
The Most Common VHL Point Mutation R167Q in Hereditary VHL Disease Interferes with Cell Plasticity Regulation


 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mutated VHL Cell Lines
2.2. Western Blots
2.3. Phase Contrast and Fluorescence Microscopy
2.4. Clonogenic Assay
2.5. Immunohistochemistry Staining for AXL
2.6. Flow Cytometry Analysis
2.7. RNA-Sequencing and Gene Set Enrichment Analysis
2.8. Analysis of Gene-Expression Data Derived from The Cancer Genome Atlas
3. Results
3.1. VHL-R167Q and WT VHL Display Similar Slight Effect on Cell Morphology and Colony Formation, Whereas VHL-R167Q Promotes HIF-2 Expression
3.2. The R167Q VHL Mutation Interferes with Epithelial–Mesenchymal Plasticity
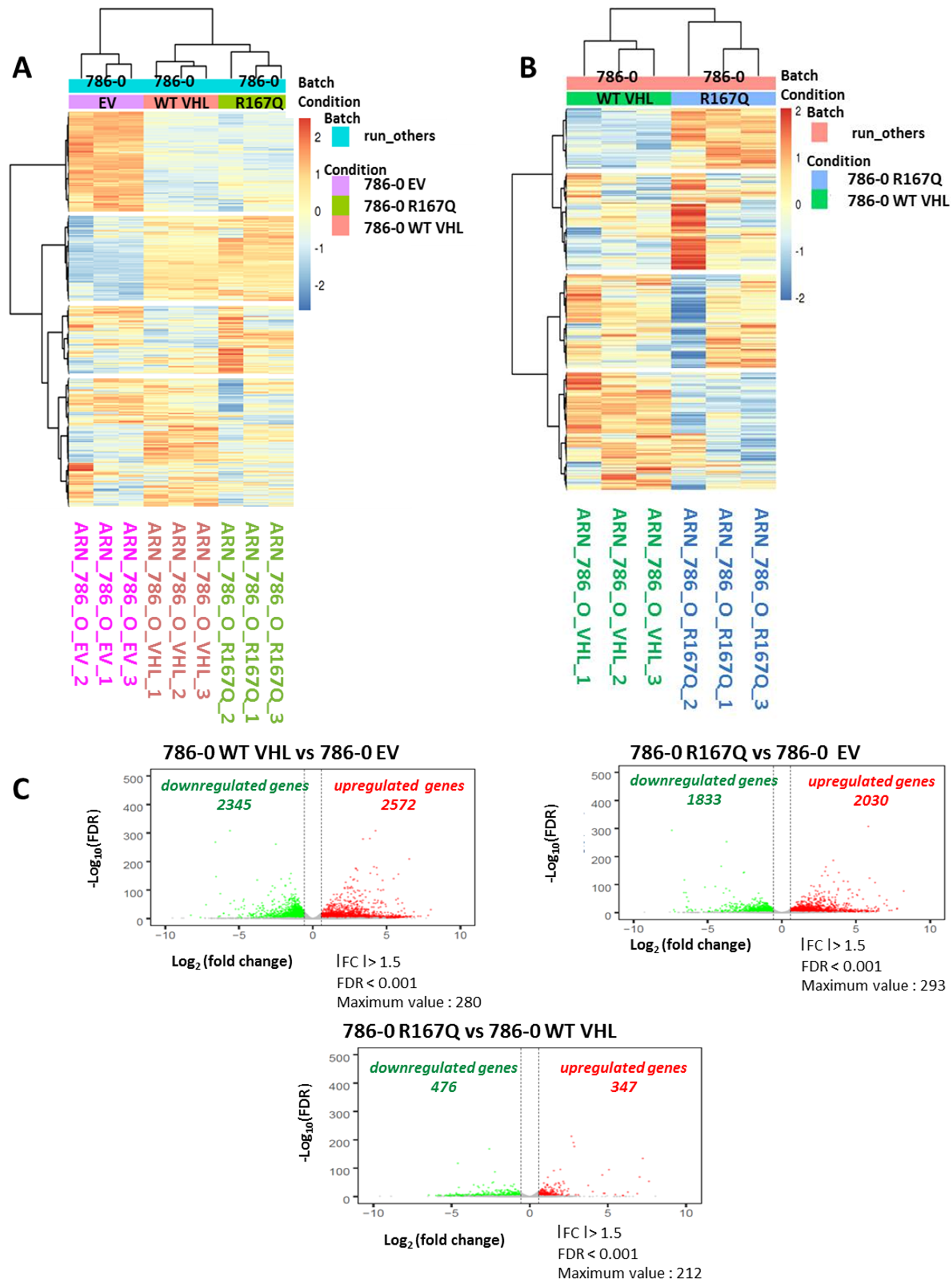
3.3. Comparison of Transcriptional Profile of WT VHL and VHL-R167Q Showed Discrete Differences
3.4. VHL-R167Q Mutation Modulates the Expression of Genes Belonging to Hypoxia and Stemness Pathways
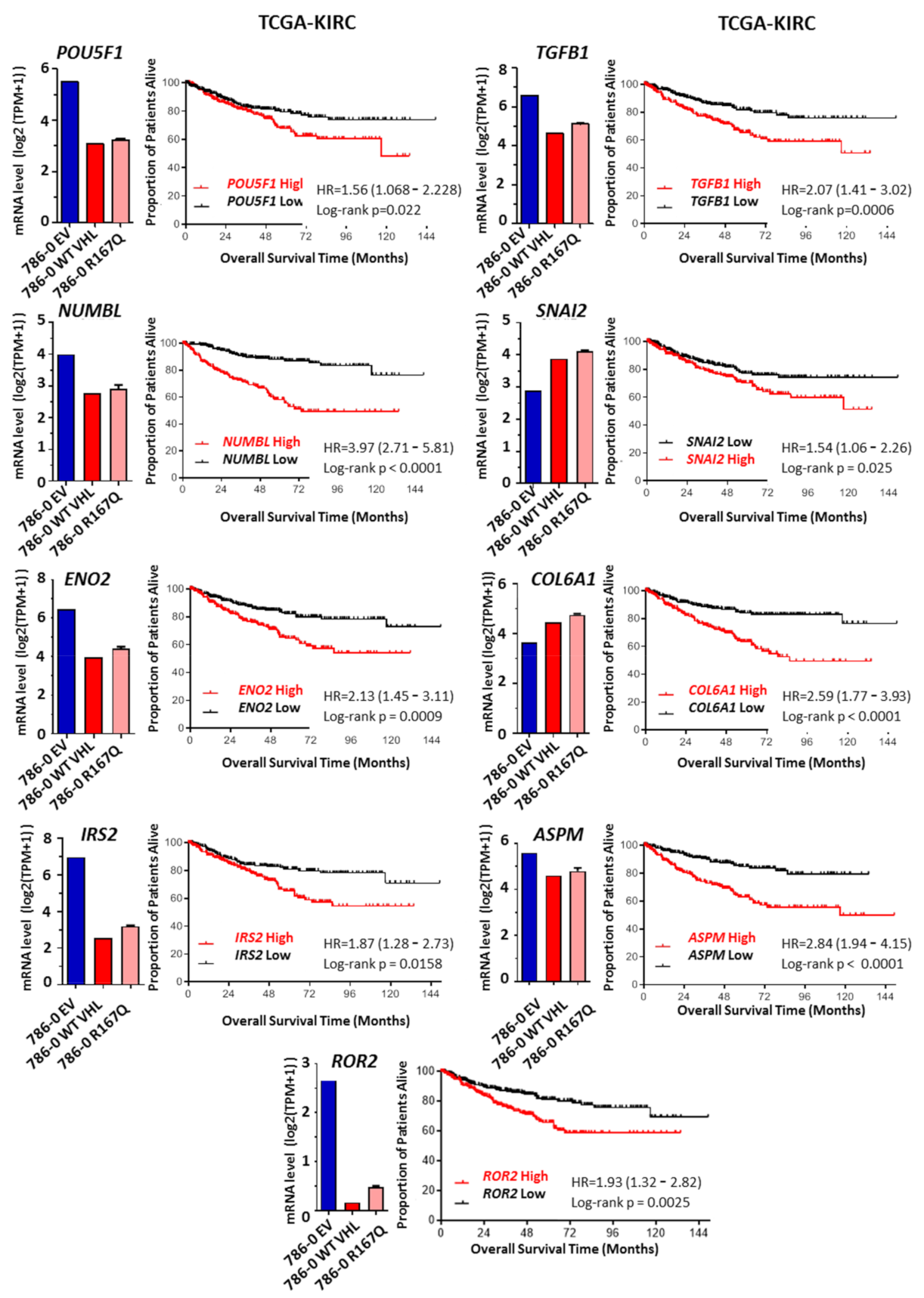
3.5. R167Q VHL Mutation Regulates Genes Associated with Poor Prognostic and Overall Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zbar, B.; Kishida, T.; Chen, F.; Schmidt, L.; Maher, E.R.; Richards, F.M.; Crossey, P.A.; Webster, A.R.; Affara, N.A.; Ferguson-Smith, M.A.; et al. Germline mutations in the Von Hippel-Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum. Mutat. 1996, 8, 348–357. [Google Scholar] [CrossRef]
- Lane, B.R.; Kattan, M.W. Predicting outcomes in renal cell carcinoma. Curr. Opin. Urol. 2005, 15, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef] [PubMed]
- Gnarra, J.R.; Tory, K.; Weng, Y.; Schmidt, L.; Wei, M.H.; Li, H.; Latif, F.; Liu, S.; Chen, F.; Duh, F.M. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat. Genet. 1994, 7, 85–90. [Google Scholar] [CrossRef]
- Salceda, S.; Caro, J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J. Biol. Chem. 1997, 272, 22642–22647. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Tanimoto, K.; Makino, Y.; Pereira, T.; Poellinger, L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000, 19, 4298–4309. [Google Scholar] [CrossRef] [Green Version]
- Trio Wins Nobel for Hypoxia Discoveries. Cancer Discov. 2019, 9, 1636–1637. [CrossRef] [Green Version]
- Cowey, C.L.; Rathmell, W.K. VHL gene mutations in renal cell carcinoma: Role as a biomarker of disease outcome and drug efficacy. Curr. Oncol. Rep. 2009, 11, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Patard, J.-J.; Rioux-Leclercq, N.; Masson, D.; Zerrouki, S.; Jouan, F.; Collet, N.; Dubourg, C.; Lobel, B.; Denis, M.; Fergelot, P. Absence of VHL gene alteration and high VEGF expression are associated with tumour aggressiveness and poor survival of renal-cell carcinoma. Br. J. Cancer 2009, 101, 1417–1424. [Google Scholar] [CrossRef]
- Rathmell, W.K.; Chen, S. VHL inactivation in renal cell carcinoma: Implications for diagnosis, prognosis and treatment. Expert Rev. Anticancer Ther. 2008, 8, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Brodaczewska, K.K.; Szczylik, C.; Kieda, C. Immune consequences of anti-angiogenic therapyin renal cell carcinoma. Contemp. Oncol. 2018, 22, 14–22. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–C579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordstrom-O’Brien, M.; van der Luijt, R.B.; van Rooijen, E.; van den Ouweland, A.M.; Majoor-Krakauer, D.F.; Lolkema, M.P.; van Brussel, A.; Voest, E.E.; Giles, R.H. Genetic analysis of von Hippel-Lindau disease. Hum. Mutat. 2010, 31, 521–537. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; German, P.; Bai, S.; Reddy, A.S.; Liu, X.-D.; Sun, M.; Zhou, L.; Chen, X.; Zhao, X.; Wu, C.; et al. Genetic and pharmacological strategies to refunctionalize the von Hippel Lindau R167Q mutant protein. Cancer Res. 2014, 74, 3127–3136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couvé, S.; Ladroue, C.; Laine, E.; Mahtouk, K.; Guégan, J.; Gad, S.; Le Jeune, H.; Le Gentil, M.; Nuel, G.; Kim, W.Y.; et al. Genetic evidence of a precisely tuned dysregulation in the hypoxia signaling pathway during oncogenesis. Cancer Res. 2014, 74, 6554–6564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.D.; Elliott, A.Y.; Stein, N.; Fraley, E.E. In vitro cultivation of human renal cell cancer. I. Establishment of cells in culture. Vitro 1976, 12, 623–627. [Google Scholar] [CrossRef]
- Corgnac, S.; Malenica, I.; Mezquita, L.; Auclin, E.; Voilin, E.; Kacher, J.; Halse, H.; Grynszpan, L.; Signolle, N.; Dayris, T.; et al. CD103+CD8+ TRM Cells Accumulate in Tumors of Anti-PD-1-Responder Lung Cancer Patients and Are Tumor-Reactive Lymphocytes Enriched with Tc17. Cell Rep. Med. 2020, 1, 100127. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pimentel, H.; Bray, N.L.; Puente, S.; Melsted, P.; Pachter, L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods 2017, 14, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gjerdrum, C.; Tiron, C.; Hoiby, T.; Stefansson, I.; Haugen, H.; Sandal, T.; Collett, K.; Li, S.; McCormack, E.; Gjertsen, B.T.; et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc. Natl. Acad. Sci. USA 2010, 107, 1124–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotsberg, M.L.; Rayford, A.; Thiery, J.P.; Belleggia, G.; Peters, S.D.; Lorens, J.B.; Chouaib, S.; Terry, S.; Engelsen, A.S.T. Decoding cancer’s camouflage: Epithelial-mesenchymal plasticity in resistance to immune checkpoint blockade. CDR 2020, 3, 832–853. [Google Scholar] [CrossRef]
- Antony, J.; Huang, R.Y. AXL-Driven EMT State as a Targetable Conduit in Cancer. Cancer Res. 2017, 77, 3725–3732. [Google Scholar] [CrossRef] [Green Version]
- Hasmim, M.; Noman, M.Z.; Lauriol, J.; Benlalam, H.; Mallavialle, A.; Rosselli, F.; Mami-Chouaib, F.; Alcaide-Loridan, C.; Chouaib, S. Hypoxia-dependent inhibition of tumor cell susceptibility to CTL-mediated lysis involves NANOG induction in target cells. J. Immunol. 2011, 187, 4031–4039. [Google Scholar] [CrossRef]
- Wang, K.; Chen, X.; Zhan, Y.; Jiang, W.; Liu, X.; Wang, X.; Wu, B. Increased expression of ALDH1A1 protein is associated with poor prognosis in clear cell renal cell carcinoma. Med. Oncol. 2013, 30, 574. [Google Scholar] [CrossRef]
- Ciccone, V.; Morbidelli, L.; Ziche, M.; Donnini, S. How to conjugate the stemness marker ALDH1A1 with tumor angiogenesis, progression, and drug resistance. Cancer Drug Resist. 2020, 3, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Midwood, K.S.; Chiquet, M.; Tucker, R.P.; Orend, G. Tenascin-C at a glance. J. Cell Sci. 2016, 129, 4321–4327. [Google Scholar] [CrossRef] [Green Version]
- Jachetti, E.; Caputo, S.; Mazzoleni, S.; Brambillasca, C.S.; Parigi, S.M.; Grioni, M.; Piras, I.S.; Restuccia, U.; Calcinotto, A.; Freschi, M.; et al. Tenascin-C Protects Cancer Stem-like Cells from Immune Surveillance by Arresting T-cell Activation. Cancer Res. 2015, 75, 2095–2108. [Google Scholar] [CrossRef] [Green Version]
- Ghaleb, A.M.; Yang, V.W. Krüppel-like factor 4 (KLF4): What we currently know. Gene 2017, 611, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.-M.; Luo, Y.-L.; Li, S.; Li, Z.-X.; Jiang, L.; Zhang, G.-X.; Owusu, L.; Chen, H.-L. Multifunctional neuron-specific enolase: Its role in lung diseases. Biosci. Rep. 2019, 39, BSR20192732. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Niu, X.; Liao, L.; Cho, E.-A.; Yang, H. The contributions of HIF-target genes to tumor growth in RCC. PLoS ONE 2013, 8, e80544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, H.-L.; Liu, T.-T.; Weng, S.-W.; Chen, C.-H.; Wei, Y.-C.; Eng, H.-L.; Huang, W.-T. Association of IRS2 overexpression with disease progression in intrahepatic cholangiocarcinoma. Oncol. Lett. 2018, 16, 5505–5511. [Google Scholar] [CrossRef]
- Piper, A.J.; Clark, J.L.; Mercado-Matos, J.; Matthew-Onabanjo, A.N.; Hsieh, C.-C.; Akalin, A.; Shaw, L.M. Insulin Receptor Substrate-1 (IRS-1) and IRS-2 expression levels are associated with prognosis in non-small cell lung cancer (NSCLC). PLoS ONE 2019, 14, e0220567. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, M.P.; Marchbank, K.; Webster, M.R.; Valiga, A.A.; Kaur, A.; Vultur, A.; Li, L.; Herlyn, M.; Villanueva, J.; Liu, Q.; et al. Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ROR1 and ROR2. Cancer Discov. 2013, 3, 1378–1393. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, N.R.; Debebe, Z.; Wright, T.M.; Brooks, S.A.; Sendor, A.B.; Brannon, A.R.; Hakimi, A.A.; Hsieh, J.J.; Choueiri, T.K.; Tamboli, P.; et al. Expression of Ror2 mediates invasive phenotypes in renal cell carcinoma. PLoS ONE 2014, 9, e116101. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-M.; Ji, S.; Li, Y.; Fu, L.-Y.; Jiang, T.; Meng, F.-D. Ror2, a Developmentally Regulated Kinase, Is Associated With Tumor Growth, Apoptosis, Migration, and Invasion in Renal Cell Carcinoma. Oncol. Res. 2017, 25, 195–205. [Google Scholar] [CrossRef]
- Covello, K.L.; Kehler, J.; Yu, H.; Gordan, J.D.; Arsham, A.M.; Hu, C.-J.; Labosky, P.A.; Simon, M.C.; Keith, B. HIF-2alpha regulates Oct-4: Effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006, 20, 557–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Ro, J.Y.; Kim, S.; Cho, Y.M. Expression of stem-cell markers OCT-4 and CD133: Important prognostic factors in papillary renal cell carcinoma. Hum. Pathol. 2012, 43, 2109–2116. [Google Scholar] [CrossRef]
- Koo, B.S.; Lee, S.H.; Kim, J.M.; Huang, S.; Kim, S.H.; Rho, Y.S.; Bae, W.J.; Kang, H.J.; Kim, Y.S.; Moon, J.H.; et al. Oct4 is a critical regulator of stemness in head and neck squamous carcinoma cells. Oncogene 2015, 34, 2317–2324. [Google Scholar] [CrossRef] [PubMed]
- Zeineddine, D.; Hammoud, A.A.; Mortada, M.; Boeuf, H. The Oct4 protein: More than a magic stemness marker. Am. J. Stem Cells 2014, 3, 74–82. [Google Scholar] [PubMed]
- Bocci, F.; Jolly, M.K.; Tripathi, S.C.; Aguilar, M.; Hanash, S.M.; Levine, H.; Onuchic, J.N. Numb prevents a complete epithelial-mesenchymal transition by modulating Notch signalling. J. R. Soc. Interface 2017, 14, 20170512. [Google Scholar] [CrossRef] [Green Version]
- García-Heredia, J.M.; Verdugo Sivianes, E.M.; Lucena-Cacace, A.; Molina-Pinelo, S.; Carnero, A. Numb-like (NumbL) downregulation increases tumorigenicity, cancer stem cell-like properties and resistance to chemotherapy. Oncotarget 2016, 7, 63611–63628. [Google Scholar] [CrossRef] [Green Version]
- Mallikarjuna, P.; Sitaram, R.T.; Landström, M.; Ljungberg, B. VHL status regulates transforming growth factor-β signaling pathways in renal cell carcinoma. Oncotarget 2018, 9, 16297–16310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Feldkoren, B.; Hutchinson, R.; Rapoport, Y.; Mahajan, A.; Margulis, V. Integrin signaling potentiates transforming growth factor-beta 1 (TGF-β1) dependent down-regulation of E-Cadherin expression—Important implications for epithelial to mesenchymal transition (EMT) in renal cell carcinoma. Exp. Cell Res. 2017, 355, 57–66. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Nassour, M.; Idoux-Gillet, Y.; Selmi, A.; Côme, C.; Faraldo, M.-L.M.; Deugnier, M.-A.; Savagner, P. Slug controls stem/progenitor cell growth dynamics during mammary gland morphogenesis. PLoS ONE 2012, 7, e53498. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Xu, W.-H.; Ren, F.; Wang, J.; Wang, H.-K.; Cao, D.-L.; Shi, G.-H.; Qu, Y.-Y.; Zhang, H.-L.; Ye, D.-W. Prognostic value of epithelial-mesenchymal transition markers in clear cell renal cell carcinoma. Aging 2020, 12, 866–883. [Google Scholar] [CrossRef]
- Chen, P.; Cescon, M.; Bonaldo, P. Collagen VI in cancer and its biological mechanisms. Trends Mol. Med. 2013, 19, 410–417. [Google Scholar] [CrossRef]
- Karabulut, Y.Y.; Köse, E.Ç.; Bozlu, M.; Tuncel, F.; Yüksek, G.E.; Etit, D.; Toru, H.S.; Akkaya, B.; Çelik, Z.E.; Öznur, M.; et al. The role of COL6A1and PD-1 expressions in renal cell carcinoma. Turk. J. Urol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Fish, J.L.; Kosodo, Y.; Enard, W.; Pääbo, S.; Huttner, W.B. Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc. Natl. Acad. Sci. USA 2006, 103, 10438–10443. [Google Scholar] [CrossRef] [Green Version]
- Capecchi, M.R.; Pozner, A. ASPM regulates symmetric stem cell division by tuning Cyclin E ubiquitination. Nat. Commun. 2015, 6, 8763. [Google Scholar] [CrossRef] [PubMed]
- Bangiyeva, V.; Rosenbloom, A.; Alexander, A.E.; Isanova, B.; Popko, T.; Schoenfeld, A.R. Differences in regulation of tight junctions and cell morphology between VHL mutations from disease subtypes. BMC Cancer 2009, 9, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.; Roberts, A.M.; Chow, J.; Coady-Osberg, N.; Ohh, M. Homotypic association between tumour-associated VHL proteins leads to the restoration of HIF pathway. Oncogene 2006, 25, 3079–3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brauch, H.; Weirich, G.; Brieger, J.; Glavac, D.; Rödl, H.; Eichinger, M.; Feurer, M.; Weidt, E.; Puranakanitstha, C.; Neuhaus, C.; et al. VHL alterations in human clear cell renal cell carcinoma: Association with advanced tumor stage and a novel hot spot mutation. Cancer Res. 2000, 60, 1942–1948. [Google Scholar] [PubMed]
- Barnabas, N.; Amin, M.B.; Pindolia, K.; Nanavati, R.; Amin, M.B.; Worsham, M.J. Mutations in the von Hippel-Lindau (VHL) gene refine differential diagnostic criteria in renal cell carcinoma. J. Surg. Oncol. 2002, 80, 52–60. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buart, S.; Terry, S.; Diop, M.K.; Dessen, P.; Couvé, S.; Abdou, A.; Adam, J.; Thiery, J.; Savagner, P.; Chouaib, S. The Most Common VHL Point Mutation R167Q in Hereditary VHL Disease Interferes with Cell Plasticity Regulation. Cancers 2021, 13, 3897. https://doi.org/10.3390/cancers13153897
Buart S, Terry S, Diop MK, Dessen P, Couvé S, Abdou A, Adam J, Thiery J, Savagner P, Chouaib S. The Most Common VHL Point Mutation R167Q in Hereditary VHL Disease Interferes with Cell Plasticity Regulation. Cancers. 2021; 13(15):3897. https://doi.org/10.3390/cancers13153897
Chicago/Turabian StyleBuart, Stéphanie, Stéphane Terry, M’boyba Khadija Diop, Philippe Dessen, Sophie Couvé, Abdérémane Abdou, Julien Adam, Jérôme Thiery, Pierre Savagner, and Salem Chouaib. 2021. "The Most Common VHL Point Mutation R167Q in Hereditary VHL Disease Interferes with Cell Plasticity Regulation" Cancers 13, no. 15: 3897. https://doi.org/10.3390/cancers13153897
APA StyleBuart, S., Terry, S., Diop, M. K., Dessen, P., Couvé, S., Abdou, A., Adam, J., Thiery, J., Savagner, P., & Chouaib, S. (2021). The Most Common VHL Point Mutation R167Q in Hereditary VHL Disease Interferes with Cell Plasticity Regulation. Cancers, 13(15), 3897. https://doi.org/10.3390/cancers13153897