
Macrophages Impair TLR9 Agonist Antitumor Activity through Interacting with the Anti-PD-1 Antibody Fc Domain

 ,
,  ,
, 
 , , , , ,
, , , , ,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture
2.3. Mice and In Vivo Studies
2.4. Flow Cytometry Analysis
2.5. Immunohistochemistry and Immunofluorescence
2.6. In Vitro Studies
2.7. Quantitative PCR Analysis
2.8. Generation of Bone Marrow-Derived Macrophages (BMDMs) and Isolation of NK Cells
2.9. 51Cr Release Assay
2.10. Statistical Analysis
3. Results
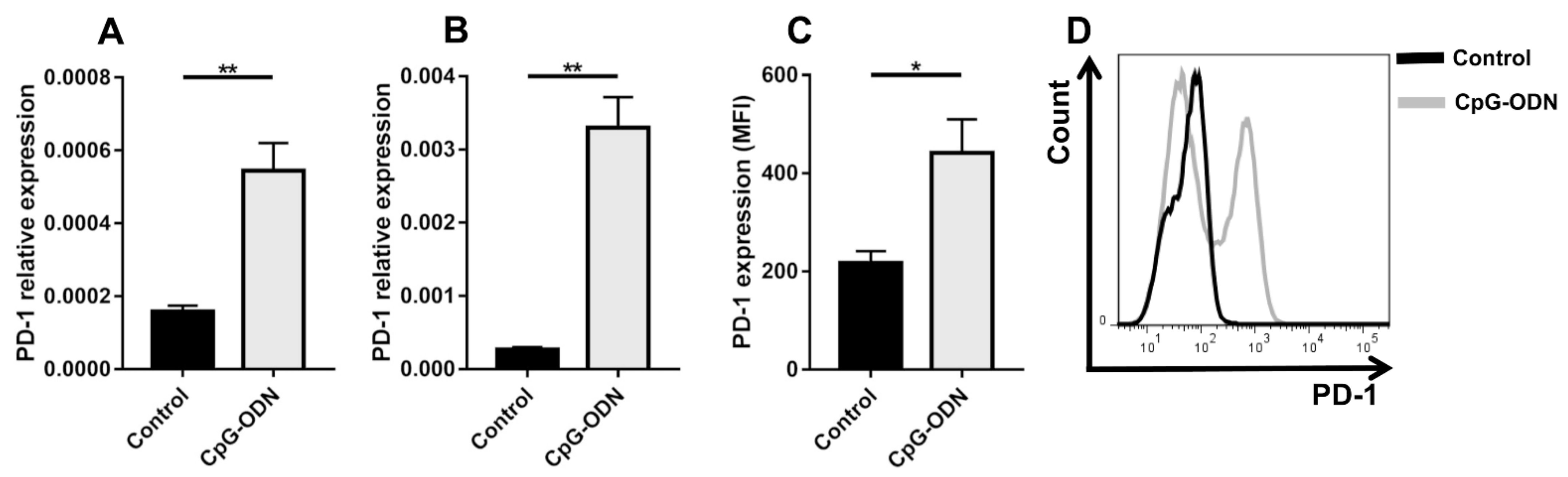
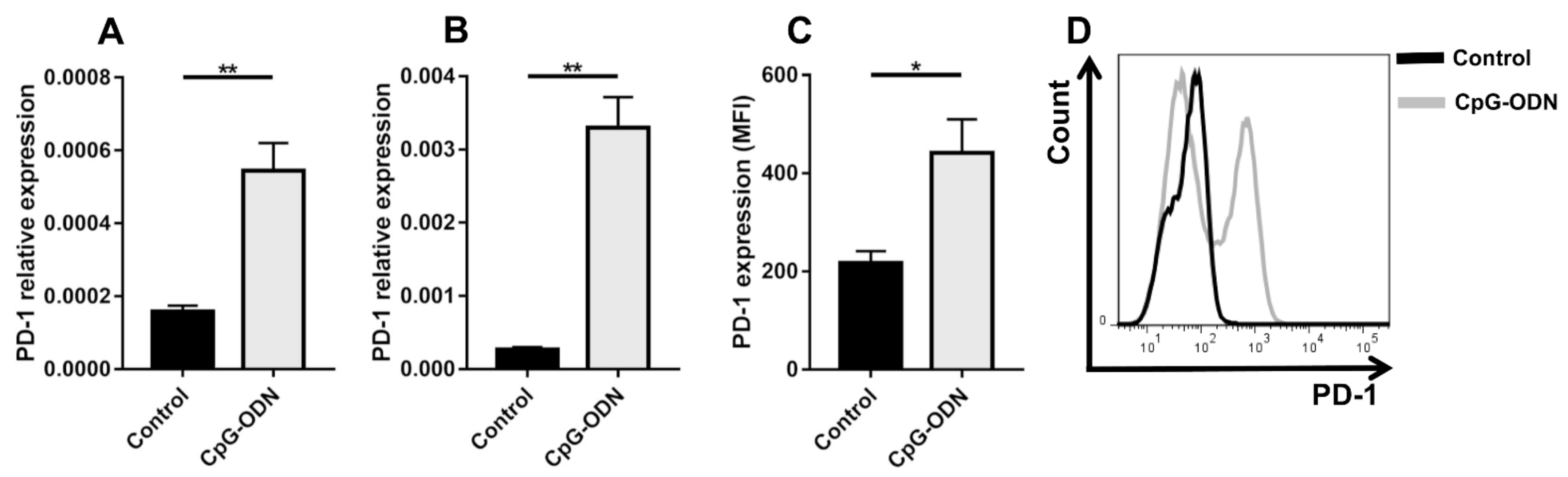
3.1. TLR9 Stimulation by CpG-ODNs Enhances PD-1 Expression on Peritoneal Immune Cells
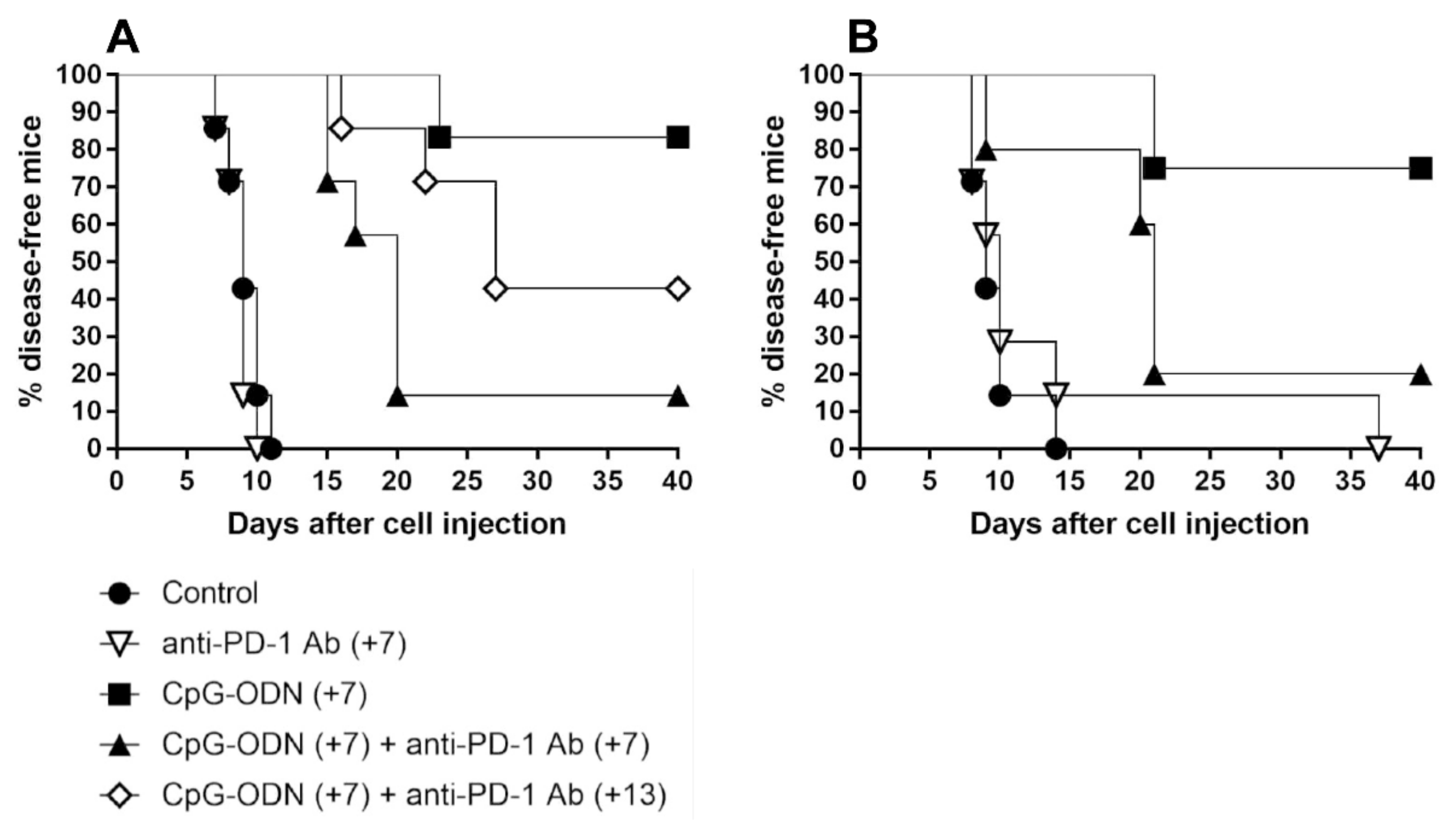
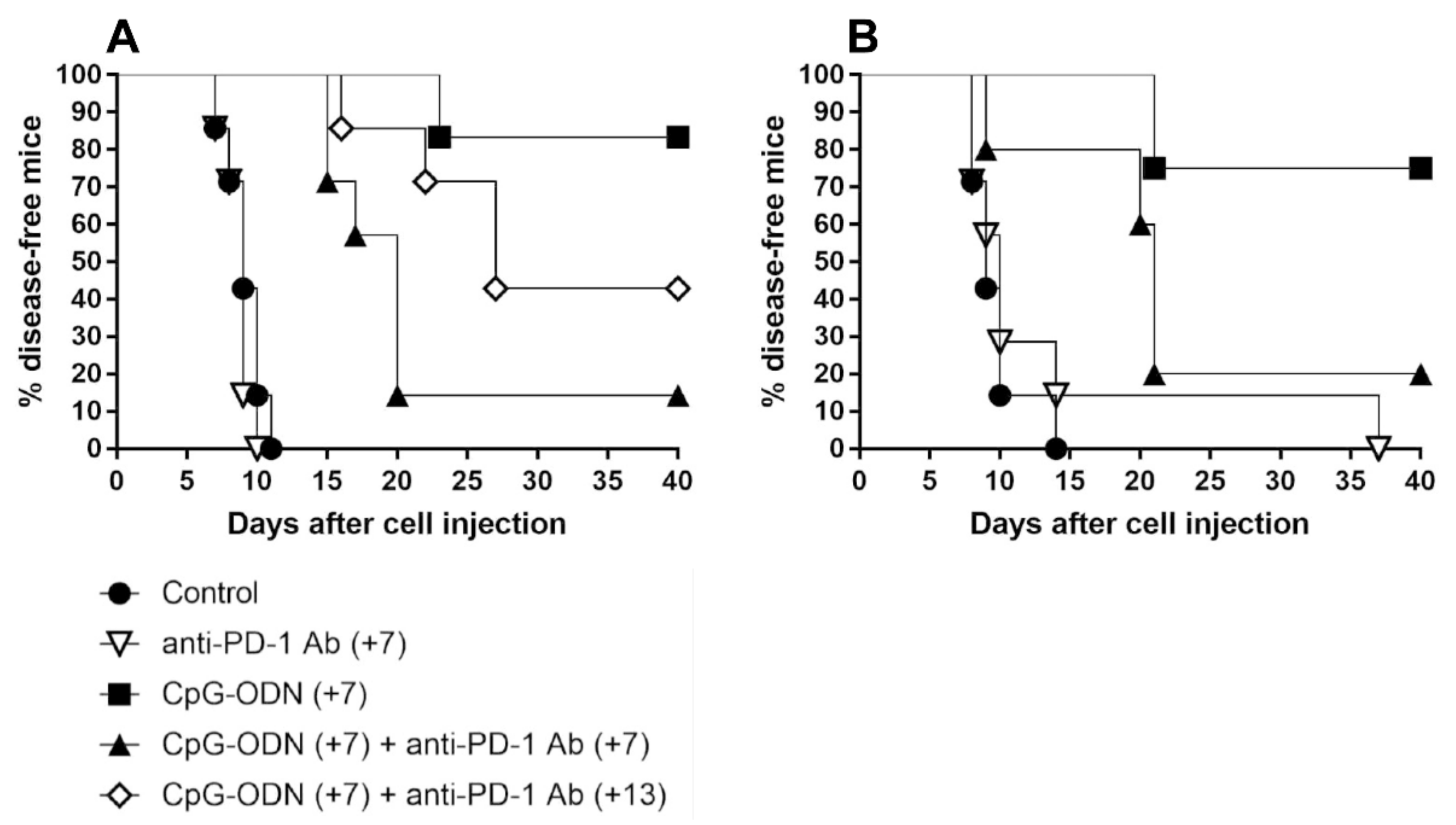
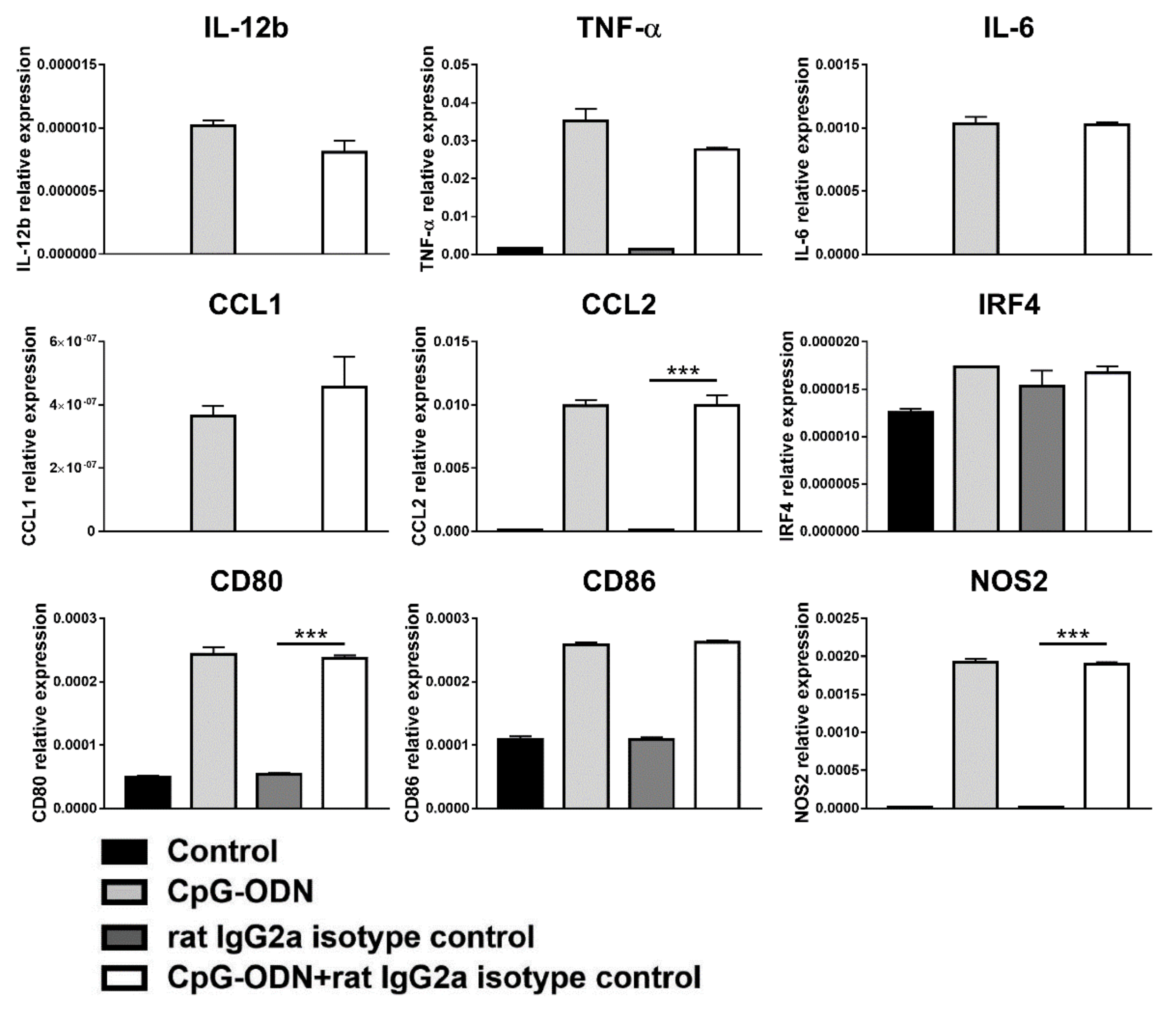
3.2. An Anti-PD-1 Antibody Reduces the Antitumor Activity of CpG-ODNs in Athymic Nude Mice Bearing Human Ovarian Cancer Xenografts
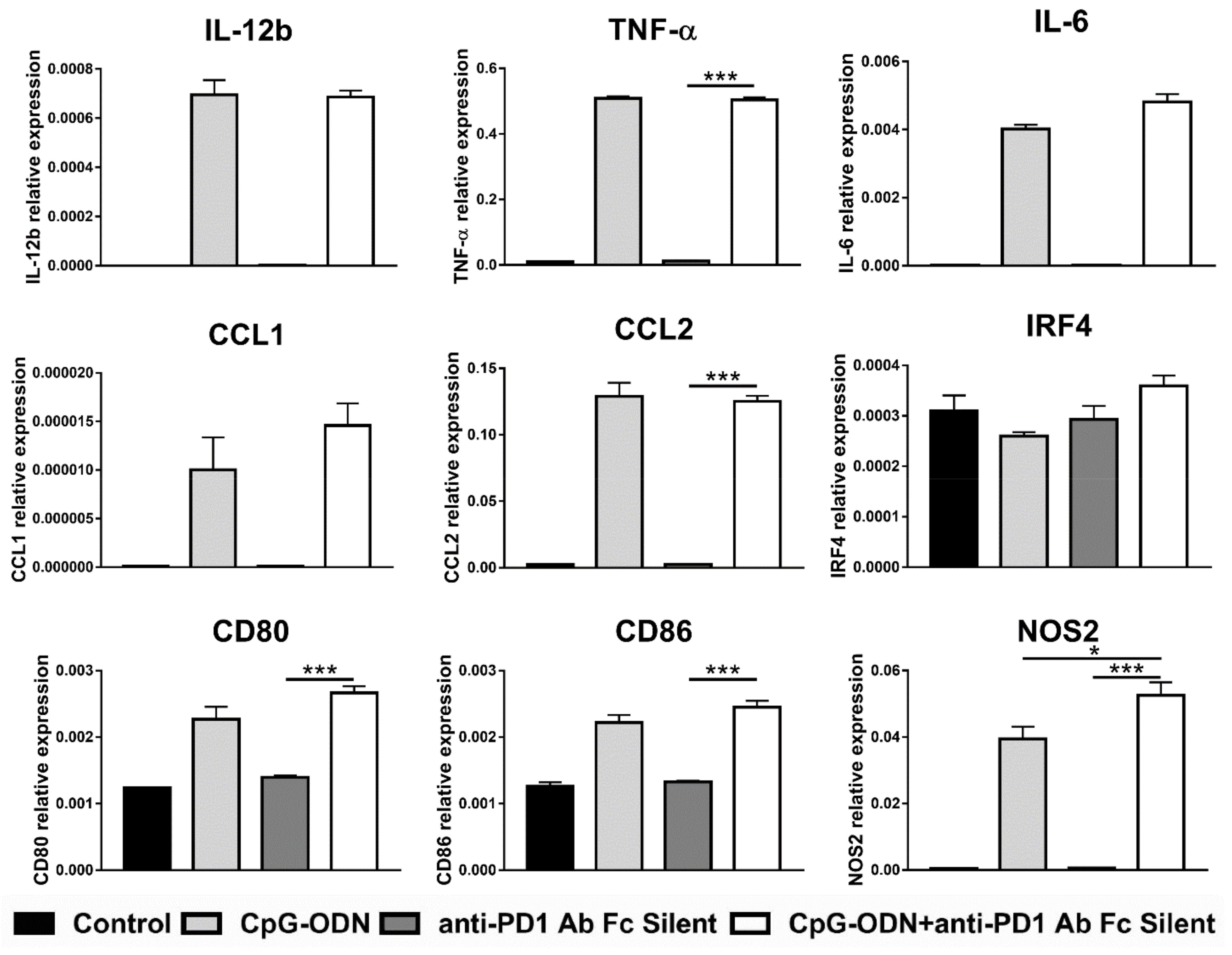
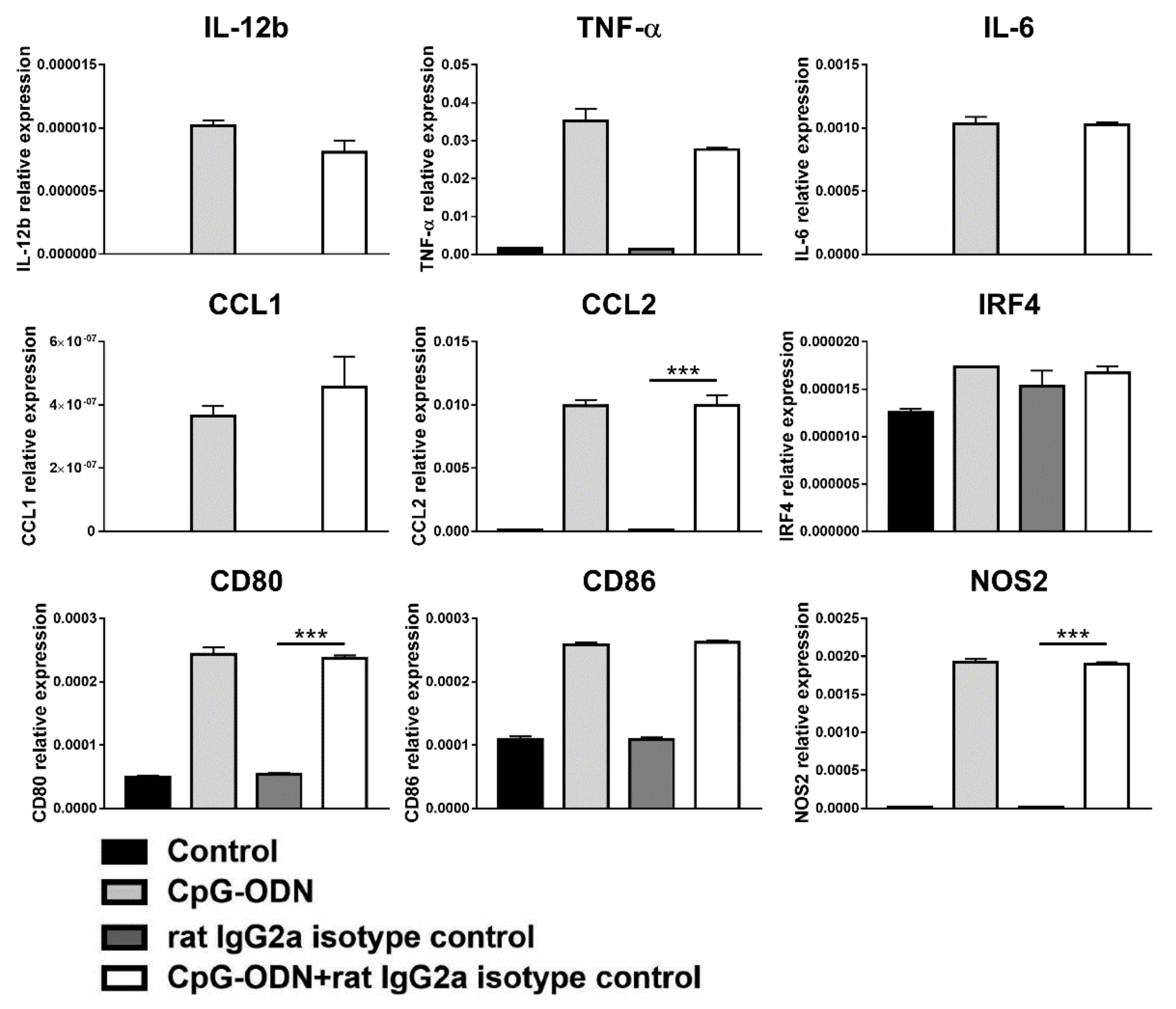
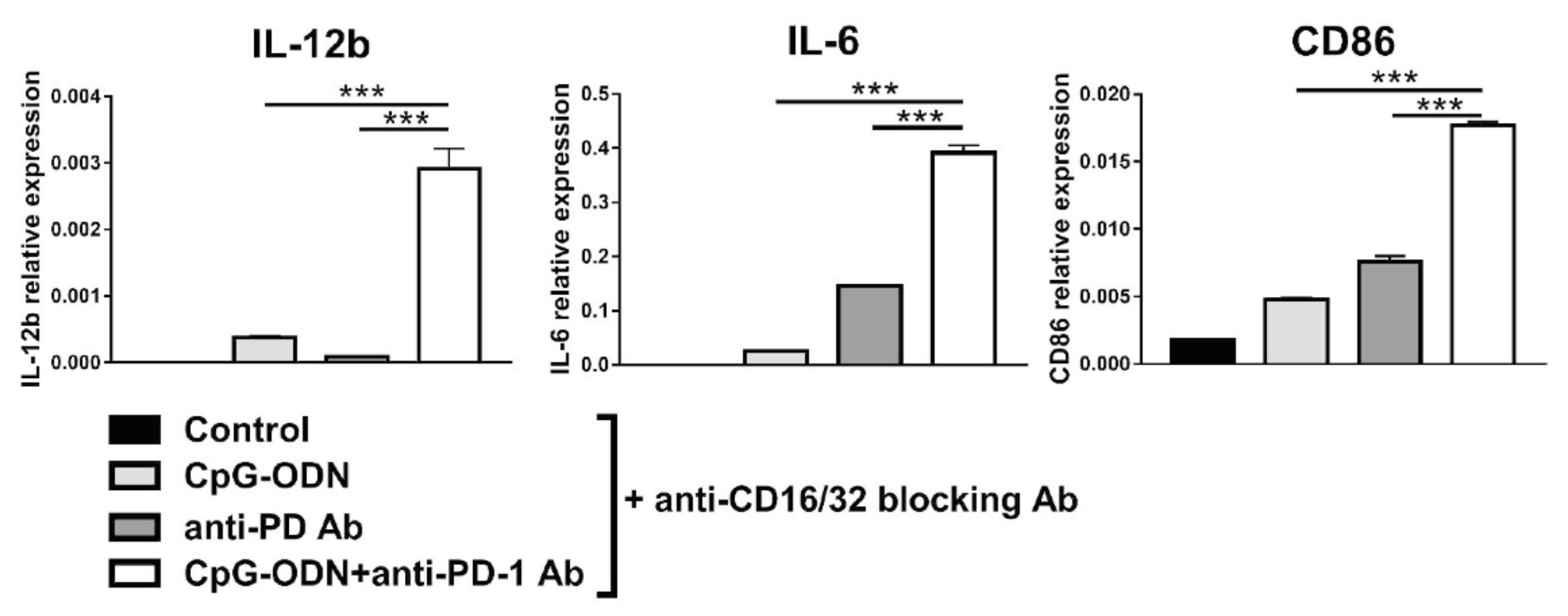
3.3. Macrophages Are the Innate Immune Cells Responsible for the Impairment of the Antitumor Activity of CpG-ODNs
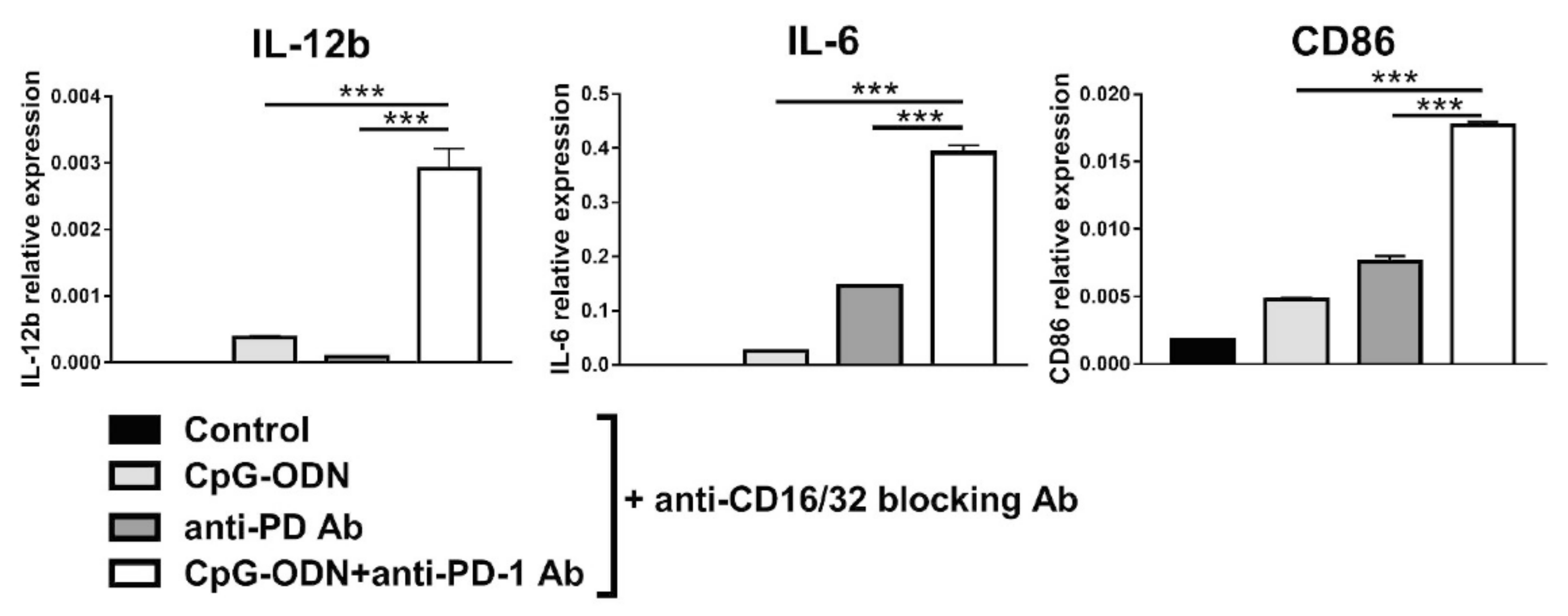
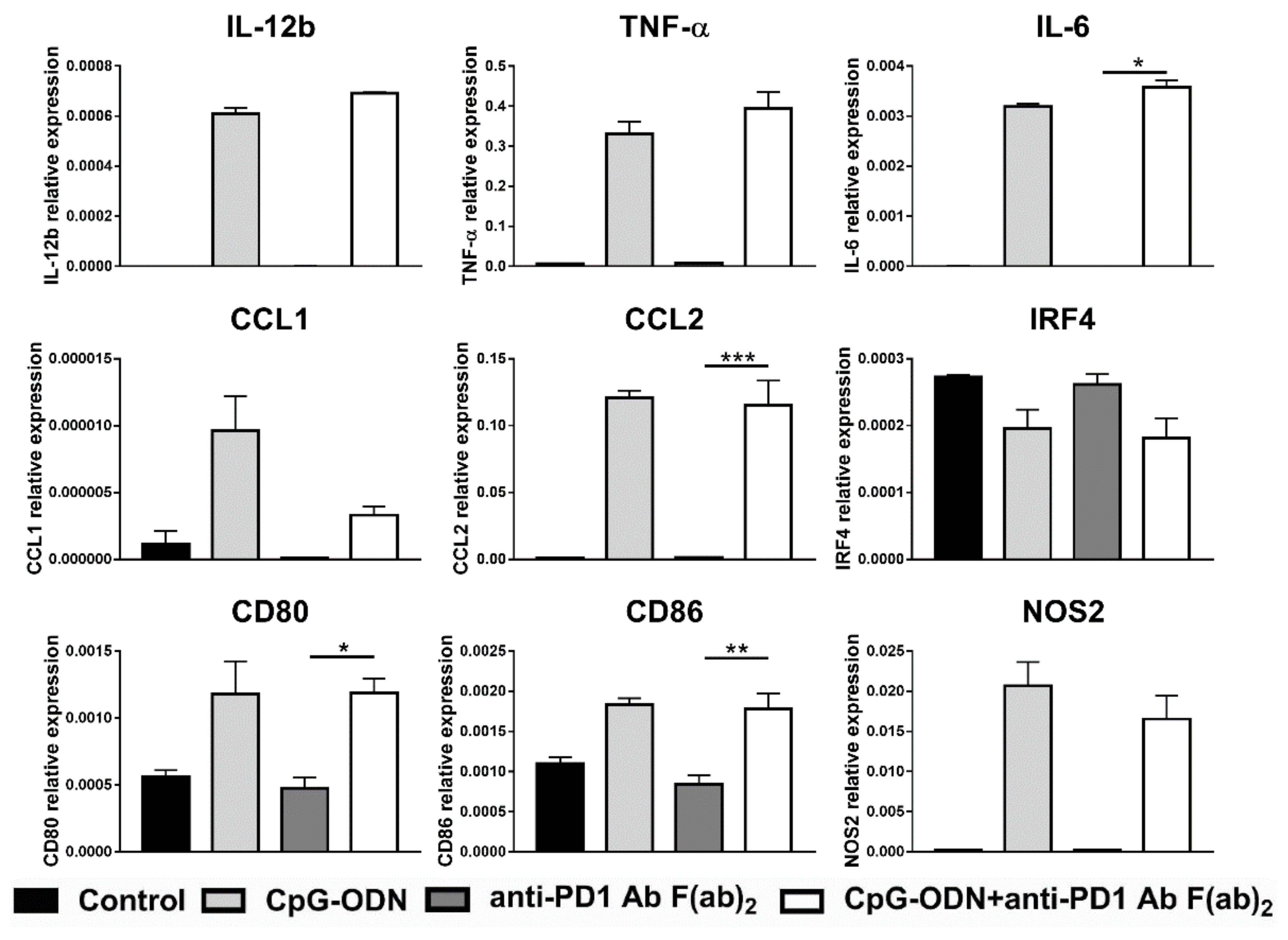
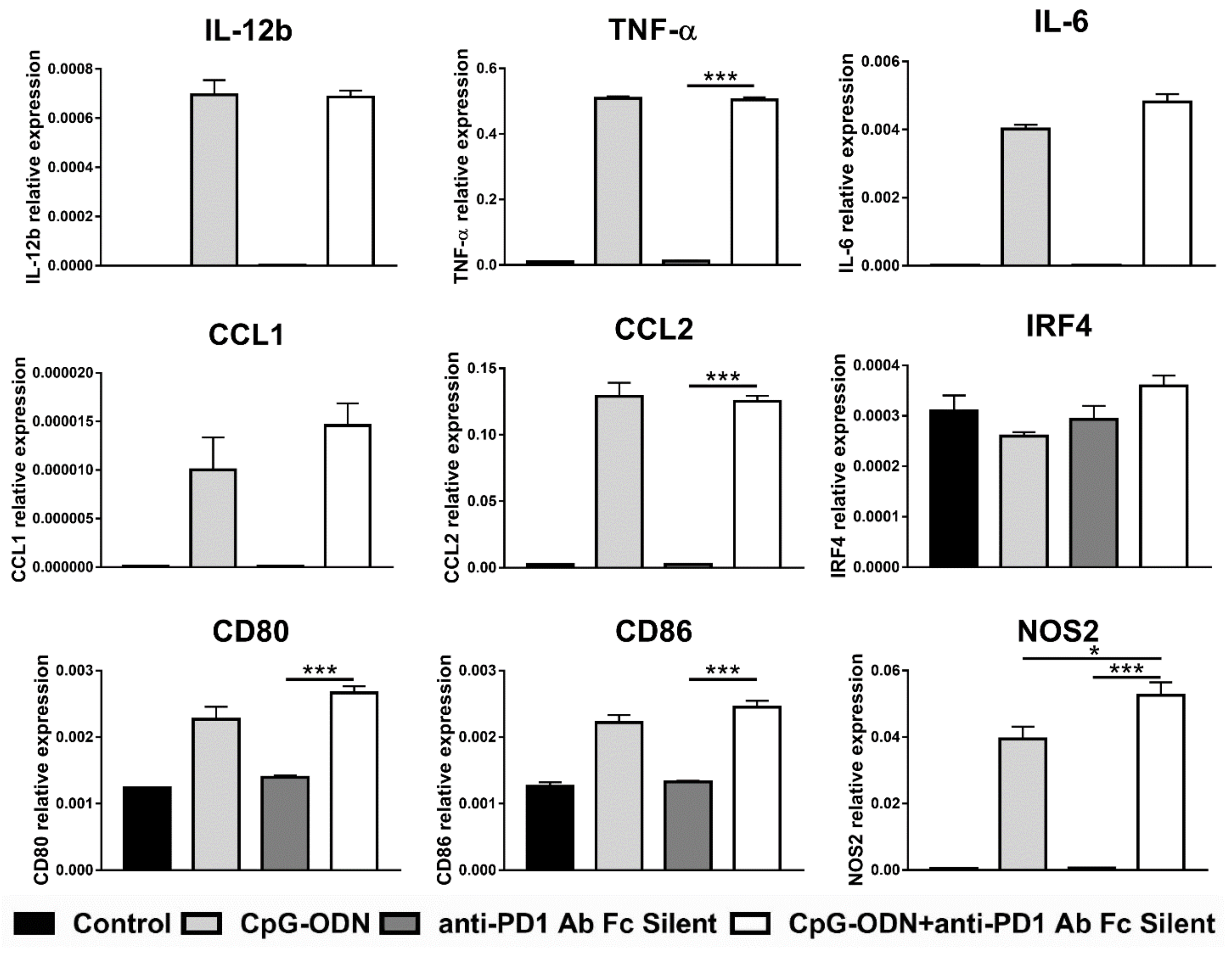
3.4. Effect of the Interaction between the Anti-PD-1 Antibody Fc Domain and Macrophages on the Antitumor Efficacy of CpG-ODNs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Zindel, J.; Kubes, P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu. Rev. Pathol. 2020, 15, 493–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunstein, M.J.; Kucharczyk, J.; Adams, S. Targeting Toll-Like Receptors for Cancer Therapy. Target. Oncol. 2018, 13, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.-C.; Tseng, J.-C.; Huang, L.-R.; Huang, C.-M.; Huang, C.-Y.F.; Chuang, T.-H. Adjuvant Effect of Toll-Like Receptor 9 Activation on Cancer Immunotherapy Using Checkpoint Blockade. Front. Immunol. 2020, 11, 1075. [Google Scholar] [CrossRef] [PubMed]
- Adamus, T.; Kortylewski, M. The revival of CpG oligonucleotide-based cancer immunotherapies. Contemp. Oncol. 2018, 22, 56–60. [Google Scholar] [CrossRef] [PubMed]
- De Cesare, M.; Calcaterra, C.; Pratesi, G.; Gatti, L.; Zunino, F.; Mènard, S.; Balsari, A. Eradication of ovarian tumor xenografts by locoregional administration of targeted immunotherapy. Clin. Cancer Res. 2008, 14, 5512–5518. [Google Scholar] [CrossRef] [Green Version]
- De Cesare, M.; Sfondrini, L.; Campiglio, M.; Sommariva, M.; Bianchi, F.; Perego, P.; van Rooijen, N.; Supino, R.; Rumio, C.; Zunino, F.; et al. Ascites regression and survival increase in mice bearing advanced-stage human ovarian carcinomas and repeatedly treated intraperitoneally with CpG-ODN. J. Immunother. 2010, 33, 8–15. [Google Scholar] [CrossRef]
- Sommariva, M.; de Cecco, L.; de Cesare, M.; Sfondrini, L.; Ménard, S.; Melani, C.; Delia, D.; Zaffaroni, N.; Pratesi, G.; Uva, V.; et al. TLR9 agonists oppositely modulate DNA repair genes in tumor versus immune cells and enhance chemotherapy effects. Cancer Res. 2011, 71, 6382–6390. [Google Scholar] [CrossRef] [Green Version]
- Sommariva, M.; de Cesare, M.; Meini, A.; Cataldo, A.; Zaffaroni, N.; Tagliabue, E.; Balsari, A. High efficacy of CpG-ODN, cetuximab and cisplatin combination for very advanced ovarian xenograft tumors. J. Transl. Med. 2013, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.A.; Basith, S.; Choi, S. Negative regulatory approaches to the attenuation of Toll-like receptor signaling. Exp. Mol. Med. 2013, 45, e11. [Google Scholar] [CrossRef] [PubMed]
- Terme, M.; Ullrich, E.; Aymeric, L.; Meinhardt, K.; Desbois, M.; Delahaye, N.; Viaud, S.; Ryffel, B.; Yagita, H.; Kaplanski, G.; et al. IL-18 induces PD-1-dependent immunosuppression in cancer. Cancer Res. 2011, 71, 5393–5399. [Google Scholar] [CrossRef] [Green Version]
- Said, E.A.; Dupuy, F.P.; Trautmann, L.; Zhang, Y.; Shi, Y.; El-Far, M.; Hill, B.J.; Noto, A.; Ancuta, P.; Peretz, Y.; et al. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat. Med. 2010, 16, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Lao, X.-M.; Chen, M.-M.; Liu, R.-X.; Wei, Y.; Ouyang, F.-Z.; Chen, D.-P.; Zhao, X.-Y.; Zhao, Q.; Li, X.-F.; et al. PD-1hi Identifies a Novel Regulatory B-cell Population in Human Hepatoma That Promotes Disease Progression. Cancer Discov. 2016, 6, 546–559. [Google Scholar] [CrossRef] [Green Version]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y. PD-1 and PD-L1 in cancer immunotherapy: Clinical implications and future considerations. Hum. Vaccin. Immunother. 2019, 15, 1111–1122. [Google Scholar] [CrossRef]
- Wu, X.; Gu, Z.; Chen, Y.; Chen, B.; Chen, W.; Weng, L.; Liu, X. Application of PD-1 Blockade in Cancer Immunotherapy. Comput. Struct. Biotechnol. J. 2019, 17, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Patsoukis, N.; Wang, Q.; Strauss, L.; Boussiotis, V.A. Revisiting the PD-1 pathway. Sci. Adv. 2020, 6, eabd2712. [Google Scholar] [CrossRef]
- Kleffel, S.; Posch, C.; Barthel, S.R.; Mueller, H.; Schlapbach, C.; Guenova, E.; Elco, C.P.; Lee, N.; Juneja, V.R.; Zhan, Q.; et al. Melanoma Cell-Intrinsic PD-1 Receptor Functions Promote Tumor Growth. Cell 2015, 162, 1242–1256. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cheng, Y.; Xu, Y.; Wang, Z.; Du, X.; Li, C.; Peng, J.; Gao, L.; Liang, X.; Ma, C. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene 2017, 36, 6143–6153. [Google Scholar] [CrossRef] [Green Version]
- Sato-Kaneko, F.; Yao, S.; Ahmadi, A.; Zhang, S.S.; Hosoya, T.; Kaneda, M.M.; Varner, J.A.; Pu, M.; Messer, K.S.; Guiducci, C.; et al. Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight 2017, 2, e93397. [Google Scholar] [CrossRef] [PubMed]
- Mangsbo, S.M.; Sandin, L.C.; Anger, K.; Korman, A.J.; Loskog, A.; Tötterman, T.H. Enhanced tumor eradication by combining CTLA-4 or PD-1 blockade with CpG therapy. J. Immunother. 2010, 33, 225–235. [Google Scholar] [CrossRef]
- Wang, S.; Campos, J.; Gallotta, M.; Gong, M.; Crain, C.; Naik, E.; Coffman, R.L.; Guiducci, C. Intratumoral injection of a CpG oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional CD8+ T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7240–E7249. [Google Scholar] [CrossRef] [Green Version]
- Borella, F.; Ghisoni, E.; Giannone, G.; Cosma, S.; Benedetto, C.; Valabrega, G.; Katsaros, D. Immune Checkpoint Inhibitors in Epithelial Ovarian Cancer: An Overview on Efficacy and Future Perspectives. Diagnostics 2020, 10, 146. [Google Scholar] [CrossRef] [Green Version]
- Lo Russo, G.; Moro, M.; Sommariva, M.; Cancila, V.; Boeri, M.; Centonze, G.; Ferro, S.; Ganzinelli, M.; Gasparini, P.; Huber, V.; et al. Antibody-Fc/FcR Interaction on Macrophages as a Mechanism for Hyperprogressive Disease in Non-small Cell Lung Cancer Subsequent to PD-1/PD-L1 Blockade. Clin. Cancer Res. 2019, 25, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Workman, P.; Aboagye, E.O.; Balkwill, F.; Balmain, A.; Bruder, G.; Chaplin, D.J.; Double, J.A.; Everitt, J.; Farningham, D.A.H.; Glennie, M.J.; et al. Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer 2010, 102, 1555–1577. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; Dittel, B.N. Isolation of mouse peritoneal cavity cells. J. Vis. Exp. 2010, 35, e1488. [Google Scholar] [CrossRef]
- Polesso, F.; Munks, M.W.; Rott, K.H.; Smart, S.; Hill, A.B.; Moran, A.E. PD-1-specific “Blocking” antibodies that deplete PD-1+ T cells present an inconvenient variable in preclinical immunotherapy experiments. Eur. J. Immunol. 2021, 51, 1473–1481. [Google Scholar] [CrossRef]
- Busnelli, M.; Manzini, S.; Bonacina, F.; Soldati, S.; Barbieri, S.S.; Amadio, P.; Sandrini, L.; Arnaboldi, F.; Donetti, E.; Laaksonen, R.; et al. Fenretinide treatment accelerates atherosclerosis development in apoE-deficient mice in spite of beneficial metabolic effects. Br. J. Pharmacol. 2020, 177, 328–345. [Google Scholar] [CrossRef] [Green Version]
- Mancini, C.; Hoxha, E.; Iommarini, L.; Brussino, A.; Richter, U.; Montarolo, F.; Cagnoli, C.; Parolisi, R.; Gondor Morosini, D.I.; Nicolò, V.; et al. Mice harbouring a SCA28 patient mutation in AFG3L2 develop late-onset ataxia associated with enhanced mitochondrial proteotoxicity. Neurobiol. Dis. 2019, 124, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Rojas, A.R.; Kelsey, I.; Pappalardo, J.L.; Chen, M.; Miller-Jensen, K. Co-stimulation with opposing macrophage polarization cues leads to orthogonal secretion programs in individual cells. Nat. Commun. 2021, 12, 301. [Google Scholar] [CrossRef]
- Sommariva, M.; Le Noci, V.; Storti, C.; Bianchi, F.; Tagliabue, E.; Balsari, A.; Sfondrini, L. Activation of NK cell cytotoxicity by aerosolized CpG-ODN/poly(I:C) against lung melanoma metastases is mediated by alveolar macrophages. Cell. Immunol. 2017, 313, 52–58. [Google Scholar] [CrossRef]
- Wang, C.; Yu, X.; Cao, Q.; Wang, Y.; Zheng, G.; Tan, T.K.; Zhao, H.; Zhao, Y.; Wang, Y.; Harris, D.C. Characterization of murine macrophages from bone marrow, spleen and peritoneum. BMC Immunol. 2013, 14, 6. [Google Scholar] [CrossRef] [Green Version]
- Toda, G.; Yamauchi, T.; Kadowaki, T.; Ueki, K. Preparation and culture of bone marrow-derived macrophages from mice for functional analysis. STAR Protoc. 2021, 2, 100246. [Google Scholar] [CrossRef]
- Lázár-Molnár, E.; Yan, Q.; Cao, E.; Ramagopal, U.; Nathenson, S.G.; Almo, S.C. Crystal structure of the complex between programmed death-1 (PD-1) and its ligand PD-L2. Proc. Natl. Acad. Sci. USA 2008, 105, 10483–10488. [Google Scholar] [CrossRef] [Green Version]
- Gallotta, M.; Assi, H.; Degagné, É.; Kannan, S.K.; Coffman, R.L.; Guiducci, C. Inhaled TLR9 Agonist Renders Lung Tumors Permissive to PD-1 Blockade by Promoting Optimal CD4+ and CD8+ T-cell Interplay. Cancer Res. 2018, 78, 4943–4956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reilley, M.J.; Morrow, B.; Ager, C.R.; Liu, A.; Hong, D.S.; Curran, M.A. TLR9 activation cooperates with T cell checkpoint blockade to regress poorly immunogenic melanoma. J. Immunother. Cancer 2019, 7, 323. [Google Scholar] [CrossRef]
- Lamichhane, P.; Karyampudi, L.; Shreeder, B.; Krempski, J.; Bahr, D.; Daum, J.; Kalli, K.R.; Goode, E.L.; Block, M.S.; Cannon, M.J.; et al. IL10 Release upon PD-1 Blockade Sustains Immunosuppression in Ovarian Cancer. Cancer Res. 2017, 77, 6667–6678. [Google Scholar] [CrossRef] [Green Version]
- Colvin, E.K. Tumor-associated macrophages contribute to tumor progression in ovarian cancer. Front. Oncol. 2014, 4, 137. [Google Scholar] [CrossRef] [Green Version]
- Batchu, R.B.; Gruzdyn, O.V.; Kolli, B.K.; Dachepalli, R.; Umar, P.S.; Rai, S.K.; Singh, N.; Tavva, P.S.; Weaver, D.W.; Gruber, S.A. IL-10 Signaling in the Tumor Microenvironment of Ovarian Cancer. Adv. Exp. Med. Biol. 2021, 1290, 51–65. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Wang, L.-X.; Zhang, S.-X.; Wu, H.-J.; Rong, X.-L.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef] [Green Version]
- Brüne, B.; Courtial, N.; Dehne, N.; Syed, S.N.; Weigert, A. Macrophage NOS2 in Tumor Leukocytes. Antioxid. Redox Signal. 2017, 26, 1023–1043. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Myasoedova, V.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. The impact of interferon-regulatory factors to macrophage differentiation and polarization into M1 and M2. Immunobiology 2018, 223, 101–111. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Camelliti, S.; Le Noci, V.; Bianchi, F.; Moscheni, C.; Arnaboldi, F.; Gagliano, N.; Balsari, A.; Garassino, M.C.; Tagliabue, E.; Sfondrini, L.; et al. Mechanisms of hyperprogressive disease after immune checkpoint inhibitor therapy: What we (don’t) know. J. Exp. Clin. Cancer Res. 2020, 39, 236. [Google Scholar] [CrossRef] [PubMed]
- Dahan, R.; Sega, E.; Engelhardt, J.; Selby, M.; Korman, A.J.; Ravetch, J.V. FcγRs Modulate the Anti-tumor Activity of Antibodies Targeting the PD-1/PD-L1 Axis. Cancer Cell 2015, 28, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Vonderheide, R.H.; Glennie, M.J. Agonistic CD40 antibodies and cancer therapy. Clin. Cancer Res. 2013, 19, 1035–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattiola, I.; Pesant, M.; Tentorio, P.F.; Molgora, M.; Marcenaro, E.; Lugli, E.; Locati, M.; Mavilio, D. Priming of Human Resting NK Cells by Autologous M1 Macrophages via the Engagement of IL-1β, IFN-β, and IL-15 Pathways. J. Immunol. 2015, 195, 2818–2828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Harrison, D.L.; Song, Y.; Ji, J.; Huang, J.; Hui, E. Antigen-Presenting Cell-Intrinsic PD-1 Neutralizes PD-L1 in cis to Attenuate PD-1 Signaling in T Cells. Cell Rep. 2018, 24, 379–390.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Ji, X.; Kang, N.; Zhou, J.; Liang, X.; Li, J.; Han, T.; Zhao, C.; Yang, T. Tumor necrosis factor α inhibition overcomes immunosuppressive M2b macrophage-induced bevacizumab resistance in triple-negative breast cancer. Cell Death Dis. 2020, 11, 993. [Google Scholar] [CrossRef]
- Asai, A.; Tsuchimoto, Y.; Ohama, H.; Fukunishi, S.; Tsuda, Y.; Kobayashi, M.; Higuchi, K.; Suzuki, F. Host antitumor resistance improved by the macrophage polarization in a chimera model of patients with HCC. Oncoimmunology 2017, 6, e1299301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browning, L.; Patel, M.R.; Horvath, E.B.; Tawara, K.; Jorcyk, C.L. IL-6 and ovarian cancer: Inflammatory cytokines in promotion of metastasis. Cancer Manag. Res. 2018, 10, 6685–6693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masoumi-Moghaddam, S.; Amini, A.; Wei, A.-Q.; Robertson, G.; Morris, D.L. Intratumoral interleukin-6 predicts ascites formation in patients with epithelial ovarian cancer: A potential tool for close monitoring. J. Ovarian Res. 2015, 8, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, T.; Matsuoka, K.; Sheikh, S.Z.; Russo, S.M.; Mishima, Y.; Collins, C.; de Zoeten, E.F.; Karp, C.L.; Ting, J.P.Y.; Sartor, R.B.; et al. IL-10 regulates Il12b expression via histone deacetylation: Implications for intestinal macrophage homeostasis. J. Immunol. 2012, 189, 1792–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yogo, R.; Yamaguchi, Y.; Watanabe, H.; Yagi, H.; Satoh, T.; Nakanishi, M.; Onitsuka, M.; Omasa, T.; Shimada, M.; Maruno, T.; et al. The Fab portion of immunoglobulin G contributes to its binding to Fcγ receptor III. Sci. Rep. 2019, 9, 11957. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, T.A.; Lang, P.A.; Lang, K.S. The Diverse Functions of the Ubiquitous Fcγ Receptors and Their Unique Constituent, FcRγ Subunit. Pathogens 2020, 9, 140. [Google Scholar] [CrossRef] [Green Version]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Wong-Rolle, A.; Wei, H.K.; Zhao, C.; Jin, C. Unexpected guests in the tumor microenvironment: Microbiome in cancer. Protein Cell 2020, 12, 426–435. [Google Scholar] [CrossRef]
- Sommariva, M.; Le Noci, V.; Bianchi, F.; Camelliti, S.; Balsari, A.; Tagliabue, E.; Sfondrini, L. The lung microbiota: Role in maintaining pulmonary immune homeostasis and its implications in cancer development and therapy. Cell. Mol. Life Sci. 2020, 77, 2739–2749. [Google Scholar] [CrossRef] [Green Version]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Byrd, A.L.; Belkaid, Y.; Segre, J.A. The human skin microbiome. Nat. Rev. Microbiol. 2018, 16, 143–155. [Google Scholar] [CrossRef]
- Emani, R.; Alam, C.; Pekkala, S.; Zafar, S.; Emani, M.R.; Hänninen, A. Peritoneal cavity is a route for gut-derived microbial signals to promote autoimmunity in non-obese diabetic mice. Scand. J. Immunol. 2015, 81, 102–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousefi, M.; Dehghani, S.; Nosrati, R.; Ghanei, M.; Salmaninejad, A.; Rajaie, S.; Hasanzadeh, M.; Pasdar, A. Current insights into the metastasis of epithelial ovarian cancer—Hopes and hurdles. Cell. Oncol. 2020, 43, 515–538. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camelliti, S.; Le Noci, V.; Bianchi, F.; Storti, C.; Arnaboldi, F.; Cataldo, A.; Indino, S.; Jachetti, E.; Figini, M.; Colombo, M.P.; et al. Macrophages Impair TLR9 Agonist Antitumor Activity through Interacting with the Anti-PD-1 Antibody Fc Domain. Cancers 2021, 13, 4081. https://doi.org/10.3390/cancers13164081
Camelliti S, Le Noci V, Bianchi F, Storti C, Arnaboldi F, Cataldo A, Indino S, Jachetti E, Figini M, Colombo MP, et al. Macrophages Impair TLR9 Agonist Antitumor Activity through Interacting with the Anti-PD-1 Antibody Fc Domain. Cancers. 2021; 13(16):4081. https://doi.org/10.3390/cancers13164081
Chicago/Turabian StyleCamelliti, Simone, Valentino Le Noci, Francesca Bianchi, Chiara Storti, Francesca Arnaboldi, Alessandra Cataldo, Serena Indino, Elena Jachetti, Mariangela Figini, Mario Paolo Colombo, and et al. 2021. "Macrophages Impair TLR9 Agonist Antitumor Activity through Interacting with the Anti-PD-1 Antibody Fc Domain" Cancers 13, no. 16: 4081. https://doi.org/10.3390/cancers13164081
APA StyleCamelliti, S., Le Noci, V., Bianchi, F., Storti, C., Arnaboldi, F., Cataldo, A., Indino, S., Jachetti, E., Figini, M., Colombo, M. P., Balsari, A., Gagliano, N., Tagliabue, E., Sfondrini, L., & Sommariva, M. (2021). Macrophages Impair TLR9 Agonist Antitumor Activity through Interacting with the Anti-PD-1 Antibody Fc Domain. Cancers, 13(16), 4081. https://doi.org/10.3390/cancers13164081