
Ovarian Cancer in the Era of Immune Checkpoint Inhibitors: State of the Art and Future Perspectives

 ,
, 

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Anti PD1 Agents
3.1.1. Pembrolizumab
3.1.2. Nivolumab
3.2. Anti-PD-L1 Agents
3.2.1. Avelumab
3.2.2. Atezolizumab
3.2.3. Durvalumab
3.3. Anti CTLA-4
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Globocan 2020. Ovary. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/25-Ovary-fact-sheet.pdf (accessed on 15 June 2021).
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian Cancer Statistics, 2018. CA A Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Prat, J.; D’Angelo, E.; Espinosa, I. Ovarian Carcinomas: At Least Five Different Diseases with Distinct Histological Features and Molecular Genetics. Hum. Pathol. 2018, 80, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Piccart, M.J. Randomized Intergroup Trial of Cisplatin-Paclitaxel vs. Cisplatin-Cyclophosphamide in Women with Advanced Epithelial Ovarian Cancer: Three-Year Results. J. Natl. Cancer Inst. 2000, 92, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Ozols, R.F.; Bundy, B.N.; Greer, B.E.; Fowler, J.M.; Clarke-Pearson, D.; Burger, R.A.; Mannel, R.S.; DeGeest, K.; Hartenbach, E.M.; Baergen, R. Phase III Trial of Carboplatin and Paclitaxel Compared with Cisplatin and Paclitaxel in Patients with Optimally Resected Stage III Ovarian Cancer: A Gynecologic Oncology Group Study. JCO 2003, 21, 3194–3200. [Google Scholar] [CrossRef] [PubMed]
- Neijt, J.P.; Engelholm, S.A.; Tuxen, M.K.; Sørensen, P.G.; Hansen, M.; Sessa, C.; de Swart, C.A.M.; Hirsch, F.R.; Lund, B.; van Houwelingen, H.C. Exploratory Phase III Study of Paclitaxel and Cisplatin vs. Paclitaxel and Carboplatin in Advanced Ovarian Cancer. JCO 2000, 18, 3084–3092. [Google Scholar] [CrossRef] [PubMed]
- McGuire, W.P.; Hoskins, W.J.; Brady, M.F.; Kucera, P.R.; Partridge, E.E.; Look, K.Y.; Clarke-Pearson, D.L.; Davidson, M. Cyclophosphamide and Cisplatin Compared with Paclitaxel and Cisplatin in Patients with Stage III and Stage IV Ovarian Cancer. N. Engl. J. Med. 1996, 334, 1–6. [Google Scholar] [CrossRef]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A Phase 3 Trial of Bevacizumab in Ovarian Cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Robert, C. A Decade of Immune-Checkpoint Inhibitors in Cancer Therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. JCO 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [Green Version]
- Matulonis, U.A.; Shapira-Frommer, R.; Santin, A.D.; Lisyanskaya, A.S.; Pignata, S.; Vergote, I.; Raspagliesi, F.; Sonke, G.S.; Birrer, M.; Provencher, D.M.; et al. Antitumor Activity and Safety of Pembrolizumab in Patients with Advanced Recurrent Ovarian Cancer: Results from the Phase II KEYNOTE-100 Study. Ann. Oncol. 2019, 30, 1080–1087. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Shapira, R.; Santin, A.; Lisyanskaya, A.S.; Pignata, S.; Vergote, S.; Raspagliesi, F.; Sonke, G.S.; Birrer, M.; Sehouli, J.; et al. Final Results from the KEYNOTE-100 Trial of Pembrolizumab in Patients with Advanced Recurrent Ovarian Cancer. JCO 2020, 38, 6005. [Google Scholar] [CrossRef]
- Varga, A.; Piha-Paul, S.; Ott, P.A.; Mehnert, J.M.; Berton-Rigaud, D.; Morosky, A.; Yang, P.; Ruman, J.; Matei, D. Pembrolizumab in Patients with Programmed Death Ligand 1–Positive Advanced Ovarian Cancer: Analysis of KEYNOTE-028. Gynecol. Oncol. 2019, 152, 243–250. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Barry, W.; Penson, R.T.; Campos, S.M.; Krasner, C.; Liu, J.F. Phase II Study of Pembrolizumab (Pembro) Combined with Pegylated Liposomal Doxorubicin (PLD) for Recurrent Platinum-Resistant Ovarian, Fallopian Tube or Peritoneal Cancer. Gynecol. Oncol. 2018, 149, 24. [Google Scholar] [CrossRef]
- Zsiros, E.; Lynam, S.; Attwood, K.M.; Wang, C.; Chilakapati, S.; Gomez, E.C.; Liu, S.; Akers, S.; Lele, S.; Frederick, P.J.; et al. Efficacy and Safety of Pembrolizumab in Combination with Bevacizumab and Oral Metronomic Cyclophosphamide in the Treatment of Recurrent Ovarian Cancer: A Phase 2 Nonrandomized Clinical Trial. JAMA Oncol. 2021, 7, 78. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination with Pembrolizumab in Patients with Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141. [Google Scholar] [CrossRef] [Green Version]
- Hamanishi, J.; Mandai, M.; Ikeda, T.; Minami, M.; Kawaguchi, A.; Murayama, T.; Kanai, M.; Mori, Y.; Matsumoto, S.; Chikuma, S.; et al. Safety and Antitumor Activity of Anti–PD-1 Antibody, Nivolumab, in Patients with Platinum-Resistant Ovarian Cancer. JCO 2015, 33, 4015–4022. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Burger, R.A.; Sill, M.W.; Powell, D.J.; Lankes, H.A.; Feldman, M.D.; Zivanovic, O.; Gunderson, C.; Ko, E.; Mathews, C.; et al. Randomized Phase II Trial of Nivolumab vs. Nivolumab and Ipilimumab for Recurrent or Persistent Ovarian Cancer: An NRG Oncology Study. JCO 2020, 38, 1814–1823. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Herold, C.; Gray, K.P.; Penson, R.T.; Horowitz, N.; Konstantinopoulos, P.A.; Castro, C.M.; Hill, S.J.; Curtis, J.; Luo, W.; et al. Assessment of Combined Nivolumab and Bevacizumab in Relapsed Ovarian Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 1731. [Google Scholar] [CrossRef]
- Omatsu, K.; Hamanishi, J.; Katsumata, N.; Nishio, S.; Sawada, K.; Takeuchi, S.; Aoki, D.; Fujiwara, K.; Sugiyama, T.; Konishi, I. 807O Nivolumab vs. Gemcitabine or Pegylated Liposomal Doxorubicin for Patients with Platinum-Resistant (Advanced or Recurrent) Ovarian Cancer: Open-Label, Randomized Trial in Japan (NINJA Trial). Ann. Oncol. 2020, 31, 611. [Google Scholar] [CrossRef]
- Disis, M.L.; Taylor, M.H.; Kelly, K.; Beck, J.T.; Gordon, M.; Moore, K.M.; Patel, M.R.; Chaves, J.; Park, H.; Mita, A.C.; et al. Efficacy and Safety of Avelumab for Patients with Recurrent or Refractory Ovarian Cancer: Phase 1b Results from the JAVELIN Solid Tumor Trial. JAMA Oncol. 2019, 5, 393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledermann, J.A.; Colombo, N.; Oza, A.M.; Fujiwara, K.; Birrer, M.J.; Randall, L.M.; Poddubskaya, E.V.; Scambia, G.; Shparyk, Y.V.; Lim, M.C.; et al. Avelumab in Combination with and/or Following Chemotherapy vs Chemotherapy Alone in Patients with Previously Untreated Epithelial Ovarian Cancer: Results from the Phase 3 Javelin Ovarian 100 Trial. Gynecol. Oncol. 2020, 159, 13–14. [Google Scholar] [CrossRef]
- NCT03642132: Avelumab and Talazoparib in Untreated Advanced Ovarian Cancer (JAVELIN OVARIAN PARP 100). Available online: https://clinicaltrials.gov/ct2/show/NCT03642132 (accessed on 15 June 2021).
- Pujade-Lauraine, E.; Fujiwara, K.; Ledermann, J.A.; Oza, A.M.; Kristeleit, R.S.; Ray-Coquard, I.L.; Richardson, G.E.; Sessa, C.; Yonemori, K.; Banerjee, S.; et al. Avelumab Alone or in Combination with Pegylated Liposomal Doxorubicin vs. Pegylated Liposomal Doxorubicin Alone in Platinum-Resistant or Refractory Epithelial Ovarian Cancer: Primary and Biomarker Analysis of the Phase III JAVELIN Ovarian 200 Trial. Gynecol. Oncol. 2019, 154, 21–22. [Google Scholar] [CrossRef]
- Cadoo, K.A.; Meyers, M.L.; Burger, R.A.; Armstrong, D.K.; Penson, R.T.; Gordon, M.S.; Fleming, G.F.; Moroney, J.W.; Hamilton, E.P.; Duska, L.R.; et al. A Phase II Randomized Study of Avelumab plus Entinostat vs. Avelumab plus Placebo in Patients (Pts) with Advanced Epithelial Ovarian Cancer (EOC). JCO 2019, 37, 5511. [Google Scholar] [CrossRef]
- Moore, K.N.; Bookman, M.; Sehouli, J.; Miller, A.; Anderson, C.; Scambia, G.; Myers, T.; Taskiran, C.; Robison, K.; Mäenpää, J.; et al. Atezolizumab, Bevacizumab, and Chemotherapy for Newly Diagnosed Stage III or IV Ovarian Cancer: Placebo-Controlled Randomized Phase III Trial (IMagyn050/GOG 3015/ENGOT-OV39). JCO 2021, 39, 1842–1855. [Google Scholar] [CrossRef] [PubMed]
- O’Cearbhaill, R.E.; Wolfer, A.; Disilvestro, P.; O’Malley, D.M.; Sabbatini, P.; Shohara, L.; Schwarzenberger, P.O.; Ricciardi, T.; Macri, M.; Ryan, A.; et al. A Phase I/II Study of Chemo-Immunotherapy with Durvalumab (Durva) and Pegylated Liposomal Doxorubicin (PLD) in Platinum-Resistant Recurrent Ovarian Cancer (PROC). Ann. Oncol. 2018, 29, 337. [Google Scholar] [CrossRef]
- Drew, Y.; de Jonge, M.; Hong, S.H.; Park, Y.H.; Wolfer, A.; Brown, J.; Ferguson, M.; Gore, M.E.; Alvarez, R.H.; Gresty, C.; et al. An Open-Label, Phase II Basket Study of Olaparib and Durvalumab (MEDIOLA): Results in Germline BRCA -Mutated ( GBRCA m) Platinum-Sensitive Relapsed (PSR) Ovarian Cancer (OC). Gynecol. Oncol. 2018, 149, 246–247. [Google Scholar] [CrossRef]
- Drew, Y.; Kaufman, B.; Banerjee, S.; Lortholary, A.; Hong, S.H.; Park, Y.H.; Zimmermann, S.; Roxburgh, P.; Ferguson, M.; Alvarez, R.H.; et al. Phase II study of olaparib 1 durvalumab (MEDIOLA): Updated Results in Germline BRCA-Mutated Platinum-Sensitive Relapsed (PSR) Ovarian Cancer (OC). Ann. Oncol. 2019, 30, 485–486. [Google Scholar] [CrossRef]
- Drew, Y.; Penson, R.T.; O’Malley, D.M.; Kim, J.-W.; Zimmermann, S.; Roxburgh, P.; Sohn, J.; Stemmer, S.M.; Bastian, S.; Ferguson, M.; et al. 814MO Phase II Study of Olaparib (O) plus Durvalumab (D) and Bevacizumab (B) (MEDIOLA): Initial Results in Patients (Pts) with Non-Germline BRCA-Mutated (Non-GBRCAm) Platinum Sensitive Relapsed (PSR) Ovarian Cancer (OC). Ann. Oncol. 2020, 31, 615–616. [Google Scholar] [CrossRef]
- Lee, J.M.; Cimino-Mathews, A.; Peer, C.J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Cao, L.; Harrell, M.I.; Swisher, E.M.; Houston, N.; et al. Safety and Clinical Activity of the Programmed Death-Ligand 1 Inhibitor Durvalumab in Combination with Poly (ADP-Ribose) Polymerase Inhibitor Olaparib or Vascular Endothelial Growth Factor Receptor 1–3 Inhibitor Cediranib in Women’s Cancers: A Dose-Escalation, Phase I Study. JCO 2017, 35, 2193–2202. [Google Scholar] [CrossRef]
- Lampert, E.J.; Zimmer, A.; Padget, M.; Cimino-Mathews, A.; Nair, J.R.; Liu, Y.; Swisher, E.M.; Hodge, J.W.; Nixon, A.B.; Nichols, E.; et al. Combination of PARP Inhibitor Olaparib, and PD-L1 Inhibitor Durvalumab, in Recurrent Ovarian Cancer: A Proof-of-Concept Phase II Study. Clin. Cancer Res. 2020, 26, 4268–4279. [Google Scholar] [CrossRef] [PubMed]
- NCT01611558: Phase II Study of Ipilimumab Monotherapy in Recurrent Platinum-Sensitive Ovarian Cancer-Study Results. Available online: https://clinicaltrials.gov/ct2/show/results/NCT01611558 (accessed on 15 June 2021).
- Gaillard, S.; Berg, M.; Harrison, J.; Huang, P.; Leatherman, J.M.; Doucet, M.; Sen, R.; Suru, A.; Cai, H.; Durham, J.N.; et al. A Clinical Study of Tremelimumab Alone or in Combination with Olaparib in Patients with Advanced Epithelial Ovarian Cancer. JCO 2020, 38, 6045. [Google Scholar] [CrossRef]
- Adams, S.F.; Rixe, O.; Lee, J.-H.; McCance, D.J.; Westgate, S.; Eberhardt, S.C.; Rutledge, T.; Muller, C. Phase I Study Combining Olaparib and Tremelimumab for the Treatment of Women with BRCA-Deficient Recurrent Ovarian Cancer. JCO 2017, 35, 17052. [Google Scholar] [CrossRef]
- Hartnett, E.G.; Knight, J.; Radolec, M.; Buckanovich, R.J.; Edwards, R.P.; Vlad, A.M. Immunotherapy Advances for Epithelial Ovarian Cancer. Cancers 2020, 12, 3733. [Google Scholar] [CrossRef]
- Ning, F.; Cole, C.B.; Annunziata, C.M. Driving Immune Responses in the Ovarian Tumor Microenvironment. Front. Oncol. 2021, 10, 604084. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, Y.; Yang, J.; Zhao, X.; Wei, X. Tumor Microenvironment in Ovarian Cancer: Function and Therapeutic Strategy. Front. Cell Dev. Biol. 2020, 8, 758. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Cannistra, S.A. Immune Checkpoint Inhibitors in Ovarian Cancer: Can We Bridge the Gap Between IMagynation and Reality? JCO 2021, 10, 1833–1838. [Google Scholar] [CrossRef]
- Kapka-Skrzypczak, L.; Wolinska, E.; Szparecki, G.; Czajka, M.; Skrzypczak, M. The Immunohistochemical Analysis of Membrane-Bound CD55, CD59 and Fluid-Phase FH and FH-like Complement Inhibitors in Cancers of Ovary and Corpus Uteri Origin. Cent. Eur. J. Immunol. 2015, 3, 349–353. [Google Scholar] [CrossRef] [Green Version]
- Wouters, M.; Dijkgraaf, E.; Kuijjer, M.; Jordanova, E.; Hollema, H.; Welters, M.; van der Hoeven, J.; Daemen, T.; Kroep, J.; Nijman, H.; et al. Interleukin-6 Receptor and Its Ligand Interleukin-6 Are Opposite Markers for Survival and Infiltration with Mature Myeloid Cells in Ovarian Cancer. OncoImmunology 2014, 3, e962397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolomeyevskaya, N.; Eng, K.H.; Khan, A.N.H.; Grzankowski, K.S.; Singel, K.L.; Moysich, K.; Segal, B.H. Cytokine Profiling of Ascites at Primary Surgery Identifies an Interaction of Tumor Necrosis Factor-α and Interleukin-6 in Predicting Reduced Progression-Free Survival in Epithelial Ovarian Cancer. Gynecol. Oncol. 2015, 138, 352–357. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.L.; Obermajer, N.; Odunsi, K.; Edwards, R.P.; Kalinski, P. Synergistic COX2 Induction by IFNγ and TNFα Self-Limits Type-1 Immunity in the Human Tumor Microenvironment. Cancer Immunol. Res. 2016, 4, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motz, G.T.; Santoro, S.P.; Wang, L.-P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor Endothelium FasL Establishes a Selective Immune Barrier Promoting Tolerance in Tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef]
- Pollard, J.W. Tumour-Educated Macrophages Promote Tumour Progression and Metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Reinartz, S.; Schumann, T.; Finkernagel, F.; Wortmann, A.; Jansen, J.M.; Meissner, W.; Krause, M.; Schwörer, A.; Wagner, U.; Müller-Brüsselbach, S.; et al. Mixed-polarization Phenotype of Ascites-associated Macrophages in Human Ovarian Carcinoma: Correlation of CD163 Expression, Cytokine Levels and Early Relapse. Int. J. Cancer 2014, 134, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Li, X.; Tan, S.; Zhou, H.J.; Ji, W.; Bellone, S.; Xu, X.; Zhang, H.; Santin, A.D.; Lou, G.; et al. Tumor-Associated Macrophages Drive Spheroid Formation during Early Transcoelomic Metastasis of Ovarian Cancer. J. Clin. Investig. 2016, 126, 4157–4173. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. A High M1/M2 Ratio of Tumor-Associated Macrophages Is Associated with Extended Survival in Ovarian Cancer Patients. J. Ovarian Res. 2014, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Lan, C.; Huang, X.; Lin, S.; Huang, H.; Cai, Q.; Wan, T.; Lu, J.; Liu, J. Expression of M2-Polarized Macrophages Is Associated with Poor Prognosis for Advanced Epithelial Ovarian Cancer. Technol. Cancer Res. Treat. 2013, 12, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Zhang, J.; Li, D.; Mao, Y.; Mo, F.; Du, W.; Ma, X. Prognostic Significance of Tumor-Associated Macrophages in Ovarian Cancer: A Meta-Analysis. Gynecol. Oncol. 2017, 147, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Alternative Activation of Macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Li, H.; Fan, X.; Houghton, J. Tumor Microenvironment: The Role of the Tumor Stroma in Cancer. J. Cell. Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific Recruitment of Regulatory T Cells in Ovarian Carcinoma Fosters Immune Privilege and Predicts Reduced Survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular Mechanisms of Treg-Mediated T Cell Suppression. Front. Immun. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obermajer, N.; Muthuswamy, R.; Lesnock, J.; Edwards, R.P.; Kalinski, P. Positive Feedback between PGE2 and COX2 Redirects the Differentiation of Human Dendritic Cells toward Stable Myeloid-Derived Suppressor Cells. Blood 2011, 118, 5498–5505. [Google Scholar] [CrossRef] [PubMed]
- Howitt, B.E.; Strickland, K.C.; Sholl, L.M.; Rodig, S.; Ritterhouse, L.L.; Chowdhury, D.; D’Andrea, A.D.; Matulonis, U.A.; Konstantinopoulos, P.A. Clear Cell Ovarian Cancers with Microsatellite Instability: A Unique Subset of Ovarian Cancers with Increased Tumor-Infiltrating Lymphocytes and PD-1/PD-L1 Expression. OncoImmunology 2017, 6, e1277308. [Google Scholar] [CrossRef] [Green Version]
- Oda, K.; Hamanishi, J.; Matsuo, K.; Hasegawa, K. Genomics to Immunotherapy of Ovarian Clear Cell Carcinoma: Unique Opportunities for Management. Gynecol. Oncol. 2018, 151, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Tamayo, P.; Yang, J.-Y.; Hubbard, D.; Zhang, H.; Creighton, C.J.; Fereday, S.; Lawrence, M.; Carter, S.L.; Mermel, C.H.; et al. Prognostically Relevant Gene Signatures of High-Grade Serous Ovarian Carcinoma. J. Clin. Invest. 2012. [Google Scholar] [CrossRef]
- Przybytkowski, E.; Davis, T.; Hosny, A.; Eismann, J.; Matulonis, U.A.; Wulf, G.M.; Nabavi, S. An Immune-Centric Exploration of BRCA1 and BRCA2 Germline Mutation Related Breast and Ovarian Cancers. BMC Cancer 2020, 20, 197. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, T.; Zhang, Z.; Payne, S.H.; Zhang, B.; McDermott, J.E.; Zhou, J.Y.; Petyuk, V.A.; Chen, L.; Ray, D.; et al. CPTAC Investigators. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 2016, 166, 755–765. [Google Scholar] [CrossRef] [Green Version]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and Prognostic Significance of BRCA1/2-Mutation Status with Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes and Expression of PD-1/PD-L1 in High Grade Serous Ovarian Cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef] [Green Version]
- Morse, C.B.; Toukatly, M.N.; Kilgore, M.R.; Agnew, K.J.; Bernards, S.S.; Norquist, B.M.; Pennington, K.P.; Garcia, R.L.; Liao, J.B.; Swisher, E.M. Tumor Infiltrating Lymphocytes and Homologous Recombination Deficiency Are Independently Associated with Improved Survival in Ovarian Carcinoma. Gynecol. Oncol. 2019, 153, 217–222. [Google Scholar] [CrossRef]
- McAlpine, J.N.; Porter, H.; Köbel, M.; Nelson, B.H.; Prentice, L.M.; Kalloger, S.E.; Senz, J.; Milne, K.; Ding, J.; Shah, S.P.; et al. BRCA1 and BRCA2 Mutations Correlate with TP53 Abnormalities and Presence of Immune Cell Infiltrates in Ovarian High-Grade Serous Carcinoma. Mod. Pathol. 2012, 25, 740–750. [Google Scholar] [CrossRef] [Green Version]
- Gadducci, A.; Guerrieri, M.E. PARP Inhibitors Alone and in Combination with Other Biological Agents in Homologous Recombination Deficient Epithelial Ovarian Cancer: From the Basic Research to the Clinic. Crit. Rev. Oncol. Hematol. 2017, 114, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, M.; Romanski, P.A.; Rosenwaks, Z.; Gerhardt, J. Gynecological Cancers Caused by Deficient Mismatch Repair and Microsatellite Instability. Cancers 2020, 12, 3319. [Google Scholar] [CrossRef]
- Helder-Woolderink, J.M.; Blok, E.A.; Vasen, H.F.A.; Hollema, H.; Mourits, M.J.; De Bock, G.H. Ovarian Cancer in Lynch Syndrome; a Systematic Review. Eur. J. Cancer 2016, 55, 65–73. [Google Scholar] [CrossRef]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A Deficiency Promotes Mutability and Potentiates Therapeutic Antitumor Immunity Unleashed by Immune Checkpoint Blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef]
- Wang, Y.; Hoang, L.; Ji, J.X.; Huntsman, D.G. SWI/SNF Complex Mutations in Gynecologic Cancers: Molecular Mechanisms and Models. Annu. Rev. Pathol. 2020, 15, 467–492. [Google Scholar] [CrossRef] [Green Version]
- Van Zyl, B.; Tang, D.; Bowden, N.A. Biomarkers of Platinum Resistance in Ovarian Cancer: What Can We Use to Improve Treatment. Endocr. Relat. Cancer 2018, 25, 303–318. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Vikas, P.; Borcherding, N.; Chennamadhavuni, A.; Garje, R. Therapeutic Potential of Combining PARP Inhibitor and Immunotherapy in Solid Tumors. Front. Oncol. 2020, 10, 570. [Google Scholar] [CrossRef] [PubMed]
- Rébé, C.; Demontoux, L.; Pilot, T.; Ghiringhelli, F. Platinum Derivatives Effects on Anticancer Immune Response. Biomolecules 2019, 10, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabosch, S.; Bulatovic, M.; Zeng, F.; Ma, T.; Zhang, L.; Ross, M.; Brozick, J.; Fang, Y.; Tseng, G.; Kim, E.; et al. Cisplatin-Induced Immune Modulation in Ovarian Cancer Mouse Models with Distinct Inflammation Profiles. Oncogene 2019, 38, 2380–2393. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Feng, Q.-M.; Wang, Y.; Shi, J.; Ge, H.-L.; Di, W. The Immunologic Aspects in Advanced Ovarian Cancer Patients Treated with Paclitaxel and Carboplatin Chemotherapy. Cancer Immunol. Immunother. 2010, 59, 279–291. [Google Scholar] [CrossRef]
- De Biasi, A.R.; Villena-Vargas, J.; Adusumilli, P.S. Cisplatin-Induced Antitumor Immunomodulation: A Review of Preclinical and Clinical Evidence. Clin. Cancer Res. 2014, 20, 5384–5391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Yu, X.; Wang, Z.; Wu, P.; Huang, J. Anthracyclines Potentiate Anti-Tumor Immunity: A New Opportunity for Chemoimmunotherapy. Cancer Letters 2015, 369, 331–335. [Google Scholar] [CrossRef]
- Madondo, M.T.; Quinn, M.; Plebanski, M. Low Dose Cyclophosphamide: Mechanisms of T-Cell Modulation. Cancer Treat. Rev. 2016, 42, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Mkrtichyan, M.; Najjar, Y.G.; Raulfs, E.C.; Abdalla, M.Y.; Samara, R.; Rotem-Yehudar, R.; Cook, L.; Khleif, S.N. Anti-PD-1 Synergizes with Cyclophosphamide to Induce Potent Anti-Tumor Vaccine Effects through Novel Mechanisms. Eur. J. Immunol. 2011, 41, 2977–2986. [Google Scholar] [CrossRef]
- Ziogas, A.C.; Gavalas, N.G.; Tsiatas, M.; Tsitsilonis, O.; Politi, E.; Terpos, E.; Rodolakis, A.; Vlahos, G.; Thomakos, N.; Haidopoulos, D.; et al. VEGF Directly Suppresses Activation of T-Cells from Ovarian Cancer Patients and Healthy Individuals via VEGF Receptor Type 2. Int. J. Cancer 2012, 130, 857–864. [Google Scholar] [CrossRef]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic Agents Can Increase Lymphocyte Infiltration into Tumor and Enhance the Effectiveness of Adoptive Immunotherapy of Cancer. Cancer Res. 2010, 70, 6171–6180. [Google Scholar] [CrossRef] [Green Version]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing Cancer Immunotherapy Using Antiangiogenics: Opportunities and Challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef]
- Chow, S.; Berek, J.S.; Dorigo, O. Development of Therapeutic Vaccines for Ovarian Cancer. Vaccines 2020, 8, 657. [Google Scholar] [CrossRef]
- Shih, I.; Kurman, R.J. Ovarian Tumorigenesis: A Proposed Model Based on Morphological and Molecular Genetic Analysis. Am. J. Pathol. 2004, 164, 1511–1518. [Google Scholar] [CrossRef]
- Peoples, G.E.; Anderson, B.W.; Fisk, B.; Kudelka, A.P.; Wharton, J.T.; Ioannides, C.G. Ovarian Cancer-Associated Lymphocyte Recognition of Folate Binding Protein Peptides. Ann. Surg. Oncol. 1998, 5, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Odunsi, K.; Jungbluth, A.A.; Stockert, E.; Qian, F.; Gnjatic, S.; Tammela, J.; Intengan, M.; Beck, A.; Keitz, B.; Santiago, D.; et al. NY-ESO-1 and LAGE-1 Cancer-Testis Antigens Are Potential Targets for Immunotherapy in Epithelial Ovarian Cancer. Cancer Res. 2003, 63, 6076–6083. [Google Scholar] [PubMed]
- Felder, M.; Kapur, A.; Gonzalez-Bosquet, J.; Horibata, S.; Heintz, J.; Albrecht, R.; Fass, L.; Kaur, J.; Hu, K.; Shojaei, H.; et al. MUC16 (CA125): Tumor Biomarker to Cancer Therapy, a Work in Progress. Mol. Cancer. 2014, 13, 129. [Google Scholar] [CrossRef] [Green Version]
- Parvathareddy, S.K.; Siraj, A.K.; Al-Badawi, I.A.; Tulbah, A.; Al-Dayel, F.; Al-Kuraya, K.S. Differential Expression of PD-L1 between Primary and Metastatic Epithelial Ovarian Cancer and Its Clinico-Pathological Correlation. Sci. Rep. 2021, 11, 3750. [Google Scholar] [CrossRef]
- Wang, Q.; Lou, W.; Di, W.; Wu, X. Prognostic Value of Tumor PD-L1 Expression Combined with CD8 + Tumor Infiltrating Lymphocytes in High Grade Serous Ovarian Cancer. Int. Immunopharmacol. 2017, 52, 7–14. [Google Scholar] [CrossRef]
- Zhu, J.; Wen, H.; Bi, R.; Wu, Y.; Wu, X. Prognostic Value of Programmed Death-Ligand 1 (PD-L1) Expression in Ovarian Clear Cell Carcinoma. J. Gynecol. Oncol. 2017, 28, e77. [Google Scholar] [CrossRef] [Green Version]
- Webb, J.R.; Milne, K.; Kroeger, D.R.; Nelson, B.H. PD-L1 Expression Is Associated with Tumor-Infiltrating T Cells and Favorable Prognosis in High-Grade Serous Ovarian Cancer. Gynecol. Oncol. 2016, 141, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Mesnage, S.J.L.; Auguste, A.; Genestie, C.; Dunant, A.; Pain, E.; Drusch, F.; Gouy, S.; Morice, P.; Bentivegna, E.; Lhomme, C.; et al. Neoadjuvant Chemotherapy (NACT) Increases Immune Infiltration and Programmed Death-Ligand 1 (PD-L1) Expression in Epithelial Ovarian Cancer (EOC). Ann. Oncol. 2017, 28, 651–657. [Google Scholar] [CrossRef]
- Kim, H.-S.; Kim, J.-Y.; Lee, Y.J.; Kim, S.H.; Lee, J.-Y.; Nam, E.J.; Kim, S.; Kim, S.W.; Kim, Y.T. Expression of Programmed Cell Death Ligand 1 and Immune Checkpoint Markers in Residual Tumors after Neoadjuvant Chemotherapy for Advanced High-Grade Serous Ovarian Cancer. Gynecol. Oncol. 2018, 151, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Lim, S.O.; Xia, W.; Lee, H.H.; Chan, L.C.; Kuo, C.W.; Khoo, K.H.; Chang, S.S.; Cha, J.H.; Kim, T.; et al. Glycosylation and Stabilization of Programmed Death Ligand-1 Suppresses T-Cell Activity. Nat. Commun. 2016, 30, 12632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.N.; Lee, H.H.; Hsu, J.L.; Yu, D.; Hung, M. The Impact of PD-L1 N-Linked Glycosylation on Cancer Therapy and Clinical Diagnosis. J. Biomed. Sci. 2020, 27, 77. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Hu, Y.; Sun, S.; Chen, L.; Schnaubelt, M.; Clark, D.; Ao, M.; Zhang, Z.; Chan, D.; Qian, J.; et al. Glycoproteomics-Based Signatures for Tumor Subtyping and Clinical Outcome Prediction of High-Grade Serous Ovarian Cancer. Nat. Commun. 2020, 11, 6139. [Google Scholar] [CrossRef]
- Lee, H.H.; Wang, Y.N.; Xia, W.; Chen, C.H.; Rau, K.M.; Ye, L.; Wei, Y.; Chou, C.K.; Wang, S.C.; Yan, M.; et al. Removal of N-Linked Glycosylation Enhances PD-L1 Detection and Predicts Anti-PD-1/PD-L1 Therapeutic Efficacy. Cancer Cell 2019, 36, 168–178. [Google Scholar] [CrossRef] [PubMed]
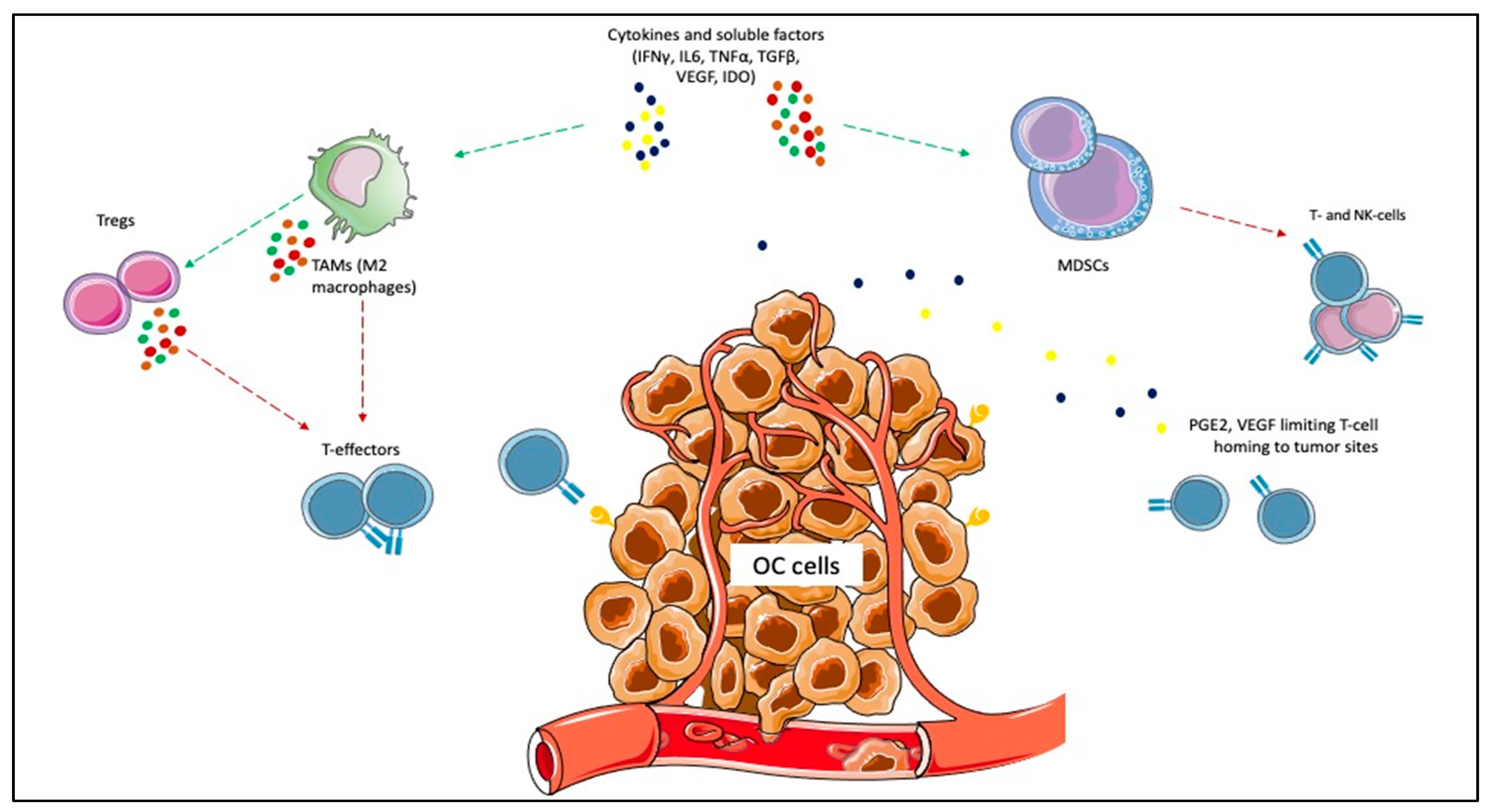

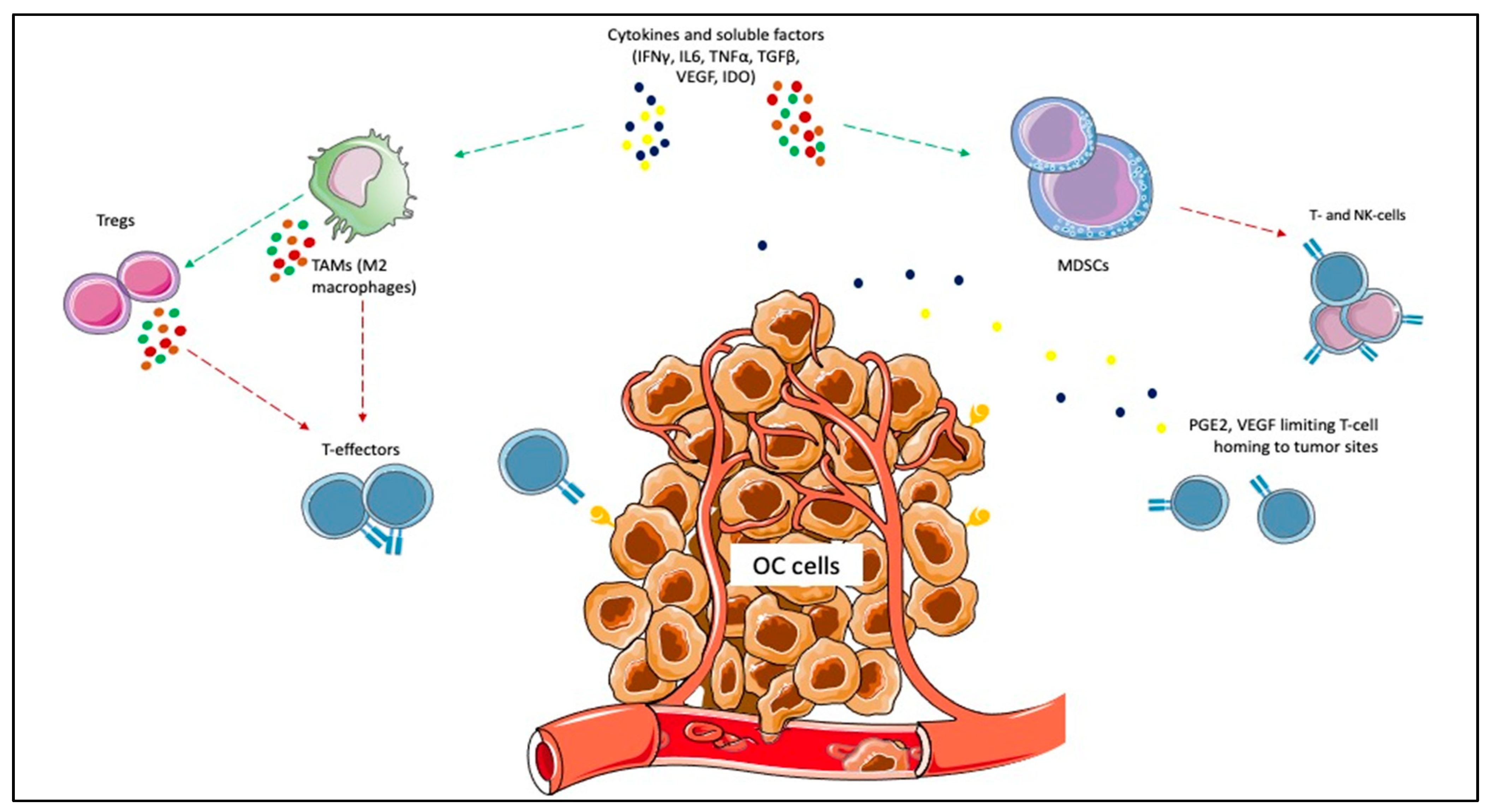{kind=link}
| Study Name | Phase | Target Population (Number of Pts) | Administered Drugs | Primary EP | Results |
|---|---|---|---|---|---|
| Keynote-100 (NCT02674061) [12,13] | II | Cohort A ROC after 1–3 therapies, TFI 3–12 mos (n = 285); Cohort B ROC after 6 lines, TFI >3 mos (n = 91) | Pembrolizumab | ORR (RECIST and by PD-L1) | ORR 8.0%, DC R 37% (overall), 1/3 responses > 6 mos; mDOR (A), NR (B); mPFS 2.1 mos; PD-L1+: ORR 17.1%, OS 21.9 (cohort A) and 24.0 mos (B); ≥G3 AEs: 19.7%; 2 treatment-related deaths. |
| Keynote-028 (NCT02054806) [14] | Ib | PD-L1+ ROC (n = 26) | Pembrolizumab | ORR | ORR 11.5%, mPFS 1.9 mos, mOS 13.8 mos AEs 73.1% (1 G3) |
| NCT02865811 [15] | II | PR-ROC, fallopian tube or peritoneal cancer (n = 26) | Pembrolizumab + PLD | Clinical Benefit Rate (CR, PR, SD) | ORR 19%, 3 PR, 1 SD > 24 w) ≥G3 AEs: rash (19%), ↑ALT (8%) |
| NCT02853318 [16] | II | PS- and PR-ROC (n = 40) | Pembrolizumab + bevacizumab + cyclophosphamide | ORR, mPFS | ORR 47.5%, mPFS 10 mos; 6 mos PFS: 100% (PS-ROC), 59% (PR-ROC) (p = 0.024) <G3 AEs: fatigue (45.0%), diarrhea (32.5%), hypertension (27.5%); ≥G3: hypertension (15.0%), lymphopenia (7.5%). |
| TOPACIO/Keynote-162 (NCT02657889) [17] | I–II | PR-ROC (n = 62) | Pembrolizumab + niraparib | ORR | ORR 25%, DCR 68%; BRCAm: ORR 45%, DCR 73% ≥G3 AEs: anemia (21%), thrombocytopenia (9%). |
| UMIN000005714 [18] | II | PR-ROC (n = 20) | Nivolumab (1 and 3 mg/kg) | BOR | 2 CR; DCR 45%; mPFS 3.5 mos; mOS 20 mos ≥G3 AEs 40%, 2 SAEs, 11% discontinuation. |
| NRG-GY003 (NCT02498600) [19] | II | PS- and PR-ROC (n = 100) | Nivolumab vs. nivolumab + ipilimumab → nivolumab maintenance | ORR | ORR 31.4% (N + I) vs. 12.2% (N) (p = 0.034); PFS 3.9 (N + I) vs. 2 (N) mos (HR = 0.53); OS 28.1 (N + I) vs. 21.8 (N) mos (HR = 0.79); responses not associated with PD-L1 ≥G3 AEs: 33% (N), 49% (N + I). |
| NCT02873962 [20] | II | PS- and PR-EOC (n = 38) | Nivolumab + bevacizumab | ORR | ORR: 28.9% (40% PS-ROC, PR-ROC 16.7%); mPFS 9.4 mos (12.1 mos PS-ROC); PD-L1- better than PD-L1+ pts. AEs 89.5%: fatigue (47.4%), headache (28.9%), myalgia (28.9%), ↑amylase (28.9%), ↑AST (26.3%), hypertension (26.3%); pneumonitis (10.5%), colitis (5.3%). ≥G3 AEs 23.7%. |
| NINJA [21] | III | PR-ROC (Japanese population, n = 316) | Nivolumab vs. gemcitabine or PLD | OS | No OS differences (HR = 1.03); PFS 2 (N) vs. 3.8 mos (gem/PLD) (HR = 1.46; p = 0.002) ≥G3 AEs: 22.4% (N), 68.4% (gem/PLD) |
| JAVELIN (NCT01772004) [22] | Ib | PR-ROC (n = 125) | Avelumab | BOR | 1/125 CR, 11/125 PR; 1 yr PFS: 10.2%; mOS: 11.2 mos; mPFS: 2.6 mos ≥G3 AEs 7.2% (↑lipase 2.4%) |
| JAVELIN 200 (NCT02580058) [25] | III | PR-ROC (n = 566) | Avelumab vs. avelumab + PLD vs. PLD | PFS, OS | Ave + PLD: PFS 3.7 mos (HR vs. PLD = 0.78, p = 0.03), OS 15.7 mos (HR vs. PLD = 0.89, p = 0.2); avelumab vs. PLD HR for OS = 1.14, HR for PFS = 1.68 PD-L1+: trend for longer PFS and OS Ave+PLD vs. PLD ≥G3 AEs: 49.7% (Ave), 68.7% (Ave + PLD), 59.3% (PLD) |
| ENCORE-603 (NCT02915523) [26] | II | PR-ROC (n = 126) | Avelumab + Entinostat vs. avelumab + PBO | PFS | mPFS = 1.64 (A + E) vs. 1.51 mos (A + P) (p = 0.031). No differences in ORR (6% vs. 5%), or OS (NE vs. 11.3 mos) AEs: 93% (A + E), 78% (A + P); ≥G3 AEs: 41% (A + E), 10% (A + P) |
| IMagyn050 (NCT03038100) [27] | III | First-line OC (n = 1301) | CHT (CBDCA + paclitaxel) + bevacizumab + atezolizumab vs. CHT + beva + PBO | PFS/OS in ITT and PD-L1+ population | PFS 19.5 vs. 18.4 mos (HR = 0.92; p = 0.28); PD-L1 + PFS: 20.8 vs. 18.5 mos (HR = 0.8; p = 0.38); no OS advantage. ≥G3: neutropenia, hypertension, anemia. |
| NCT02431559 [28] | I–II | PR-ROC (n = 40) | Durvalumab + PLD | PFS6 | PFS6: 47.7%; ORR 15% G3 Aes in ≥2 pts: lymphopenia, anemia, lipase increased, rash, stomatitis |
| MEDIOLA (NCT02734004) [29,30,31] | II | gBRCAm (n = 32) and BRCAwt (n = 63) PS-ROC | gBRCAm group: olaparib (4 w) → durvalumab + olaparib BRCAwt group: durvalumab + olaparib (D + O; n = 32), durvalumab + olaparib + bevacizumab (D + O + B; n = 31) | 12 w DCR, safety | gBRCAm group: 12 w DCR 81%, mPFS 11.1 mos, ORR 71.9% BRCAwt D + O group: ORR 31.3%, mPFS 5.5 mos, 24 w DCR 28.1%; 6% discontinuation BRCAwt D + O + B group: ORR 77.4%, mPFS 14.7 mos, 24 w DCR 77.4%; 16% discontinuation ≥G3 AEs: anemia, lipase increased, neutropenia; + hypertension, fatigue (O + D + B cohort) |
| NCT02484404 [32] | I–II | PS/PR-ROC (n = 35) | Durvalumab + olaparib or cediranib | RP2D | 5 PR, 13 SD, DCR 53% ≥G3 AEs: anemia (26%), lymphopenia (14%) |
| NCT02484404 [33] | II | PR-ROC (n = 35) | Durvalumab + olaparib | ORR | ORR 14%; longer PFS with ↑IFNγ (p = 0.023), shorter PFS with ↑VEGFR3 (p = 0.017) ≥G3 AE: anemia (31%). |
| NCT02485990 [35] | I–II | PR-ROC (n = 24) | Tremelimumab vs. tremelimumab + olaparib | RP2D, ORR | 1 PR, 9 SD ≥G3 AEs: rash (13%), hepatitis (8%), colitis (8%); no ≥ G4 AEs. |
| NCT02571725 [36] | I–II | BRCAm OC (n = 3) | Tremelimumab + olaparib | RP2D | G1/2 AEs, decreased tumor size after 3 cycles |
| Clinicaltrials.gov Registration Number (Name) | Phase | ICI Combinations (Drug Class) |
|---|---|---|
| NCT03508570 | I | Nivolumab + Ipilimumab |
| NCT03355976 | II | |
| NCT02834013 | II | |
| NCT03959761 | I–II | Nivolumab (IP) + Surgery plus HIPEC |
| NCT02737787 | I | Nivolumab + WT1 or NY-ESO-1 (vaccine) |
| NCT03522246 (ATHENA) | III | Nivolumab + Rucaparib (PARPi) |
| NCT02873962 | II | Nivolumab + Bevacizumab (anti-VEGF) ± Rucaparib |
| NCT04611126 | I–II | Nivolumab + Relatimab (anti-LAG-3) + Ipilimumab + ACT |
| NCT03100006 | IB-IIA | Nivolumab + Oregovomab (anti-Ca125) |
| NCT04620954 (ORION-02) | I–II | Nivolumab + Oregovomab + PLD + CBDCA |
| NCT03667716 | I | Nivolumab + COM701 (PVRIG inhibitor) |
| NCT04570839 | I–II | Nivolumab + COM701 + BMS-986207 (anti-TIGIT) |
| NCT04514484 | I | Nivolumab + Cabozantinib (TKI) |
| NCT02335918 | I–II | Nivolumab, Varlilumab (anti-CD27) |
| NCT02526017 | I | Nivolumab + Cabiralizumab (anti-CSF1R) |
| NCT02440425 | II | Pembrolizumab + Paclitaxel |
| NCT03029598 | I–II | Pembrolizumab + CBDCA |
| NCT04387227 | II | |
| NCT04575961 (PERCEPTION) | II | Pembrolizumab + Platinum-based CTx |
| NCT02766582 | II | Pembrolizumab + CBDCA + Paclitaxel |
| NCT02834975 | II | |
| NCT03410784 (MITO28MaNGOov4) | II | |
| NCT02520154 | II | |
| NCT03126812 | I–II | |
| NCT03539328 | II | Pembrolizumab + Gemcitabine or Paclitaxel or PLD vs. CTx |
| NCT02900560 | II | Pembrolizumab ± Azacitidine |
| NCT02901899 | II | Pembrolizumab + Guadecitabine |
| NCT03596281 (PEMBOV) | I | Pembrolizumab + Bevacizumab + PLD |
| NCT04417192 (OLAPem) | II | Pembrolizumab + Olaparib |
| NCT03740165 (MK-7339-001/KEYLYNK-001/ENGOT-ov43/GOG-3036) | III | CBDCA + Paclitaxel → Pembrolizumab + Olaparib vs. Pembrolizumab + PBO vs. PBO + Olaparib |
| NCT04519151 | II | Pembrolizumab + Lenvatinib |
| NCT03797326 (MK-7902-005/E7080-G000-224/LEAP-005) | II | |
| NCT04781088 | II | Pembrolizumab + Lenvatinib + Paclitaxel |
| NCT02606305 | I–II | Pembrolizumab + Mirvetuximab soravtansine (anti-FRα ADC) |
| NCT03734692 | I–II | Pembrolizumab + IP Rintatolimod (anti-TLR3) + Cisplatin |
| NCT03158935 (ACTIVATE) | Pembrolizumab + Cyclophosphamide + autologous TILs + IL-2 | |
| NCT03029403 | II | Pembrolizumab + DPX-Survivac (vaccine) + Cyclophosphamide |
| NCT03113487 | II | Pembrolizumab + p53 MVA (vaccine) |
| NCT04713514 (TEDOVA) | II | Pembrolizumab + OSE2101 vs. OSE2101 (multi-epitope vaccine) |
| NCT03558139 | I | Avelumab + Magrolimab (anti-CD47) |
| NCT04510584 | II | Atezolizumab + Bevacizumab |
| NCT02891824 (ATALANTE) | III | Atezolizumab + Bevacizumab + platinum-based Ctx vs. PBO + Bevacizumab + platinum-based Ctx |
| NCT03353831 | III | Atezolizumab + Bevacizumab + Ctx vs. Bevacizumab + Ctx |
| NCT02839707 | II–III | Atezolizumab + Bevacizumab + PLD |
| NCT02659384 | II | Atezolizumab + Bevacizumab ± acetylsalicylic acid |
| NCT03363867 (BEACON) | II | Atezolizumab + Bevacizumab + Cobimetinib |
| NCT03695380 | I | Atezolizumab + Cobimetinib (anti-MEK) + Niraparib |
| NCT03598270 (ANITA) | III | Atezolizumab + Platinum-based Ctx vs. platinum-based Ctx → Niraparib ± Atezolizumab maintenance |
| NCT02914470 (PROLOG) | I | Atezolizumab + CBDCA + Cyclophosphamide |
| NCT03206047 | I–II | Atezolizumab + Guadecitabine + CDX-1401 (vaccine) |
| NCT03073525 | II | Atezolizumab + Vigil (cancer cell therapy) |
| NCT01975831 | I | Durvalumab + Tremelimumab |
| NCT02953457 | II | |
| NCT03026062 | II | |
| NCT04644289 (WoO) | II | Durvalumab + Olaparib |
| NCT04742075 (DOVACC) | II | Durvalumab + Olaparib + UV-1 |
| NCT04015739 (BOLD) | II | Durvalumab + Olaparib + Bevacizumab |
| NCT03737643 (DUO-O) | III | Durvalumab + platinum-based Ctx + Bevazicumab vs. PBO + platinum-based Ctx + Bevacizumab → Durvalumab + Bevacizumab + Olaparib maintenance |
| NCT03699449 (AMBITION) | II | Durvalumab + Tremelimumab or Olaparib or Cediranib or Ctx |
| NCT02726997 (N-Dur) | I–II | Durvalumab + CBDCA + Paclitaxel |
| NCT03430518 | I | Durvalumab + Eribuline |
| NCT03085225 (TRAMUNE) | I | Durvalumab + Trabectedin |
| NCT02811497 (METADUR) | II | Durvalumab + Azacitidine |
| NCT02764333 | II | Durvalumab + TPIV200 (anti-FR vaccine) |
| NCT02725489 | II | Durvalumab + Vigil |
| NCT03267589 | II | Durvalumab + Tremelimumab + MEDI 9447 (anti-CD73 Ab) + MEDI 0562 (anti-OX40) |
| NCT04019288 | I–II | Durvalumab + AVB-S6-500 (Anti-AXL Fusion Protein) |
| NCT03277482 | I | Durvalumab + Tremelimumab + RT |
| NCT02571725 | II | Tremelimumab + Olaparib |
| NCT03602859 | III | Dostarlimab (anti-PD1) + Ctx vs. Ctx + Niraparib vs. Ctx + PBO |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiorano, B.A.; Maiorano, M.F.P.; Lorusso, D.; Maiello, E. Ovarian Cancer in the Era of Immune Checkpoint Inhibitors: State of the Art and Future Perspectives. Cancers 2021, 13, 4438. https://doi.org/10.3390/cancers13174438
Maiorano BA, Maiorano MFP, Lorusso D, Maiello E. Ovarian Cancer in the Era of Immune Checkpoint Inhibitors: State of the Art and Future Perspectives. Cancers. 2021; 13(17):4438. https://doi.org/10.3390/cancers13174438
Chicago/Turabian StyleMaiorano, Brigida Anna, Mauro Francesco Pio Maiorano, Domenica Lorusso, and Evaristo Maiello. 2021. "Ovarian Cancer in the Era of Immune Checkpoint Inhibitors: State of the Art and Future Perspectives" Cancers 13, no. 17: 4438. https://doi.org/10.3390/cancers13174438
APA StyleMaiorano, B. A., Maiorano, M. F. P., Lorusso, D., & Maiello, E. (2021). Ovarian Cancer in the Era of Immune Checkpoint Inhibitors: State of the Art and Future Perspectives. Cancers, 13(17), 4438. https://doi.org/10.3390/cancers13174438