoma, ganglioneuroblastoma, leukemia, neuroblastoma and carcinomas (pancreatic, larynx, gastric, colon, medullary thyroid) [13,15]. In vitro studies have shown SP to prevent apoptosis of tumor cells and induce tumor cell migration [10,14,16,17,18,19,20]. Substance P (SP) is known to trigger inflammatory responses, stimulate the production of reactive oxygen species (ROS) [21,22,23]. Our prior studies demonstrate that SP receptor antagonism increases the efficacy of doxorubicin in TNBC cell lines while protecting cardiomyocytes from doxorubicin-induced oxidative stress, apoptosis and chemotherapy-induced death [24]. In the current studies, we will determine if SP receptor antagonism will increase the efficacy of cisplatin, another chemotherapeutic drug in TNBC cell lines. We will also determine if SP receptor antagonism will protect PC12, a rat neuronal cell line, from cisplatin-induced oxidative stress and apoptosis.
In the current studies, we quantitated the levels of SP and NK1R in two TNBC cell lines and in PC12 cells in the presence and absence of cisplatin. We also determined if the levels of apoptosis and ROS will be significantly altered in response to cisplatin treatment. Furthermore, we determined if treatment with an SP receptor antagonist, aprepitant (that is widely used to attenuate chemotherapy-associated nausea), would reverse these cisplatin-induced alterations. Most importantly, we determined if aprepitant would enhance the efficacy of cisplatin in TNBC cells while protecting PC12 cells from cisplatin-induced oxidative stress and apoptosis. The studies in this manuscript will determine if SP receptor antagonism in combination with cisplatin may serve as a novel, more efficacious and safer therapeutic option than existing therapies for TNBC.
2. Results
2.1. Cisplatin Increases NK1R Levels in Both PC12 Neuronal Cells and TNBC Cells
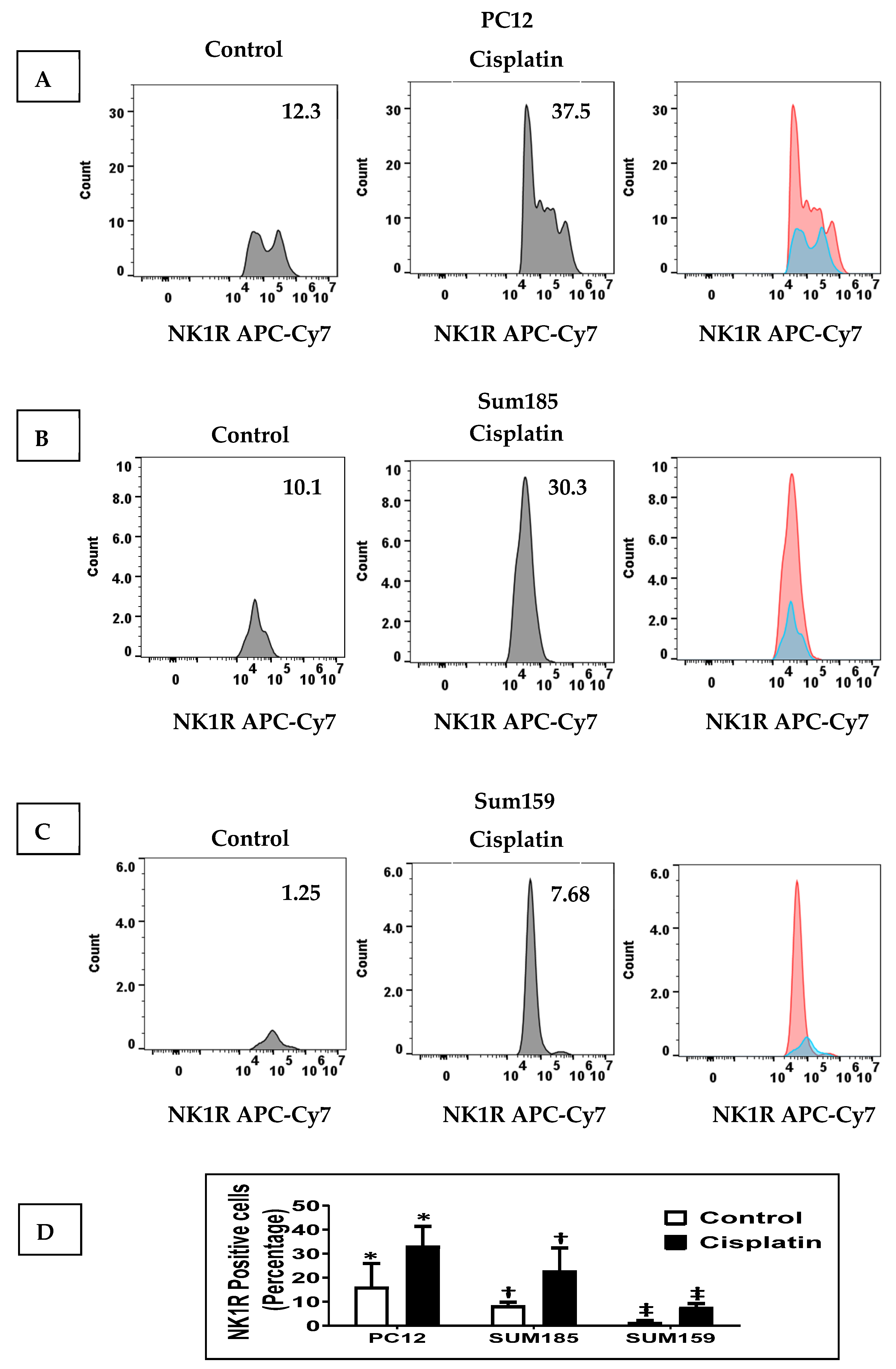
We determined whether cisplatin treatment increased NK1R levels in PC12 cells and in Sum 185 and Sum 159 TNBC cells. We treated PC12 cells, Sum 185 and Sum 159, with their respective IC50 dose of cisplatin for 48 h and then determined SP levels by flow cytometry. We determined the percentage of NK1R positive cells in all three cells in response to cisplatin was significantly higher than the untreated cells. The percentage of NK1R positive cells in the absence of cisplatin, respectively, for PC12, Sum 185 and Sum 159 cells was 16.07% ± 9.85%, 8.27% ± 1.56% and 1.41% ± 0.74%. Cisplatin treatment led to a significant increase in the percentage of NK1R positive cells to 33.1% ± 8.29%, 22.87% ± 9.53% and 7.7% ± 1.56%, respectively, for PC12, Sum 185 and Sum 159 cells. (Figure 1, p ≤ 0.05, Students t-test, n = 2, for all cells).
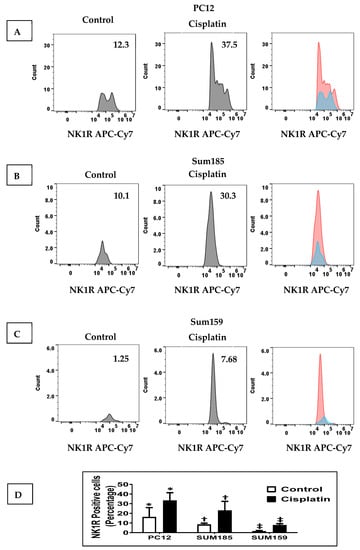
Figure 1.
Cisplatin increases NK1R Levels in both PC12 neuronal cells and TNBC Cells. NK1R positive cells as determined by flow cytometry, in cisplatin-treated versus untreated PC12 cells, Sum 185 and Sum 159 cells (A–C), gate and numbers represent NK1R positive population within live cells. Levels of NK1R positive cells in all three cells (D) (*, †, ‡, p ≤ 0.05, Student’s t-test, n = 2).
2.2. NK1R Antagonism Protects Rat Neuronal PC12 Cells from Loss of Viability Induced by Cisplatin
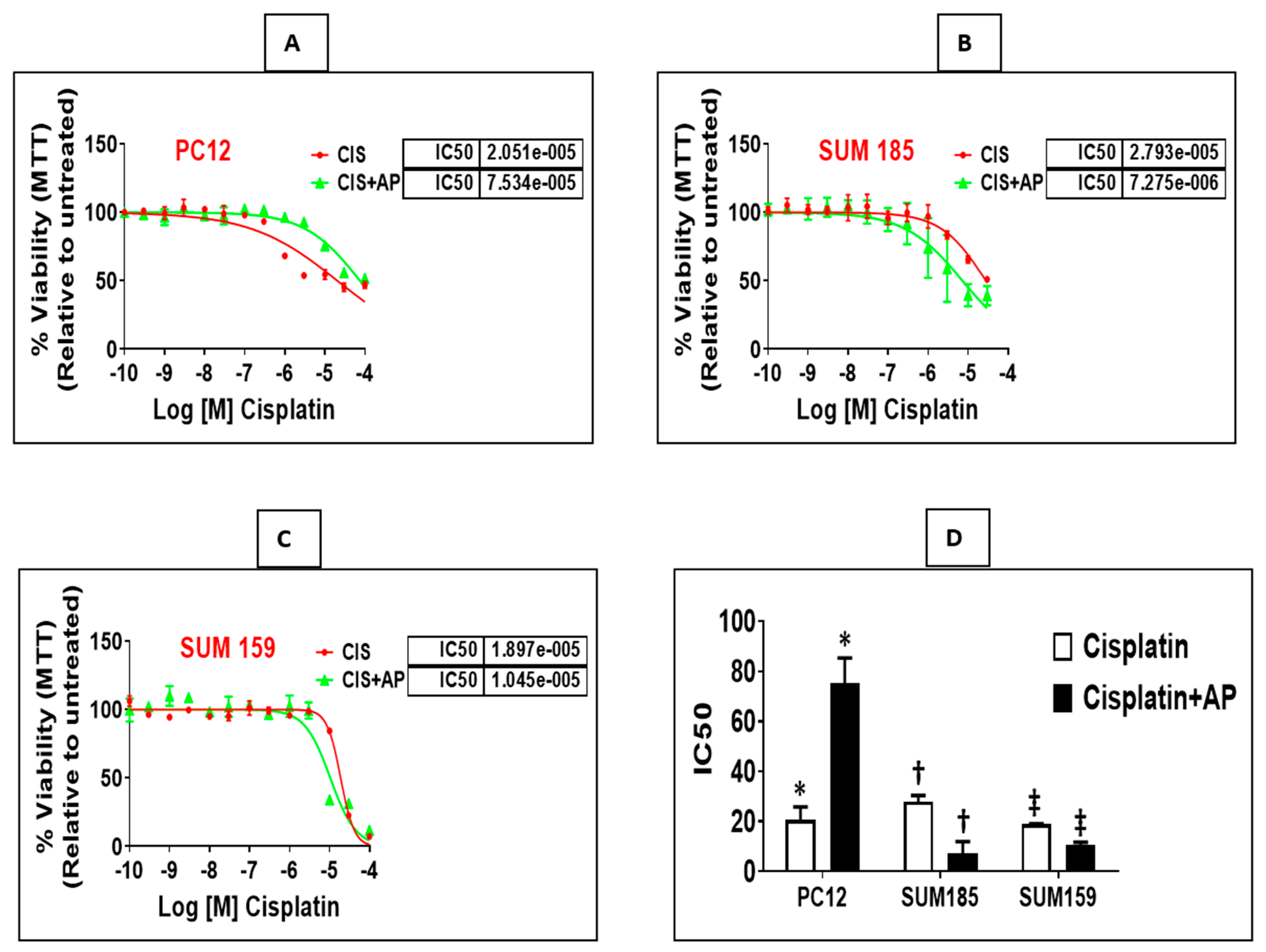
We determined if treatment with aprepitant, an NK1R antagonist prevents cisplatin-induced reduction in neuronal viability. PC12 cells were plated in a 96-well plate (3000 cells/well). Twenty-four hours later, we treated triplicate wells with concentrations of cisplatin (ranging from 0.003 μm to 100 μm) in the presence and absence of aprepitant (10 μm, treated 2 h before cisplatin treatment). Moreover, we included control wells consisting of similar concentrations of vehicle (DMSO) that were used in the reconstitution of aprepitant (0.1% DMSO in water), media alone or with aprepitant alone. Each experiment was performed at two different times, and results are represented as mean ± SEM. We elucidated that aprepitant protected PC12 cells from cisplatin-induced loss of viability. The mean inhibitory concentration (IC50) of cisplatin was 20.51 μm ± 5.28 μm; the presence of aprepitant with cisplatin led to a 3.6-fold increase in the IC50 levels to 75.34 μm ± 10.06 μm (Figure 2A,D; p = 0.01, ANOVA, n = 2).
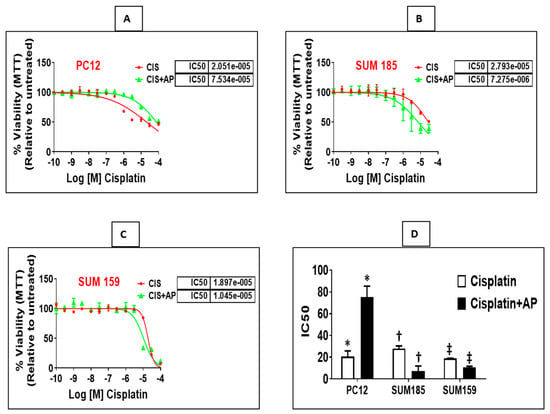
Figure 2.
NK1R Antagonism protects rat neuronal PC12 cells while causing enhancement of cisplatin-induced loss of viability in TNBC cells. Loss of viability induced by cisplatin in the presence and absence of aprepitant in rat neuronal PC12 cells, Sum 185 and Sum 159 cells (A–C) and IC50 levels of cisplatin in the presence and absence of aprepitant in PC12 and TNBC cells (D) (*, †, ‡, p ≤ 0.05, Student’s t-test, n = 2–3).
2.3. NK1R Antagonism Enhances Efficacy of Cisplatin in Two Triple Negative Breast Cancer (TNBC) Cells
We determined if treatment with aprepitant enhances cisplatin-induced reduction in viability of TNBC cells. Sum 185 and Sum 159, human TNBC cells, were plated in a 96-well plate (3000 cells/well). Twenty-four hours later, we treated triplicate wells with concentrations of cisplatin (0.003 μm to 100 μm) in the presence and absence of aprepitant (10 μm, treated 2 h before cisplatin treatment). Moreover, we included control wells consisting of treatment with similar concentrations of vehicle (DMSO) used for reconstitution of aprepitant (0.1% DMSO in water), media alone or wells treated with aprepitant alone. All experiments were performed at two different times, and results are represented as mean ± SEM. We determined that aprepitant enhanced cisplatin-induced loss of viability of both TNBC cells. The mean inhibitory concentration (IC50) of cisplatin for Sum 185 was 27.93 μm ± 2.43 μm; the presence of aprepitant with cisplatin led to a 3.84-fold decrease in the IC50 levels to 7.27 μm ± 4.67 μm (Figure 2B,D; p = 0.001, ANOVA, n = 3). The mean inhibitory concentration (IC50) of cisplatin for Sum 159 was 18.97 μm ± 0.19 μm; the presence of aprepitant with cisplatin led to a 1.8-fold decrease in the IC50 levels to 10.45 μm ± 1.27 μm (Figure 2C,D; p ≤ 0.05, ANOVA, n = 2).
2.4. NK1R Antagonism Attenuates ROS Production Induced by Cisplatin in PC12 Cells
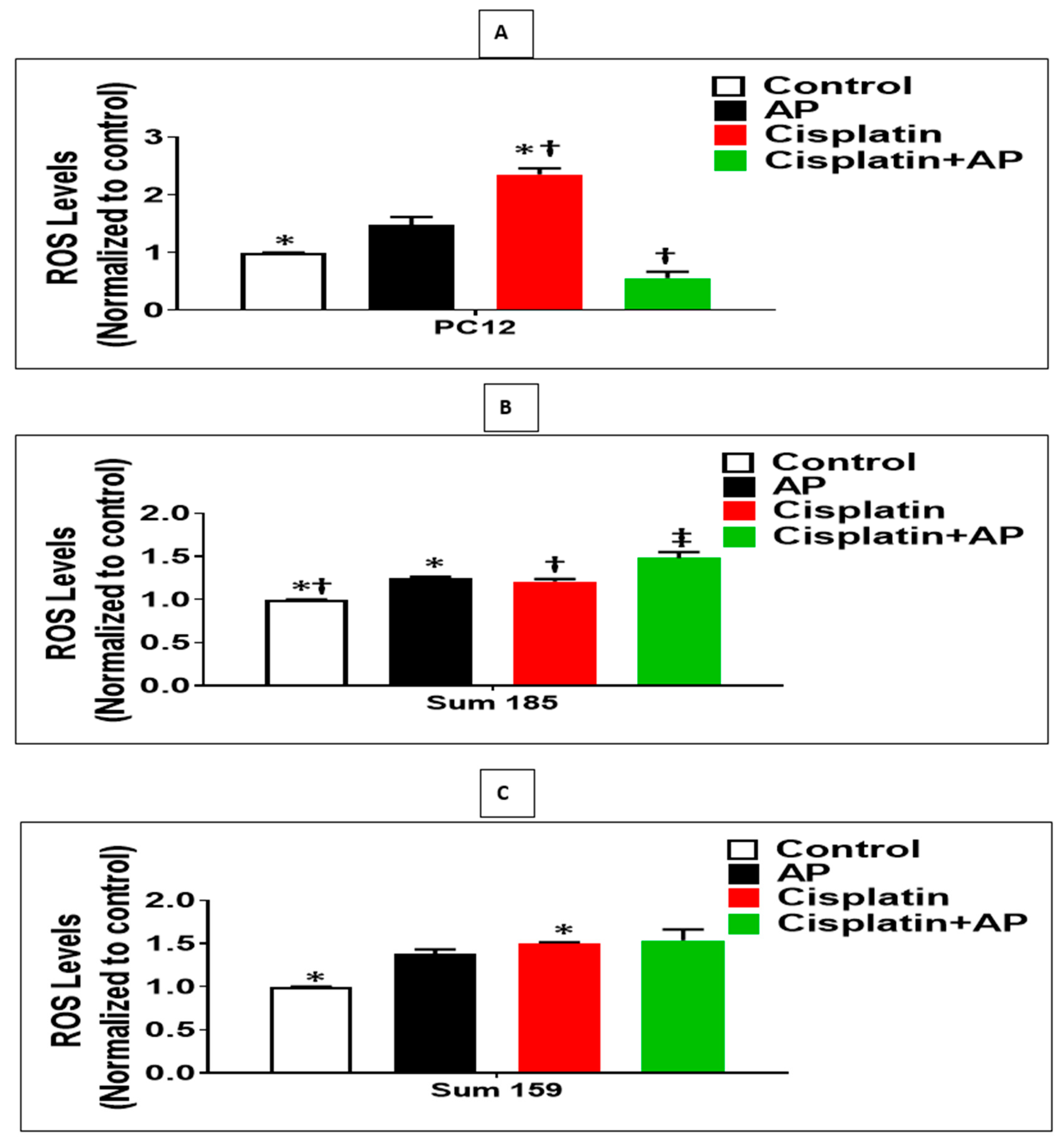
We determined whether the protective effects of SP antagonism on the cisplatin-induced loss of viability and prevention of apoptosis of PC12 cells were accompanied by decreased ROS levels. We determined the levels of cisplatin-induced ROS production in the media control group, the aprepitant alone group, and in the cisplatin group in the presence and absence of aprepitant in PC12 cells. The levels of ROS were normalized to that of the media control group. There was no significant difference in ROS levels between the media control and the aprepitant treated groups (Figure 3A; p > 0.05, ANOVA, n = 2). The percentage increase in ROS levels in response to cisplatin was significantly higher compared to media control (136% ± 5.3%; Figure 3A; p < 0.05, ANOVA, n = 2). Most importantly, the percentage increase in ROS levels in response to cisplatin-treated cells in the absence of aprepitant was significantly higher than the cisplatin-treated cells in the presence of aprepitant (79% ± 18.2%; Figure 3A; p ≤ 0.05, ANOVA, n = 2).
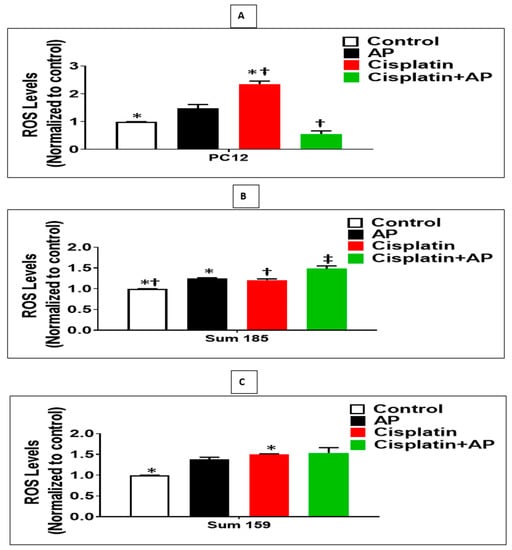
Figure 3.
NK1R Antagonism decreased cisplatin-induced ROS in rat neuronal PC12 cells while causing increased cisplatin-induced ROS levels in TNBC cells. Effect of NK1R antagonism on ROS Production induced by cisplatin in PC12 cells and TNBC cells. Levels of ROS as determined by the DCFDA assay, in untreated (media control) or treated with aprepitant or cisplatin alone or cisplatin + aprepitant, in PC12 (A), Sum 185 (B,C), Sum 159 cells (*, †, ‡, p ≤ 0.05, ANOVA, n = 2). Only statistical comparisons between control vs. aprepitant (AP) or Cis and Cis vs. Cis + AP are shown.
2.5. Effect of NK1R Antagonism on ROS Production Induced by Cisplatin in TNBC Cells
We determined if the enhanced loss of cisplatin-induced viability and increased apoptosis upon SP antagonist treatment was also accompanied by increased levels of ROS in TNBC cells. We determined the levels of cisplatin-induced ROS production in the media control group, the aprepitant alone group and in the cisplatin group in the presence and absence of aprepitant, in Sum 185 and Sum 159 TNBC cells. The levels of ROS were normalized to that of the media control group. Compared to media control, ROS levels was increased in the aprepitant treated group by 24% ± 7.2% and 38% ± 2.5%, respectively, in the Sum 185 and Sum 159 TNBC cells (Figure 4B,C; p < 0.05, ANOVA, n = 2, for both cells). The percentage increase in ROS levels in response to cisplatin was also significantly higher compared to media control (50% ± 0.7% and 21% ± 1.4%, respectively, in the Sum 185 and Sum 159 TNBC cells; Figure 3B,C; p < 0.05, ANOVA, n = 2, for both cells). The percentage increase in ROS levels in the Sum 185 cells in response to cisplatin in the absence of aprepitant was significantly lower than the cisplatin-treated cells in the presence of aprepitant (16% ± 5.1% Figure 3B; p < 0.05, ANOVA, n = 2). However, there was no significant difference in the percentage increase in ROS levels in the Sum 159 cells in response to cisplatin in the presence or absence of aprepitant (Figure 3C; p > 0.05, ANOVA, n = 2).
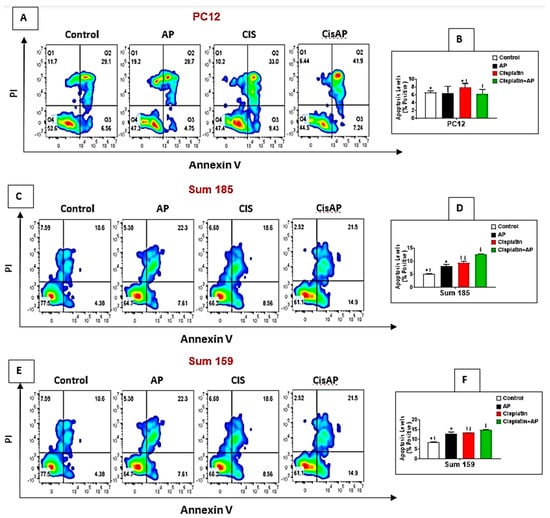
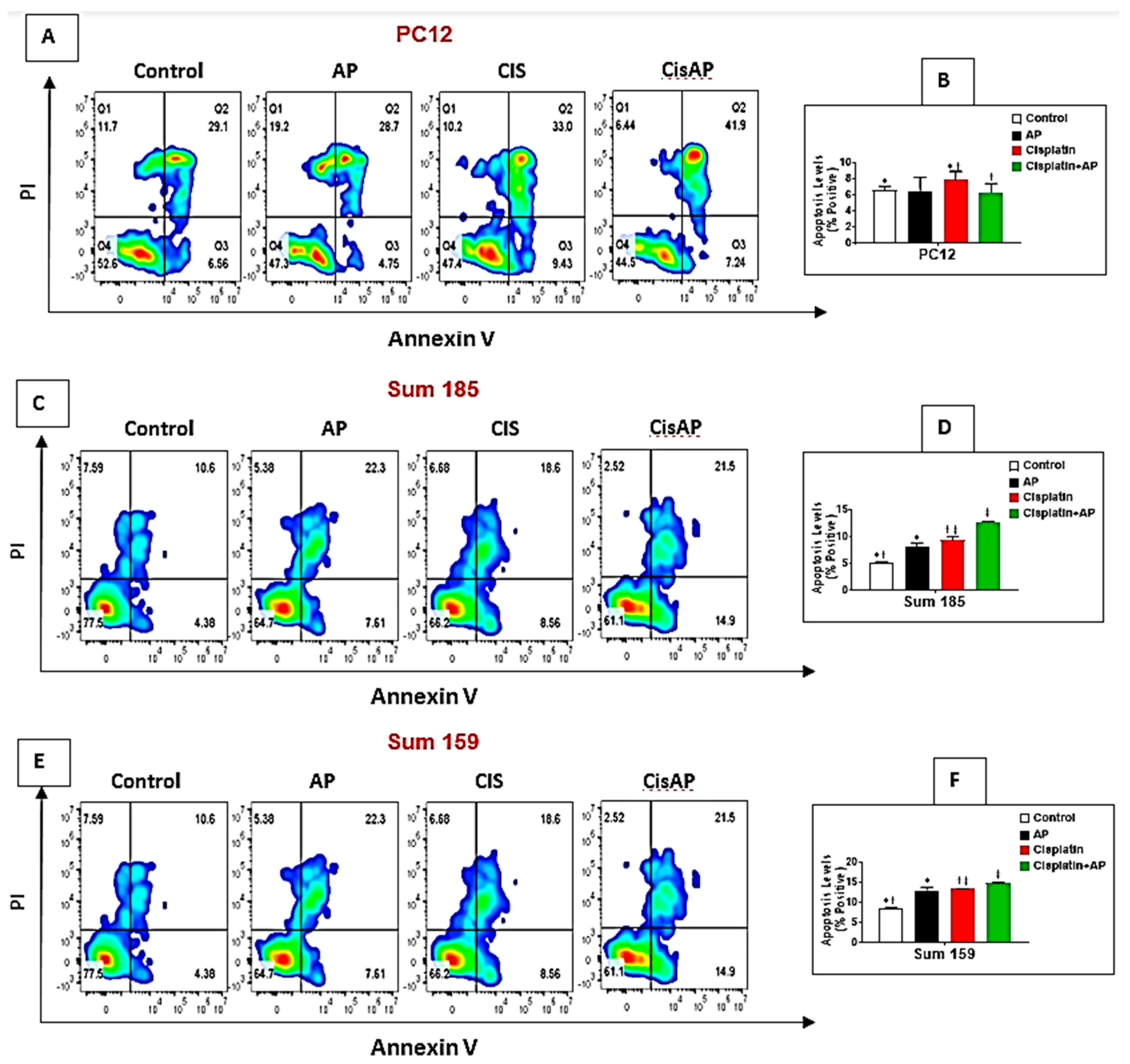
Figure 4.
NK1R Antagonism protects PC12 cells while causing enhancement of cisplatin-induced apoptosis in TNBC cells. PC12, Sum 185 and Sum 159 TNBC cells were untreated (media control) or treated with aprepitant or cisplatin alone or cisplatin + aprepitant, stained with Annexin V-FITC/PI and cell apoptosis analyzed by flow cytometry (A,C,E). Quadrant gating was made based on unstained controls. Q2 demonstrates necrotic cells, Q3 demonstrates apoptotic cells, and Q4 demonstrates viable cells. Levels of apoptosis (% positive) in the different groups in PC12 (B), Sum 185 (D) and Sum 159 cells (F) (*, †, ‡, p ≤ 0.05, ANOVA, n = 2). Only statistical comparisons between control vs. AP or Cis and Cis vs. Cis + AP are shown.
2.6. NK1R Antagonism Led to Decreased Levels of Apoptosis of PC12 Cells
We determined if NK1Rantagonist treatment could also result in the prevention of apoptosis in PC12 cells. The levels of cisplatin-induced apoptotic cells were determined in the media control group, the aprepitant alone group and in the cisplatin group in the presence and absence of aprepitant in PC12 cells by flow cytometry. The percentage of positive apoptotic cells in the media alone group was 6.64% ± 0.45%. There was no significant difference between the percentage of positive apoptotic cells in the control media group versus the aprepitant treated group (6.46% ± 1.72% (aprepitant alone) vs. 6.64% ± 0.45% (media control); Figure 4A,B; p ≥ 0.05, ANOVA, n = 2). The percentage of positive apoptotic cells was significantly higher in the cisplatin-treated group compared to media control (7.89% ± 1.04% (cisplatin alone) vs. 6.64% ± 0.45%. (media control); Figure 4A,B; p ≤ 0.05, ANOVA, n = 2). Most importantly, the percentage of positive apoptotic cells in the cisplatin-treated cells in the presence of aprepitant was significantly lower than the cisplatin-treated cells in the absence of aprepitant (6.34% ± 1.09% (cisplatin + aprepitant) vs. 7.89% ± 1.04% (cisplatin alone); Figure 4A,B; p ≤ 0.05, ANOVA, n = 2).
2.7. NK1R Antagonism Led to Increased Levels of Apoptosis of TNBC Cells
We determined if the enhanced loss of cisplatin-induced viability upon NK1R antagonist treatment was also accompanied by increased levels of apoptosis in TNBC cells. We determined the levels of cisplatin-induced apoptotic cells in the media control group, the aprepitant alone group and in the cisplatin group in the presence and absence of aprepitant in Sum 185 and Sum 159 TNBC cells. The percentage of positive apoptotic cells in the media alone group was 5.26% ± 0.15% and 8.69% ± 0.13%, respectively, for the Sum 185 and Sum 159 cells. Compared to media control, the percentage of positive apoptotic cells was significantly higher in the aprepitant treated group (8.21% ± 0.69% (aprepitant alone) vs. 5.26% ± 0.15% (media control) and 13% ± 0.84% (aprepitant alone) vs. 8.69% ± 0.13% (media control)), respectively, for the Sum 185 and Sum 159 cells (Figure 4C–F; p ≤ 0.05, ANOVA, n = 2, for both cells). The percentage of positive apoptotic cells was also significantly higher in the cisplatin-treated group compared to media control (9.41% ± 0.56% (cisplatin alone) vs. 5.26% ± 0.15% (media control) and 13.35% ± 0.17% (cisplatin alone) vs. 8.69% ± 0.13% (media control), respectively, for the Sum 185 and Sum 159 cells, (Figure 4C–F; p ≤ 0.05, ANOVA, n = 2, for both cells). Most importantly, the percentage of positive apoptotic cells in the cisplatin-treated cells in the presence of aprepitant was significantly higher than the cisplatin-treated cells in the absence of aprepitant (12.7% ± 0.14% (cisplatin + aprepitant) vs. 9.41% ± 0.56% (cisplatin alone) and 14.8% ± 0.21% (cisplatin + aprepitant) vs. 13.35% ± 0.17% (cisplatin alone), respectively, for the Sum 185 and Sum 159 cells. (Figure 4C–F; p ≤ 0.05, ANOVA, n = 2, both cells).
2.8. Effects of NK1R Antagonism on the Transcriptome in TNBC Cells
In order to determine the mechanisms by which NK1R antagonism increased the efficacy of cisplatin. We determined levels of mRNA by RNA-Seq in Sum 185 TNBC cells that were treated with and without cisplatin in the presence and absence of aprepitant.
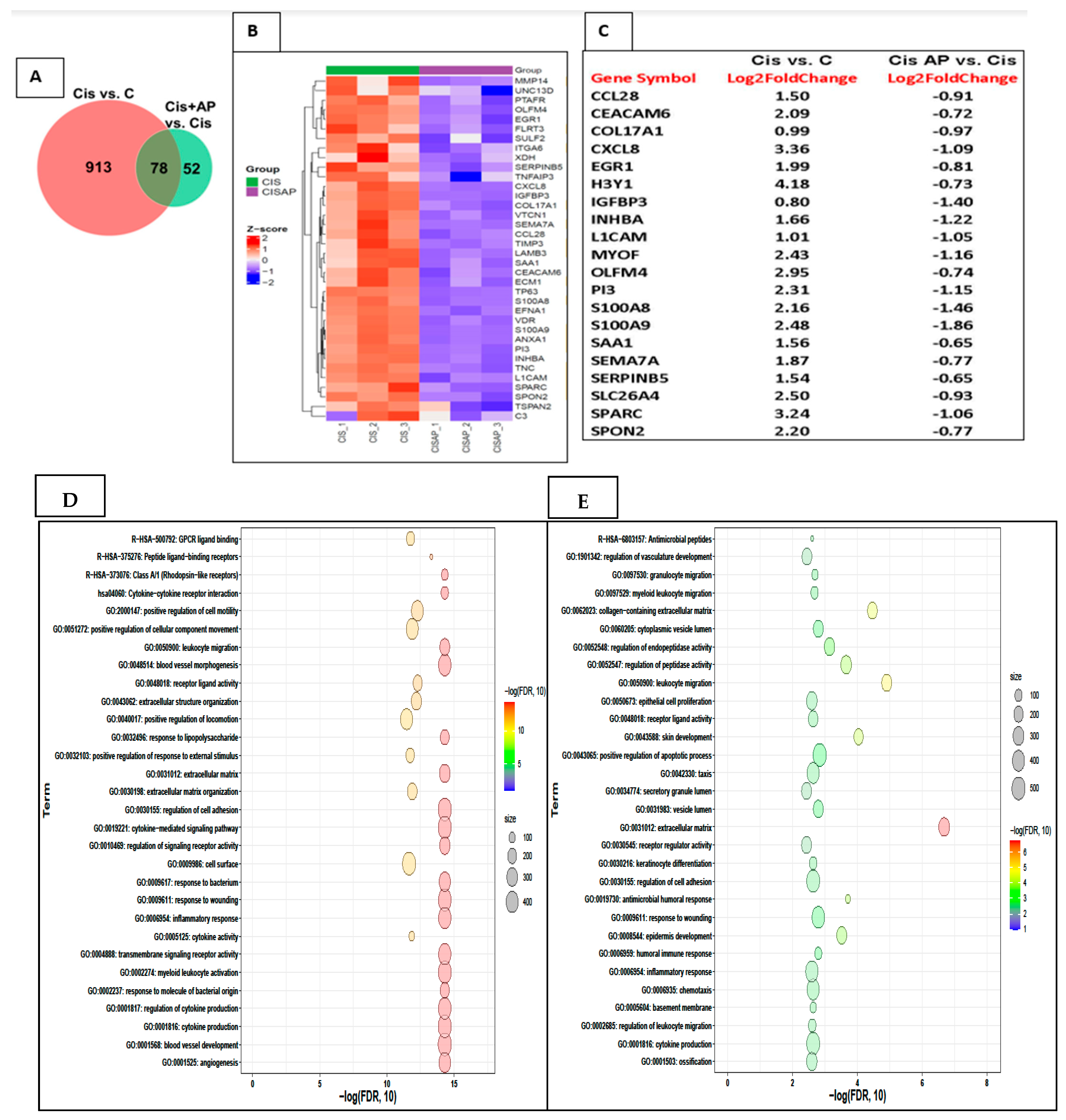
To identify candidate mRNAs that contributed to the increased efficacy of cisplatin, we performed two comparisons: CIS vs. no CIS mRNA sets and CIS + AP vs. CIS mRNA sets. In the CIS vs. no CIS comparison, the total number of genes determined was 991, of which 612 genes were upregulated and 379 genes were down-regulated. In the CIS + AP vs. CIS comparison, the total number of genes determined was 130, of which 44 genes were upregulated and 86 genes were down-regulated. We further identified genes that were differentially expressed between the two comparisons: CIS vs. No CIS mRNA sets and CIS + AP vs. CIS mRNA sets. We determined differentially expressed genes with 1.5-fold change and false discovery rate (FDR) < 0.05 (n = 3). There were 78 genes that were differentially expressed between the two comparisons (Figure 5A). Of the 78 genes, 68 genes were upregulated with CIS but downregulated in CISAP, and 10 genes were downregulated with CIS but upregulated in CISAP.
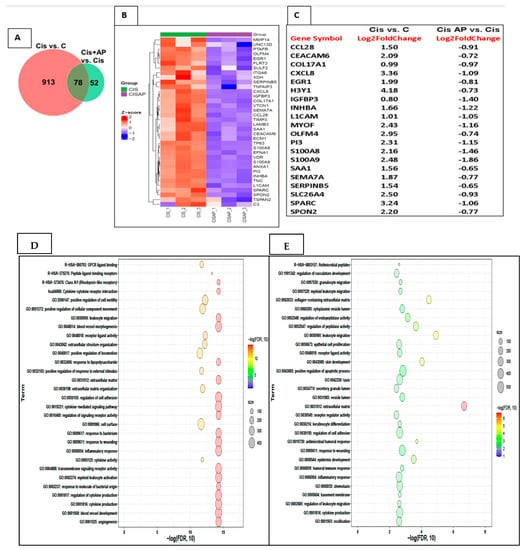
Figure 5.
Effects of NK1R antagonism on the transcriptome in Sum 185 TNBC cells. (A) Venn diagram showing differentially expressed genes between 2 comparisons CIS vs. C and CIS + AP vs. CIS (B) Heat map showing the upregulation (Z-score scaled) of genes linked to pro-inflammatory responses, extracellular matrix (ECM) and cell adhesion in CIS vs. CIS + AP. (C) Top 20 differentially expressed genes (D,E) Functional enrichment analysis of the upregulated gene sets in CIS and downregulated gene sets in CIS + AP using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) annotations showing top 30 enriched pathways.
Out of the 68 genes that were upregulated with CIS but downregulated in CISAP, 36 genes represented in the heat map in Figure 5B–E are genes associated with pathways linked to pro-inflammatory responses, extracellular matrix (ECM) and cell adhesion such as MMP14, UNC13D, PTAFR, OLFM4, EGR1, FLRT3, SULF2, ITGA6, XDH, SERPINB5, TNFAIP3, CXCL8, IGFBP3, COL17A1, VTCN1, SEMA7A, CCL28, TIMP3, LAMB3, SAA1, CEACAM6, ECM1, TP63, S100A8, EFNA1, VDR, S100A9, ANXA1, PI3, INHBA, TNC, L1CAM, SPARC, SPON2, TSPAN2 and C3.
Ten genes were downregulated with CIS but upregulated in CISAP; importantly, out of the 10 genes, only one gene, Stearoyl-CoA desaturase (SCD), was increased more than two-fold change. SCD is one of the genes that is involved in ameliorating the neuropathic phenotype induced by diabetes by restoring aberrant fatty acid biosynthesis and thereby preventing altered myelin lipid profile and ensuing myelin structural abnormalities [25]. Of special relevance is that two genes, although increased by only 1.5-fold, are genes known to be associated with better prognosis in TNBC (PTPRD and Neurl1). PTPRD is associated with negative regulation of stemness, epithelial-mesenchymal transition (EMT), and migration and invasion in breast cancer cells [26].
Neurl1 overexpression is known to downregulate Notch signaling, which is a key pathway associated with tumor growth, metastases and chemoresistance [27].
3. Material and Methods
3.1. Cell Culture
Rat neuronal cell lines, PC12, were purchased from the American Type Culture Collection (Manassas, VA, USA). Human TNBC cell lines, SUM 159 and SUM 185, were a kind courtesy of Dr. Nato Ueno, Translational Breast Cancer Research, Department of Breast Medical Oncology, MD Anderson Cancer Center, Houston, TX, USA. The SUM 159 and SUM 185 were cultured in RPMI containing 10% heat-inactivated fetal bovine serum, antibiotics (streptomycin and penicillin), an antifungal agent (amphotericin B; all from Invitrogen, Carlsbad, CA, USA) and were not passaged more than 4 weeks continuously. PC12 cells were cultured in the same media used for SUM 185 and SUM 159 cells but also contained 10% heat-inactivated horse serum and 100 ng/mL nerve growth factor (NGF). Cisplatin and aprepitant (respectively, Cat nos. S1166 and S1189) were purchased from Selleck-Chemicals, Houston, TX, USA.
3.2. Viability Assay
In order to determine the effect of aprepitant on the cellular metabolic activity as an indicator of cell viability, proliferation and cytotoxicity cell viability, we used the MTT assay. We plated in a 96-well plate (3000 cells/well; SUM 159, SUM 185 or PC12 cells per well); after 24 h, we treated triplicate wells with cisplatin (0.001 µm to 100 µm) in the presence or absence of10 µm of AP (2 h before cisplatin treatment and continuing until termination). We selected 10 µm of AP based on it being the IC10 of Sum 185 and Sum 159, and for consistency purposes, we used 10 µm for all cells. Control wells were treated with media or AP alone. Following 48 h treatment with cisplatin with or without AP, we determined the cell viability. Each well was emptied, and MTT (1 mg/mL in medium containing 1% serum) was then added to each well and incubated at 37 °C for 2 h. Following which viable cells that contain NADPH-dependent oxidoreductase enzymes reduce the MTT to formazan. The insoluble formazan crystals were solubilized in an extraction buffer containing 20% sodium dodecyl sulfate and 50% dimethylformamide. The cells were incubated overnight with the extraction buffer at 37 °C, following which the optical density was measured at 590 nm using a 96-well multi scanner (Molecular Devices, Sunnyvale, CA, USA). Data are represented as percentage viability related to untreated cells ± SEM.
3.3. ROS Measurement
ROS levels were determined by the dichlorofluorescein diacetate (DCFDA) assay kit (Cat no. C6827, Thermofisher Scientific, Waltham, MA, USA). We treated SUM 159, SUM 185 or PC12 cells with each of their respective IC50 concentrations of cisplatin (as deduced from the MTT experiments) with or without AP (10 µm, 2 h before cisplatin treatment and continuing until termination (48 h)). Controls consisted of treatment with media or AP alone. Following incubation with each of the conditions, the wells were emptied and washed once with 1× PBS, following which each of the cells was treated with CM-H2DCFDA (10 μM in 1× PBS) for 30 min at 37 °C in the dark. The wells were then read using a fluorescence spectrophotometer with maximum excitation and emission spectra of 495 nm and 517 nm, respectively. Data are represented as fluorescence intensity ± SEM for each group.
3.4. Flow Cytometry
To determine the NK1R expression, Sum 185, Sum 159 or PC12 cells cultured with cisplatin were washed with their respective complete media and stained with LIVE/DEAD Zombie Aqua™ (423101, BioLegend®, San Diego, CA, USA and NK1R APC/Cy7 (NB300-119APCCY7, Novus Biologicals®, (Littleton, CO, USA). To determine apoptosis of the cells, Annexin V/PI staining was performed using a commercially available kit (640914, BioLegend®). Briefly, after culture, the cells were washed once with the media and once with FACS buffer then suspended in Annexin V Binding Buffer at a concentration of 0.25–1.0 × 107 cells/mL. One hundred microliters of cell suspension was stained with 5 μL of FITC Annexin V and 10 μL of Propidium Iodide Solution, gently vortexed, and incubated for 15 min at room temperature (25 °C) in the dark. Subsequently, 400 μL of Annexin V Binding Buffer was added to each tube prior to the acquisition. The samples were acquired by CytoFlex™ (Indianapolis, IN, USA) and analyzed with FlowJo software version 10.7.1 (Ashland, OR, USA).
3.5. RNA Isolation and Library Preparation and RNA-Seq Data Analysis
We isolated RNA from SUM 185 cells following 48 h treatment with cisplatin in the presence and absence of 10 µm AP, and controls included media alone with and without AP. For the RNA-Seq experiments, as previously [28], RNA was briefly extracted with a Purelink Kit (Ambion, Life Technologies, Carlsbad, CA, USA) following reverse-transcription to cDNA using SuperScript III Reverse Transcriptase (Invitrogen, Waltham, MA, USA). We then performed enrichment of Poly (A)-tailed mRNA and prepared the RNA-seq library using the UT Cancer Genomics Core Center facility following manufacturer’s instructions outlined in the KAPA mRNA HyperPrep Kit (KK8581, Roche, Holding AG, city, Switzerland) and KAPA Unique Dual-indexed Adapter kit (KK8727, Roche). We then performed RNA-seq using the Illumina Nextseq550 (San Diego, CA, USA) with the 75 bp pair ended running mode.
Cutadapt v1.15 was used to preprocess the raw mRNA sequence reads to remove bases with quality scores < 20 and adapter sequences. The clean RNA-seq reads were then aligned to human genome assembly GRCh38 with STAR v2.5.3a. HTseq-count with default parameter using annotation from ENSEMBL v102 was used to count uniquely mapped reads overlapping genes. Only genes with >5 reads in at least one sample were retained. We then performed differential expression analysis only on the raw read counts of retained genes using DESeq2 software, which uses a model based on the negative binomial distribution [29]. Benjamini and Hochberg’s approach was used to attain p-values adjusted to control for false discovery rate (FDR). Genes with fold change (FC) > 1.5 and FDR < 0.05 were assigned as differentially expressed genes (DEGs). A standard gene set enrichment analysis was performed with a hypergeometric test using WebGestalt v 0.4.3 [30]. The resulting p values were also adjusted using Benjamini and Hochberg’s approach.
3.6. Statistical Analyses
Data represented are mean ± SEM of a minimum of two experiments. We determined statistical differences using analysis of variance (ANOVA), followed by Tukey’s or Dunn’s posttest as appropriate or by Student’s unpaired t-test. The threshold of statistical significance was set at p ≤ 0.05. We used the Graph Pad Prism version 6.04 for Windows, Graph Pad Software (San Diego, CA, USA) for performing data and statistical analysis.
4. Discussion
Though cisplatin is very effective as a treatment strategy in TNBC, it has unwarranted outcomes owing to recurrence, chemoresistance and detrimental side effects. There is an urgent need for effective and safe therapeutics for treating TNBC.
We determined if NK1R antagonism in combination with cisplatin may serve as a novel, more efficacious and safer therapeutic option than existing therapies for TNBC.
We treated a neuronal cell line (PC12) and two TNBC cell lines (Sum 185 and Sum 159) with aprepitant, an NK1R antagonist that is widely used to attenuate chemotherapy-associated nausea, and demonstrated the following responses. We demonstrated that (a) aprepitant decreased cisplatin-induced loss of viability, ROS production and apoptotic cell death in PC12 cells compared with cells treated with cisplatin alone and (b) aprepitant increased cisplatin-induced loss of viability, ROS production and apoptotic cell death in TNBC cells compared with cells treated with cisplatin alone.
The current studies are extremely important and timely. Treatment of TNBC is extremely challenging compared to other breast cancers that express one or more receptors such as the progesterone receptor (PR), estrogen receptor (ER) and human epidermal growth factor receptor (HER2). The expression of these receptors makes it more amenable for treatment with chemotherapeutic agents that block these receptors. TNBC cells lack these three receptors, resulting in limited treatment options [2]. Although cisplatin is very effective as a treatment strategy in TNBC, it can cause challenging side effects such as nerve damage, kidney damage, hearing loss and other manifestations [9,31]. Furthermore, recurrence and chemoresistance are commonly noted in TNBC patients treated with cisplatin. Our strategy to use NK1R antagonism in combination with cisplatin is aimed at targeting multiple pathways that are associated with TNBC growth, metastases and development of chemoresistance. Our strategy has importantly resulted in using lesser amounts of cisplatin to achieve tumor cytotoxicity, thus probably serving as an important mechanism to attenuate the side effects of cisplatin. Our studies demonstrating the ability of SP receptor antagonism to enhance the efficacy of cisplatin in TNBC cells and at the same time protect neuronal cells from cisplatin-induced toxicity addresses the challenges outlined above.
The mechanism by which cisplatin, a DNA intercalating agent, induces its anti-tumor effects is via crosslinking DNA with resultant interference with RNA transcription and DNA replication leading to cell-cycle arrest and apoptosis in tumor cells [31]. There are several mechanisms by which tumor cells attain chemoresistance to cisplatin, such as decreased levels of cisplatin being accumulated in tumor cells as a result of (a) lesser uptake or enhanced efflux of the drug, (b) detoxification of the drug by intrinsic mechanisms mediated by tumor cells, (c) enhanced repair of the DNA or (d) negative regulation of apoptotic mechanisms [31,32].
Besides the ability of cisplatin to intervene with the proliferation of tumor cells as a result of the formation of deoxyribonucleic acid (DNA)-platinum adducts, it mediates its effects on tumor cells by causing elevated production of reactive oxygen species leading to alteration of mitochondrial function and activation of apoptotic pathways [33]. We demonstrated that SP receptor antagonism enhanced the cisplatin-induced ROS production and apoptosis levels in two TNBC cancer cells. Importantly, our RNA-Seq studies determined that SP receptor antagonism attenuates the levels of several genes associated with apoptosis, such as OLFM4, CCL28, and TNFAIP3 (anti-apoptotic) [34,35].
Furthermore, we demonstrated that important genes associated with metastases are attenuated with SP receptor antagonist treatment. One of the main mechanisms by which metastases occur is via the ability of cancer cells to degrade basement membranes and spread to other tissues via blood or lymphatic vessels [36]. MMP14, a known mediator of extracellular matrix (ECM) degradation and ensuing metastases [37,38,39], is attenuated with SP receptor antagonist treatment. Other metastases genes associated with poor prognosis in breast cancer, such as CEACAM6, COL17A1, CXCL8, TNFAIP3, SEMA7A and L1CAM [31,38,39,40,41], are also attenuated with SP receptor antagonist treatment.
Studies have shown that inflammation is associated with the aggressive phenotype of TNBC [42,43]. We determined that SP receptor antagonism attenuates the levels of genes associated with inflammatory processes such as S100A8, S100A9, CXCL8, CCL28, ANXA1 and SAA1 [42,44,45,46,47].
Furthermore, genes associated with recurrence and chemoresistance that are attenuated by SP receptor antagonism include IGFBP-3, PI3 and SPARC [48,49,50]. Moreover, Histone H3Y1, an important gene that positively controls cell cycle progression and cell growth, is attenuated by SP receptor antagonism [51].
Genes that are attenuated with SP receptor antagon.ism, such as SERPINB5, are known to have a controversial role in TNBC. Studies show that the absence of SERPINB5 (Maspin) is an indicator of tumor progression and metastatic potential, while other studies show that Maspin expression correlates with an aggressive phenotype in breast cancer and with poor prognosis. The outcome varied with subcellular Maspin expression, and nuclear staining was demonstrated to be significantly associated with a better prognosis than cytoplasmic staining [52,53].
Genes that are attenuated with SP receptor antagonism, such as SPON2, are involved in cancer progression and metastasis of many tumors other than breast cancer [54]. Similarly, INHBA was used as a diagnostic marker in ovarian cancer [55], but there are no published studies that have determined its expression and/or role in breast cancer. The only gene that was downregulated by SP receptor antagonism but was beneficial in TNBC was EGR1, which is a tumor suppressor gene [56].
Cisplatin is one of the chemotherapeutic agents that causes chemotherapy-induced peripheral neuropathy (CIPN). CIPN often leads to the manifestation of ongoing pain as a result of damage to peripheral sensory and motor neurons in cancer patients that were treated with platinum-based chemotherapeutic agents such as cisplatin and oxaliplatin [31]. CIPN occurs in a dose- and time-dependent manner [57]. The initial symptoms of the onset of CIPN can vary from its occurrence as early as after the first dose or after several cycles of therapy [58,59]. One of the mechanisms leading to cisplatin-induced neuropathy is due to oxidative stress, mitochondrial dysfunction and induction of apoptosis [33]. PC12 cells have been routinely used to study cisplatin-induced neurotoxicity [60]. Our findings revealed that aprepitant attenuated cisplatin-induced neurotoxicity through inhibition of ROS-production and apoptosis. Our studies demonstrating the protective effect of aprepitant in PC12 neuronal cells are concurrent with other studies wherein aprepitant has been demonstrated to protect against cisplatin-induced nephrotoxicity and hepatotoxicity [61].
In the current studies, we determined that the levels of NK1R were significantly elevated in response to cisplatin in a rat neuronal cell line and in two TNBC cell lines. We also determined that the levels of ROS and apoptosis were significantly increased in response to cisplatin treatment. We determined that treatment with aprepitant (an NK1R antagonist that is widely used to attenuate chemotherapy-associated nausea) reversed one or more of these cisplatin-induced alterations in the rat neuronal cell line and in TNBC cells. Most importantly, we determined that aprepitant enhance the efficacy of cisplatin in TNBC cells while protecting PC12 cells from cisplatin-induced oxidative stress and apoptosis. The studies in this manuscript will determine if SP receptor antagonism in combination with cisplatin may serve as a novel, more efficacious and safer therapeutic option than existing therapies for TNBC.
5. Conclusions
These studies, if proven in animal models of chemotherapy-induced peripheral neuropathy (CIPN), could lead to the possibility of using SP antagonism as a therapeutic intervention to prevent chemotherapy-induced neurotoxicity. Most importantly, cisplatin is known to be highly effective in several cancers with increased survival rates. However, the side effects of neurotoxicity and ensuing CIPN often lead to the necessity for dose de-escalation, pausing of therapy or replacement with less effective chemotherapeutic regimens. These studies pursuant to animal studies could possibly determine the use of NK1R antagonism as a novel therapeutic strategy for the prevention of cisplatin-induced neurotoxicity and enhancement of the efficacy of chemotherapy in cancer.
Author Contributions
Conceptualization, P.R.; methodology, P.R., E.R., S.T.K., G.P., and Z.Z.; formal analyses, P.R., S.T.K., G.P., and Z.Z.; investigation, A.G.; original draft preparation, P.R.; review and editing, P.R., E.R., S.T.K., and G.P. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a Startup fund awarded to P. Robinson from the Department of Internal Medicine, University of Texas, MD Anderson Cancer Center. This work was conducted with support from the Cancer Prevention and Research Institute of Texas (CPRIT RP180734).
Institutional Review Board Statement
Ethical review and approval were waived for this study, since neither animals nor human subjects were used for this study.
Informed Consent Statement
Not Applicable.
Data Availability Statement
We are in the process of depositing our RNA-Seq data into Geo database, which can then be accessed by entering the manuscript information.
Conflicts of Interest
The authors do not have any conflict of interest to disclose.
References
- DeSantis, C.E.; Ma, J.; Goding Sauer, A.; Newman, L.A.; Jemal, A. Breast cancer statistics, 2017, racial disparity in mortality by state. CA Cancer J. Clin. 2017, 67, 439–448. [Google Scholar] [CrossRef] [Green Version]
- Collignon, J.; Lousberg, L.; Schroeder, H.; Jerusalem, G. Triple-negative breast cancer: Treatment challenges and solutions. Breast Cancer Targets Ther. 2016, 8, 93–107. [Google Scholar] [CrossRef] [Green Version]
- Parise, C.A.; Caggiano, V. Risk factors associated with the triple-negative breast cancer subtype within four race/ethnicities. Breast Cancer Res. Treat. 2017, 163, 151–158. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Z.; Hu, X.; Wang, B.; Wang, L.; Yang, W.; Liu, Y.; Liu, G.; Di, G.; Hu, Z.; et al. Cisplatin and gemcitabine as the first line therapy in metastatic triple negative breast cancer. Int. J. Cancer 2015, 136, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Kumar, S.; Tchounwou, P.B. Cisplatin-Based Chemotherapy of Human Cancers. J. Cancer Sci Ther 2019, 11, 97. [Google Scholar]
- Tsimberidou, A.M.; Braiteh, F.; Stewart, D.J.; Kurzrock, R. Ultimate fate of oncology drugs approved by the us food and drug administration without a randomized Trial. J. Clin. Oncol. 2009, 27, 6243–6250. [Google Scholar] [CrossRef]
- Dhar, S.; Kolishetti, N.; Lippard, S.J.; Farokhzad, O.C. Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 1850–1855. [Google Scholar] [CrossRef] [Green Version]
- Argyriou, A.A.; Bruna, J.; Marmiroli, P.; Cavaletti, G. Chemotherapy-induced peripheral neurotoxicity (CIPN): An update. Crit. Rev. Oncol. Hematol. 2012, 82, 51–77. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Munoz, M.; Rosso, M.; Covenas, R. A new frontier in the treatment of cancer: NK-1 receptor antagonists. Curr. Med. Chem. 2010, 17, 504–516. [Google Scholar] [CrossRef]
- Munoz, M.; Rosso, M.; Covenas, R. The NK-1 receptor: A new target in cancer therapy. Curr. Drug Targets 2011, 12, 909–921. [Google Scholar] [CrossRef]
- Hennig, I.M.; Laissue, J.A.; Horisberger, U.; Reubi, J.C. Substance-P receptors in human primary neoplasms: Tumoral and vascular localization. Int. J. Cancer 1995, 61, 786–792. [Google Scholar] [CrossRef]
- Munoz, M.; Covenas, R. Involvement of substance P and the NK-1 receptor in cancer progression. Peptides 2013, 48, 1–9. [Google Scholar] [CrossRef]
- Munoz, M.; Gonzalez-Ortega, A.; Salinas-Martin, M.V.; Carranza, A.; Garcia-Recio, S.; Almendro, V.; Covenas, R. The neurokinin-1 receptor antagonist aprepitant is a promising candidate for the treatment of breast cancer. Int. J. Oncol. 2014, 45, 1658–1672. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Moles, M.A.; Ramos-Garcia, P.; Esteban, F. Significance of the Overexpression of Substance P and Its Receptor NK-1R in Head and Neck Carcinogenesis: A Systematic Review and Meta-Analysis. Cancers 2021, 13, 1349. [Google Scholar] [CrossRef]
- Munoz, M.; Covenas, R. The Neurokinin-1 Receptor Antagonist Aprepitant: An Intelligent Bullet against Cancer? Cancers 2020, 12, 2682. [Google Scholar] [CrossRef]
- DeFea, K.A.; Vaughn, Z.D.; O’Bryan, E.M.; Nishijima, D.; Dery, O.; Bunnett, N.W. The proliferative and antiapoptotic effects of substance P are facilitated by formation of a beta -arrestin-dependent scaffolding complex. Proc. Natl. Acad. Sci. USA 2000, 97, 11086–11091. [Google Scholar] [CrossRef] [Green Version]
- Feng, F.; Yang, J.; Tong, L.; Yuan, S.; Tian, Y.; Hong, L.; Wang, W.; Zhang, H. Substance P immunoreactive nerve fibres are related to gastric cancer differentiation status and could promote proliferation and migration of gastric cancer cells. Cell Biol. Int. 2011, 35, 623–629. [Google Scholar] [CrossRef]
- Lewis, K.M.; Harford-Wright, E.; Vink, R.; Nimmo, A.J.; Ghabriel, M.N. Walker 256 tumour cells increase substance P immunoreactivity locally and modify the properties of the blood-brain barrier during extravasation and brain invasion. Clin. Exp. Metastasis 2013, 30, 1–12. [Google Scholar] [CrossRef]
- Munoz, M.; Covenas, R.; Esteban, F.; Redondo, M. The substance P/NK-1 receptor system: NK-1 receptor antagonists as anti-cancer drugs. J. Biosci. 2015, 40, 441–463. [Google Scholar] [CrossRef]
- Munoz, M.; Covenas, R. Involvement of substance P and the NK-1 receptor in human pathology. Amino Acids 2014, 46, 1727–1750. [Google Scholar] [CrossRef] [PubMed]
- Tejero-Taldo, M.I.; Kramer, J.H.; Mak Iu, T.; Komarov, A.M.; Weglicki, W.B. The nerve-heart connection in the pro-oxidant response to Mg-deficiency. Heart Fail. Rev. 2006, 11, 35–44. [Google Scholar] [CrossRef]
- Sterner-Kock, A.; Braun, R.K.; van der Vliet, A.; Schrenzel, M.D.; McDonald, R.J.; Kabbur, M.B.; Vulliet, P.R.; Hyde, D.M. Substance P primes the formation of hydrogen peroxide and nitric oxide in human neutrophils. J. Leukoc. Biol. 1999, 65, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.; Kasembeli, M.; Bharadwaj, U.; Engineer, N.; Eckols, K.T.; Tweardy, D.J. Substance P Receptor Signaling Mediates Doxorubicin-Induced Cardiomyocyte Apoptosis and Triple-Negative Breast Cancer Chemoresistance. Biomed. Res. Int. 2016, 2016, 1959270. [Google Scholar] [CrossRef] [Green Version]
- Mitro, N.; Cermenati, G.; Brioschi, E.; Abbiati, F.; Audano, M.; Giatti, S.; Crestani, M.; De Fabiani, E.; Azcoitia, I.; Garcia-Segura, L.M.; et al. Neuroactive steroid treatment modulates myelin lipid profile in diabetic peripheral neuropathy. J. Steroid Biochem. Mol. Biol. 2014, 143, 115–121. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, F.; Mao, J.; Lu, Y.; Li, J.; Ma, W.; Fan, S.; Zhang, C.; Li, Q.; Wang, B.; et al. Protein tyrosine phosphatase receptor-type delta acts as a negative regulator suppressing breast cancer. Oncotarget 2017, 8, 98798–98811. [Google Scholar] [CrossRef]
- Teider, N.; Scott, D.K.; Neiss, A.; Weeraratne, S.D.; Amani, V.M.; Wang, Y.; Marquez, V.E.; Cho, Y.J.; Pomeroy, S.L. Neuralized1 causes apoptosis and downregulates Notch target genes in medulloblastoma. Neuro Oncol. 2010, 12, 1244–1256. [Google Scholar] [CrossRef] [Green Version]
- Sopariwala, D.H.; Likhite, N.; Pei, G.; Haroon, F.; Lin, L.; Yadav, V.; Zhao, Z.; Narkar, V.A. Estrogen-related receptor alpha is involved in angiogenesis and skeletal muscle revascularization in hindlimb ischemia. FASEB J. 2021, 35, e21480. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [Green Version]
- Tchounwou, P.B.; Dasari, S.; Noubissi, F.K.; Ray, P.; Kumar, S. Advances in Our Understanding of the Molecular Mechanisms of Action of Cisplatin in Cancer Therapy. J. Exp. Pharmacol. 2021, 13, 303–328. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [Green Version]
- Starobova, H.; Vetter, I. Pathophysiology of Chemotherapy-Induced Peripheral Neuropathy. Front. Mol. Neurosci. 2017, 10, 174. [Google Scholar] [CrossRef]
- Tardaguila, M.; Mira, E.; Garcia-Cabezas, M.A.; Feijoo, A.M.; Quintela-Fandino, M.; Azcoitia, I.; Lira, S.A.; Manes, S. CX3CL1 promotes breast cancer via transactivation of the EGF pathway. Cancer Res. 2013, 73, 4461–4473. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Ouzounova, M.; Piranlioglu, R.; Ma, M.T.; Guzel, M.; Marasco, D.; Chadli, A.; Gestwicki, J.E.; Cowell, J.K.; Wicha, M.S.; et al. The pleiotropic effects of TNFalpha in breast cancer subtypes is regulated by TNFAIP3/A20. Oncogene 2019, 38, 469–482. [Google Scholar] [CrossRef]
- Bravo-Cordero, J.J.; Hodgson, L.; Condeelis, J. Directed cell invasion and migration during metastasis. Curr. Opin. Cell Biol. 2012, 24, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Venning, F.A.; Wullkopf, L.; Erler, J.T. Targeting ECM Disrupts Cancer Progression. Front. Oncol. 2015, 5, 224. [Google Scholar] [CrossRef] [Green Version]
- Fingleton, B. Matrix metalloproteinases: Roles in cancer and metastasis. Front. Biosci. 2006, 11, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Shay, G.; Lynch, C.C.; Fingleton, B. Moving targets: Emerging roles for MMPs in cancer progression and metastasis. Matrix Biol. 2015, 44-46, 200–206. [Google Scholar] [CrossRef]
- Johnson, B.; Mahadevan, D. Emerging Role and Targeting of Carcinoembryonic Antigen-related Cell Adhesion Molecule 6 (CEACAM6) in Human Malignancies. Clin. Cancer Drugs 2015, 2, 100–111. [Google Scholar] [CrossRef]
- Liu, Q.; Li, A.; Tian, Y.; Wu, J.D.; Liu, Y.; Li, T.; Chen, Y.; Han, X.; Wu, K. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. 2016, 31, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Okano, M.; Oshi, M.; Butash, A.L.; Katsuta, E.; Tachibana, K.; Saito, K.; Okayama, H.; Peng, X.; Yan, L.; Kono, K.; et al. Triple-Negative Breast Cancer with High Levels of Annexin A1 Expression Is Associated with Mast Cell Infiltration, Inflammation, and Angiogenesis. Int. J. Mol. Sci 2019, 20, 4197. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Shi, Z.; Bai, Y.; Liu, L.; Cheng, K. Prognostic significance of systemic immune-inflammation index in triple-negative breast cancer. Cancer Manag Res. 2019, 11, 4471–4480. [Google Scholar] [CrossRef] [Green Version]
- Bergenfelz, C.; Gaber, A.; Allaoui, R.; Mehmeti, M.; Jirstrom, K.; Leanderson, T.; Leandersson, K. S100A9 expressed in ER(-)PgR(-) breast cancers induces inflammatory cytokines and is associated with an impaired overall survival. Br. J. Cancer 2015, 113, 1234–1243. [Google Scholar] [CrossRef] [Green Version]
- Liubomirski, Y.; Lerrer, S.; Meshel, T.; Rubinstein-Achiasaf, L.; Morein, D.; Wiemann, S.; Korner, C.; Ben-Baruch, A. Tumor-Stroma-Inflammation Networks Promote Pro-metastatic Chemokines and Aggressiveness Characteristics in Triple-Negative Breast Cancer. Front. Immunol 2019, 10, 757. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yang, Z.; Lu, W.; Chen, Z.; Chen, L.; Han, S.; Wu, X.; Cai, T.; Cai, Y. Chemokines and chemokine receptors: A new strategy for breast cancer therapy. Cancer Med. 2020, 9, 3786–3799. [Google Scholar] [CrossRef] [PubMed]
- Ignacio, R.M.C.; Gibbs, C.R.; Kim, S.; Lee, E.S.; Adunyah, S.E.; Son, D.S. Serum amyloid A predisposes inflammatory tumor microenvironment in triple negative breast cancer. Oncotarget 2019, 10, 511–526. [Google Scholar] [CrossRef] [Green Version]
- Marzec, K.A.; Baxter, R.C.; Martin, J.L. Targeting Insulin-Like Growth Factor Binding Protein-3 Signaling in Triple-Negative Breast Cancer. Biomed. Res. Int. 2015, 2015, 638526. [Google Scholar] [CrossRef] [PubMed]
- Pascual, J.; Turner, N.C. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann. Oncol. 2019, 30, 1051–1060. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.; Yuan, P.; Du, F.; Hong, R.; Ding, X.; Shi, X.; Fan, Y.; Wang, J.; Luo, Y.; Ma, F.; et al. SPARC overexpression in primary tumors correlates with disease recurrence and overall survival in patients with triple negative breast cancer. Oncotarget 2016, 7, 76628–76634. [Google Scholar] [CrossRef]
- Wiedemann, S.M.; Mildner, S.N.; Bonisch, C.; Israel, L.; Maiser, A.; Matheisl, S.; Straub, T.; Merkl, R.; Leonhardt, H.; Kremmer, E.; et al. Identification and characterization of two novel primate-specific histone H3 variants, H3.X and H3.Y. J. Cell Biol. 2010, 190, 777–791. [Google Scholar] [CrossRef] [Green Version]
- Hojo, T.; Akiyama, Y.; Nagasaki, K.; Maruyama, K.; Kikuchi, K.; Ikeda, T.; Kitajima, M.; Yamaguchi, K. Association of maspin expression with the malignancy grade and tumor vascularization in breast cancer tissues. Cancer Lett. 2001, 171, 103–110. [Google Scholar] [CrossRef]
- Xia, W.; Lau, Y.K.; Hu, M.C.; Li, L.; Johnston, D.A.; Sheng, S.; El-Naggar, A.; Hung, M.C. High tumoral maspin expression is associated with improved survival of patients with oral squamous cell carcinoma. Oncogene 2000, 19, 2398–2403. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.G.; Kim, W.J.; Noh, M.G.; Chun, K.H.; Kim, S.J. SPON2 Is Upregulated through Notch Signaling Pathway and Promotes Tumor Progression in Gastric Cancer. Cancers 2020, 12, 1439. [Google Scholar] [CrossRef]
- Robertson, D.M.; Pruysers, E.; Jobling, T. Inhibin as a diagnostic marker for ovarian cancer. Cancer Lett. 2007, 249, 14–17. [Google Scholar] [CrossRef]
- Lasham, A.; Mehta, S.Y.; Fitzgerald, S.J.; Woolley, A.G.; Hearn, J.I.; Hurley, D.G.; Ruza, I.; Algie, M.; Shelling, A.N.; Braithwaite, A.W.; et al. A novel EGR-1 dependent mechanism for YB-1 modulation of paclitaxel response in a triple negative breast cancer cell line. Int. J. Cancer 2016, 139, 1157–1170. [Google Scholar] [CrossRef] [Green Version]
- Ozols, R.F.; Young, R.C. High-dose cisplatin therapy in ovarian cancer. Semin. Oncol. 1985, 12, 21–30. [Google Scholar]
- Seretny, M.; Currie, G.L.; Sena, E.S.; Ramnarine, S.; Grant, R.; MacLeod, M.R.; Colvin, L.A.; Fallon, M. Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: A systematic review and meta-analysis. Pain 2014, 155, 2461–2470. [Google Scholar] [CrossRef] [Green Version]
- Cersosimo, R.J. Cisplatin neurotoxicity. Cancer Treat. Rev. 1989, 16, 195–211. [Google Scholar] [CrossRef]
- Li, D.W.; Sun, J.Y.; Wang, K.; Zhang, S.; Hou, Y.J.; Yang, M.F.; Fu, X.Y.; Zhang, Z.Y.; Mao, L.L.; Yuan, H.; et al. Attenuation of Cisplatin-Induced Neurotoxicity by Cyanidin, a Natural Inhibitor of ROS-Mediated Apoptosis in PC12 Cells. Cell Mol. Neurobiol. 2015, 35, 995–1001. [Google Scholar] [CrossRef]
- Un, H.; Ugan, R.A.; Kose, D.; Bayir, Y.; Cadirci, E.; Selli, J.; Halici, Z. A novel effect of Aprepitant: Protection for cisplatin-induced nephrotoxicity and hepatotoxicity. Eur. J. Pharmacol. 2020, 880, 173168. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).