
Can the New and Old Drugs Exert an Immunomodulatory Effect in Acute Myeloid Leukemia?

 ,
,  ,
,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Conventional Chemotherapy
3. FLT3 Inhibitors
4. Hypomethylating Agents
5. IDH Inhibitors
6. BCL-2 Inhibitors
7. Hedgehog Pathway Inhibitors
8. p53 Targeted Therapies
9. Discussion
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Heuser, M.; Ofran, Y.; Boissel, N.; Brunet Mauri, S.; Craddock, C.; Janssen, J.; Wierzbowska, A.; Buske, C. Acute myeloid leukaemia in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 697–712. [Google Scholar] [CrossRef]
- Curti, A.; Pandolfi, S.; Valzasina, B.; Aluigi, M.; Isidori, A.; Ferri, E.; Salvestrini, V.; Bonanno, G.; Rutella, S.; Durelli, I.; et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25- into CD25+ T regulatory cells. Blood 2007, 109, 2871–2877. [Google Scholar] [CrossRef]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Curran, E.K.; Godfrey, J.; Kline, J. Mechanisms of Immune Tolerance in Leukemia and Lymphoma. Trends Immunol. 2017, 38, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Austin, R.; Smyth, M.J.; Lane, S.W. Harnessing the immune system in acute myeloid leukaemia. Crit. Rev. Oncol. Hematol. 2016, 103, 62–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folgiero, V.; Goffredo, B.M.; Filippini, P.; Masetti, R.; Bonanno, G.; Caruso, R.; Bertaina, V.; Mastronuzzi, A.; Gaspari, S.; Zecca, M.; et al. Indoleamine 2,3-dioxygenase 1 (IDO1) activity in leukemia blasts correlates with poor outcome in childhood acute myeloid leukemia. Oncotarget 2014, 5, 2052–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teague, R.M.; Kline, J. Immune evasion in acute myeloid leukemia: Current concepts and future directions. J. Immunother. Cancer 2013, 1, 13. [Google Scholar] [CrossRef] [Green Version]
- Barrett, A.J. Acute myeloid leukaemia and the immune system: Implications for immunotherapy. Br. J. Haematol. 2020, 188, 147–158. [Google Scholar] [CrossRef]
- Vago, L.; Gojo, I. Immune escape and immunotherapy of acute myeloid leukemia. J. Clin. Investig. 2020, 130, 1552–1564. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bewersdorf, J.P.; Stahl, M.; Zeidan, A.M. Immunotherapy in acute myeloid leukemia and myelodysplastic syndromes: The dawn of a new era? Blood Rev. 2019, 34, 67–83. [Google Scholar] [CrossRef]
- Catellani, S.; Pierri, I.; Gobbi, M.; Poggi, A.; Zocchi, M.R. Imatinib treatment induces CD5+ B lymphocytes and igm natural antibodies with anti-leukemic reactivity in patients with chronic myelogenous leukemia. PLoS ONE 2011, 6, e18925. [Google Scholar] [CrossRef] [PubMed]
- Cunha, C.; Carvalho, A.; Esposito, A.; Bistoni, F.; Romani, L. DAMP signaling in fungal infections and diseases. Front. Immunol. 2012, 3, 286. [Google Scholar] [CrossRef] [Green Version]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, N.R.; Baumgartner, F.; Braun, L.; O’Sullivan, D.; Thomas, S.; Waterhouse, M.; Müller, T.A.; Hanke, K.; Taromi, S.; Apostolova, P.; et al. Sorafenib promotes graft-versus-leukemia activity in mice and humans through IL-15 production in FLT3-ITD-mutant leukemia cells. Nat. Med. 2018, 24, 282–291. [Google Scholar] [CrossRef]
- Brunetti, C.; Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. CPX-351 in acute myeloid leukemia: Can a new formulation maximize the efficacy of old compounds? Expert Rev. Hematol. 2017, 10, 853–862. [Google Scholar] [CrossRef]
- Kiyoi, H.; Kawashima, N.; Ishikawa, Y. FLT3 mutations in acute myeloid leukemia: Therapeutic paradigm beyond inhibitor development. Cancer Sci. 2020, 111, 312–322. [Google Scholar] [CrossRef] [Green Version]
- Stone, R.M. What FLT3 inhibitor holds the greatest promise? Best Pract. Res. Clin. Haematol. 2018, 31, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chang, Y.J.; Xu, L.P.; Zhang, X.H.; Wang, Y.; Liu, K.Y.; Huang, X.J. Reversal of T Cell Exhaustion by the First Donor Lymphocyte Infusion Is Associated with the Persistently Effective Antileukemic Responses in Patients with Relapsed AML after Allo-HSCT. Biol. Blood Marrow Transplant. 2018, 24, 1350–1359. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Manero, G. Demethylating agents in myeloid malignancies. Curr. Opin. Oncol. 2008, 20, 705–710. [Google Scholar] [CrossRef] [Green Version]
- Frikeche, J.; Clavert, A.; Delaunay, J.; Brissot, E.; Grégoire, M.; Gaugler, B.; Mohty, M. Impact of the hypomethylating agent 5-azacytidine on dendritic cells function. Exp. Hematol. 2011, 39, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.N.; Lin, J.; Wang, L.; Yu, L. Demethylating treatment suppresses natural killer cell cytolytic activity. Mol. Immunol. 2009, 46, 2064–2070. [Google Scholar] [CrossRef]
- Kopp, L.M.; Ray, A.; Denman, C.J.; Senyukov, V.S.; Somanchi, S.S.; Zhu, S.; Lee, D.A. Decitabine has a biphasic effect on natural killer cell viability, phenotype, and function under proliferative conditions. Mol. Immunol. 2013, 54, 296–301. [Google Scholar] [CrossRef]
- Ando, T.; Sano, H.; Yokoo, M.; Kusaba, K.; Kidoguchi, K.; Yamaguchi, K.; Katsuya, H.; Yoshihara, S.; Kubota, Y.; Kojima, K.; et al. Durable remission of post-transplant relapsed FLT3-ITD AML in response to gilteritinib administration after a second transplant from the same donor. Int. J. Hematol. 2020, 112, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Triozzi, P.L.; Aldrich, W.; Achberger, S.; Ponnazhagan, S.; Alcazar, O.; Saunthararajah, Y. DiVerential eVects of low-dose decitabine on immune eVector and suppressor responses in melanoma-bearing mice. Cancer Immunol. Immunother. 2012, 61, 1441–1450. [Google Scholar] [CrossRef] [PubMed]
- Ørskov, A.D.; Treppendahl, M.B.; Skovbo, A.; Holm, M.S.; Friis, L.S.; Hokland, M.; Grønbæk, K. Hypomethylation and up-regulation of PD-1 in T cells by azacytidine in MDS/AML patients: A rationale for combined targeting of PD-1 and DNA methylation. Oncotarget 2015, 6, 9612–9626. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Bueso-Ramos, C.; Dinardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.W.; Kurago, Z.B.; Stewart, C.A.; Wilson, M.J.; Martin, M.P.; Mace, B.E.; Carrington, M.; Trowsdale, J.; Lutz, C.T. DNA methylation maintains allele-specific KIR gene expression in human natural killer cells. J. Exp. Med. 2003, 197, 245–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cany, J.; Roeven, M.W.; Van Evert, J.S.; Hobo, W.; Maas, F.; Fernandez, R.F.; Blijlevens, N.M.; Van Der Velden, W.J.; Huls, G.; Jansen, J.H.; et al. Decitabine enhances targeting of AML cells by CD34 + progenitor-derived NK cells in NOD/SCID/IL2Rg null mice. Blood 2018, 131, 202–214. [Google Scholar] [CrossRef]
- Cheng, E.H.; Wei, M.C.; Weiler, S.; Flavell, R.A.; Mak, T.W.; Lindsten, T.; Korsmeyer, S.J. BCL-2, BCL-XL sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 2001, 8, 705–711. [Google Scholar] [CrossRef]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; Deangelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid Leukemia. Cancer Discov. 2014, 4, 362–675. [Google Scholar] [CrossRef] [Green Version]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Béne, M.C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/measurable residual disease in AML: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, K.C.; Ruela-De-Sousa, R.R.; Fuhler, G.M.; Aberson, H.L.; Ferreira, C.V.; Peppelenbosch, M.P.; Spek, C.A. Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene 2010, 29, 6314–6322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Q.; Gu, D.; Liu, H.; Yang, L.; Zhang, X.; Yoder, M.C.; Kaplan, M.H.; Xie, J. Defective TGF-β signaling in bone marrow-derived cells prevents hedgehog-induced skin tumors. Cancer Res. 2014, 74, 471–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yánez, D.C.; Lau, C.I.; Chawda, M.M.; Ross, S.; Furmanski, A.L.; Crompton, T. Hedgehog signaling promotes TH2 differentiation in naive human CD4 T cells. J. Allergy Clin. Immunol. 2019, 144, 1419–1423.e1. [Google Scholar] [CrossRef] [Green Version]
- Sallman, D.A.; McLemore, A.F.; Aldrich, A.L.; Komrokji, R.S.; McGraw, K.L.; Dhawan, A.; Geyer, S.; Hou, H.A.; Eksioglu, E.A.; Sullivan, A.; et al. TP53 mutations in myelodysplastic syndromes and secondary AML confer an immunosuppressive phenotype. Blood 2020, 136, 2812–2823. [Google Scholar] [CrossRef]
- Sallman, D.A. To target the untargetable: Elucidation of synergy of APR-246 and azacitidine in TP53 mutant myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2020, 105, 1470–1472. [Google Scholar] [CrossRef]
- Ersvaer, E.; Brenner, A.K.; Vetås, K.; Reikvam, H.; Bruserud, Ø. Effects of cytarabine on activation of human T cells-cytarabine has concentration-dependent effects that are modulated both by valproic acid and all-trans retinoic acid. BMC Pharmacol. Toxicol. 2015, 16, 12. [Google Scholar] [CrossRef] [Green Version]
- Goswami, M.; Prince, G.; Biancotto, A.; Moir, S.; Kardava, L.; Santich, B.H.; Cheung, F.; Kotliarov, Y.; Chen, J.; Shi, R.; et al. Impaired B cell immunity in acute myeloid leukemia patients after chemotherapy. J. Transl. Med. 2017, 15, 155. [Google Scholar] [CrossRef]
- Ramakrishnan, R.; Assudani, D.; Nagaraj, S.; Hunter, T.; Cho, H.I.; Antonia, S.; Altiok, S.; Celis, E.; Gabrilovich, D.I. Chemotherapy enhances tumor cell susceptibility to CTL-mediated killing during cancer immunotherapy in mice. J. Clin. Investig. 2010, 120, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Smyth, M.J.; Kroemer, G. Mechanism of Action of Conventional and Targeted Anticancer Therapies: Reinstating Immunosurveillance. Immunity 2013, 39, 74–88. [Google Scholar] [CrossRef] [Green Version]
- Ocadlikova, D.; Lecciso, M.; Isidori, A.; Loscocco, F.; Visani, G.; Amadori, S.; Cavo, M.; Curti, A. Chemotherapy-Induced Tumor Cell Death at the Crossroads Between Immunogenicity and Immunotolerance: Focus on Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 1004. [Google Scholar] [CrossRef]
- Rusakiewicz, S.; Semeraro, M.; Sarabi, M.; Desbois, M.; Locher, C.; Mendez, R.; Vimond, N.; Concha, A.; Garrido, F.; Isambert, N.; et al. Immune in filtrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res. 2013, 73, 3499–3510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murao, A.; Aziz, M.; Wang, H.; Brenner, M.; Wang, P. Release mechanisms of major DAMPs. Apoptosis 2021, 26, 152–162. [Google Scholar] [CrossRef]
- Lecciso, M.; Ocadlikova, D.; Sangaletti, S.; Trabanelli, S.; De Marchi, E.; Orioli, E.; Pegoraro, A.; Portararo, P.; Jandus, C.; Bontadini, A.; et al. ATP release from chemotherapy-treated dying leukemia cells Elicits an immune suppressive effect by increasing regulatory T cells and Tolerogenic dendritic cells. Front. Immunol. 2017, 8, 1918. [Google Scholar] [CrossRef]
- Fredly, H.; Ersvær, E.; Gjertsen, B.T.; Bruserud, Ø. Immunogenic apoptosis in human acute myeloid leukemia (AML): Primary human AML cells expose calreticulin and release heat shock protein (HSP) 70 and HSP90 during apoptosis. Oncol. Rep. 2011, 25, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Truxova, I.; Hensler, M.; Becht, E.; Kasikova, L.; Moserova, I.; Vosahlikova, S.; Klouckova, J.; Church, S.E.; Cremer, I.; et al. Calreticulin exposure by malignant blasts correlates with robust anticancer immunity and improved clinical outcome in AML patients. Blood 2016, 128, 3113–3124. [Google Scholar] [CrossRef] [Green Version]
- Fucikova, J.; Kralikova, P.; Fialova, A.; Brtnicky, T.; Rob, L.; Bartunkova, J.; Špíšek, R. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res. 2011, 71, 4821–4833. [Google Scholar] [CrossRef] [Green Version]
- Wemeau, M.; Kepp, O.; Tesnière, A.; Panaretakis, T.; Flament, C.; De Botton, S.; Zitvogel, L.; Kroemer, G.; Chaput, N. Calreticulin exposure on malignant blasts predicts a cellular anticancer immune response in patients with acute myeloid leukemia. Cell Death Dis. 2010, 1, e104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Fosco, D.; Kline, D.E.; Kline, J. Calreticulin promotes immunity and type I interferon-dependent survival in mice with acute myeloid leukemia. Oncoimmunology 2017, 6, e1278332. [Google Scholar] [CrossRef] [Green Version]
- Oliva, E.N.; Franek, J.; Patel, D.; Zaidi, O.; Nehme, S.A.; Almeida, A.M. The Real-World Incidence of Relapse in Acute Myeloid Leukemia (AML): A Systematic Literature Review (SLR). Blood 2018, 132, 5188. [Google Scholar] [CrossRef]
- Belyaev, N.N.; Abdolla, N.; Perfilyeva, Y.V.; Ostapchuk, Y.O.; Krasnoshtanov, V.K.; Kali, A.; Tleulieva, R. Daunorubicin conjugated with alpha-fetoprotein selectively eliminates myeloid-derived suppressor cells (MDSCs) and inhibits experimental tumor growth. Cancer Immunol. Immunother. 2018, 67, 101–111. [Google Scholar] [CrossRef]
- Ogbomo, H.; Michaelis, M.; Klassert, D.; Doerr, H.W.; Cinatl, J. Resistance to cytarabine induces the up-regulation of NKG2D ligands and enhances natural killer cell lysis of leukemic cells. Neoplasia 2008, 10, 1402–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravandi, F.; Alattar, M.L.; Grunwald, M.R.; Rudek, M.A.; Rajkhowa, T.; Richie, M.A.; Pierce, S.; Daver, N.; Garcia-Manero, G.; Faderl, S.; et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood 2013, 121, 4655–4662. [Google Scholar] [CrossRef]
- Lange, A.; Jaskula, E.; Lange, J.; Dworacki, G.; Nowak, D.; Simiczyjew, A.; Mordak-Domagala, M.; Sedzimirska, M. The sorafenib anti-relapse effect after alloHSCT is associated with heightened alloreactivity and accumulation of CD8+PD-1+ (CD279+) lymphocytes in marrow. PLoS ONE 2018, 13, e0190525. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, L.; Jang, M.; Zhang, T.; Akhtari, M.; Alachkar, H. Midostaurin reduces Regulatory T cells markers in Acute Myeloid Leukemia. Sci. Rep. 2018, 8, 17544. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Wolleschak, D.; Mack, T.S.; Perner, F.; Frey, S.; Schnöder, T.M.; Wagner, M.C.; Höding, C.; Pils, M.C.; Parkner, A.; Kliche, S.; et al. Clinically relevant doses of FLT3-kinase inhibitors quizartinib and midostaurin do not impair T-cell reactivity and function. Haematologica 2014, 99, e90. [Google Scholar] [CrossRef] [Green Version]
- Du, M.; Zhou, F.; Jin, R.; Hu, Y.; Mei, H. Mutations in the DNA methylation pathway predict clinical efficacy to hypomethylating agents in myelodysplastic syndromes: A meta-analysis. Leuk. Res. 2019, 80, 11–18. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Patel, K.P.; Garcia-Manero, G.; Luthra, R.; Pierce, S.; Borthakur, G.; Jabbour, E.; Kadia, T.; Pemmaraju, N.; Konopleva, M.; et al. Lack of association of IDH1, IDH2 and DNMT3A mutations with outcome in older patients with acute myeloid leukemia treated with hypomethylating agents. Leuk. Lymphoma 2014, 55, 1925–1929. [Google Scholar] [CrossRef] [Green Version]
- Hourigan, C.S.; Karp, J.E. Development of therapeutic agents for older patients with acute myelogenous leukemia. Curr. Opin. Investig. Drugs 2010, 11, 669–677. [Google Scholar] [PubMed]
- Kwon, Y.R.; Kim, H.J.; Sohn, M.J.; Lim, J.Y.; Park, K.S.; Lee, S.; Chung, N.G.; Jeong, D.C.; Min, C.K.; Kim, Y.J. Effects of decitabine on allogeneic immune reactions of donor lymphocyte infusion via activation of dendritic cells. Exp. Hematol. Oncol. 2020, 9, 22. [Google Scholar] [CrossRef]
- Daver, N.; Boddu, P.; Garcia-Manero, G.; Yadav, S.S.; Sharman, P.; Allison, J.; Kantarjian, H. Hypomethylating agents in combination with immune checkpoint inhibitors in acute myeloid leukemia and myelodysplastic syndromes. Leukemia 2018, 32, 1094–1105. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, M.; Scarpa, M.; Castagliuolo, I.; Erroi, F.; Basato, S.; Brun, P.; Angriman, I.; Castoro, C. CD80 down-regulation is associated to aberrant DNA methylation in non-inflammatory colon carcinogenesis. BMC Cancer 2016, 16, 388. [Google Scholar] [CrossRef] [PubMed]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157.e19. [Google Scholar] [CrossRef] [Green Version]
- Costantini, B.; Kordasti, S.Y.; Kulasekararaj, A.G.; Jiang, J.; Seidl, T.; Abellan, P.P.; Mohamedali, A.; Thomas, N.S.; Farzaneh, F.; Mufti, G.J. The effects of 5-azacytidine on the function and number of regulatory T cells and T-effectors in myelodysplastic syndrome. Haematologica 2013, 98, 1196–1205. [Google Scholar] [CrossRef] [Green Version]
- Bontkes, H.J.; Ruben, J.M.; Alhan, C.; Westers, T.M.; Ossenkoppele, G.J.; van de Loosdrecht, A.A. Azacitidine differentially affects CD4 pos T-cell polarization in vitro and in vivo in high risk myelodysplastic syndromes. Leuk. Res. 2012, 36, 921–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordasti, S.Y.; Ingram, W.; Hayden, J.; Darling, D.; Barber, L.; Afzali, B.; Lombardi, G.; Wlodarski, M.W.; Maciejewski, J.P.; Farzaneh, F.; et al. CD4+CD25high Foxp3+ regulatory T cells in myelodysplastic syndrome (MDS). Blood 2007, 110, 847–850. [Google Scholar] [CrossRef] [Green Version]
- Kehrmann, J.; Tatura, R.; Zeschnigk, M.; Probst-Kepper, M.; Geffers, R.; Steinmann, J.; Buer, J. Impact of 5-aza-2’-deoxycytidine and epigallocatechin-3-gallate for induction of human regulatory T cells. Immunology 2014, 142, 384–395. [Google Scholar] [CrossRef]
- Craddock, C.; Jilani, N.; Siddique, S.; Yap, C.; Khan, J.; Nagra, S.; Ward, J.; Ferguson, P.; Hazlewood, P.; Buka, R.; et al. Tolerability and Clinical Activity of Post-Transplantation Azacitidine in Patients Allografted for Acute Myeloid Leukemia Treated on the RICAZA Trial. Biol. Blood Marrow Transplant. 2016, 22, 385–390. [Google Scholar] [CrossRef] [Green Version]
- Goodyear, O.C.; Dennis, M.; Jilani, N.Y.; Loke, J.; Siddique, S.; Ryan, G.; Nunnick, J.; Khanum, R.; Raghavan, M.; Cook, M.; et al. Azacitidine augments expansion of regulatory T cells after allogeneic stem cell transplantation in patients with acute myeloid leukemia (AML). Blood 2012, 119, 3361–3369. [Google Scholar] [CrossRef]
- Sánchez-Abarca, L.I.; Gutierrez-Cosio, S.; Santamaría, C.; Caballero-Velazquez, T.; Blanco, B.; Herrero-Sánchez, C.; García, J.L.; Carrancio, S.; Hernández-Campo, P.; González, F.J.; et al. Immunomodulatory effect of 5-azacytidine (5-azaC): Potential role in the transplantation setting. Blood 2010, 115, 107–121. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Ritchey, J.; Prior, J.L.; Holt, M.; Shannon, W.D.; Deych, E.; Piwnica-Worms, D.R.; DiPersio, J.F. In vivo administration of hypomethylating agents mitigate graft-versus-host disease without sacrificing graft-versus-leukemia. Blood 2010, 116, 129–139. [Google Scholar] [CrossRef]
- Goswami, M.; Hensel, N.; Smith, B.D.; Prince, G.T.; Qin, L.; Levitsky, H.I.; Strickland, S.A.; Jagasia, M.; Savani, B.N.; Fraser, J.W.; et al. Expression of putative targets of immunotherapy in acute myeloid leukemia and healthy tissues. Leukemia 2014, 28, 1167–1170. [Google Scholar] [CrossRef] [Green Version]
- Fozza, C.; Corda, G.; Barraqueddu, F.; Virdis, P.; Contini, S.; Galleu, A.; Isoni, A.; Dore, F.; Angelucci, E.; Longinotti, M. Azacitidine improves the T-cell repertoire in patients with myelodysplastic syndromes and acute myeloid leukemia with multilineage dysplasia. Leuk. Res. 2015, 39, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Zhang, Y.; Li, X.; Chen, M.; Liu, C.; Han, W. DNA demethylating agent decitabine broadens the peripheral T cell receptor repertoire. Oncotarget 2016, 7, 37882–37892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; He, Q.; Tao, Y.; Guo, J.; Xu, F.; Wu, L.Y.; Zhao, Y.S.; Wu, D.; Zhou, L.Y.; Su, J.Y.; et al. Decitabine treatment sensitizes tumor cells to T-cell-mediated cytotoxicity in patients with myelodysplastic syndromes. Am. J. Transl. Res. 2017, 9, 454–465. [Google Scholar] [PubMed]
- Son, C.H.; Lee, H.R.; Koh, E.K.; Shin, D.Y.; Bae, J.H.; Yang, K.; Park, Y.S. Combination treatment with decitabine and ionizing radiation enhances tumor cells susceptibility of T cells. Sci. Rep. 2016, 6, 32470. [Google Scholar] [CrossRef] [Green Version]
- Niu, C.; Li, M.; Zhu, S.; Chen, Y.; Zhou, L.; Xu, D.; Li, W.; Cui, J.; Liu, Y.; Chen, J. Decitabine inhibits gamma delta T cell cytotoxicity by promoting KIR2DL2/3 expression. Front. Immunol. 2018, 9, 617. [Google Scholar] [CrossRef] [Green Version]
- Schmiedel, B.J.; Arélin, V.; Gruenebach, F.; Krusch, M.; Schmidt, S.M.; Salih, H.R. Azacytidine impairs NK cell reactivity while decitabine augments NK cell responsiveness toward stimulation. Int. J. Cancer 2011, 128, 2911–2922. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Sánchez, A.; Raneros, A.B.; Palao, R.C.; Sanz, A.B.; Ortiz, A.; Ortega, F.; Suárez-Álvarez, B.; López-Larrea, C. DNA demethylation and histone H3K9 acetylation determine the active transcription of the NKG2D gene in human CD8+ T and NK cells. Epigenetics 2013, 8, 66–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasu, S.; He, S.; Cheney, C.; Gopalakrishnan, B.; Mani, R.; Lozanski, G.; Mo, X.; Groh, V.; Whitman, S.P.; Konopitzky, R.; et al. Decitabine enhances anti-CD33 monoclonal antibody BI 836858-mediated natural killer ADCC against AML blasts. Blood 2016, 127, 2879–2889. [Google Scholar] [CrossRef]
- Liu, X.; Gong, Y. Isocitrate dehydrogenase inhibitors in acute myeloid leukemia. Biomark. Res. 2019, 7, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talati, C.; Sweet, K. Recently approved therapies in acute myeloid leukemia: A complex treatment landscape. Leuk. Res. 2018, 73, 58–66. [Google Scholar] [CrossRef]
- Bunse, L.; Pusch, S.; Bunse, T.; Sahm, F.; Sanghvi, K.; Friedrich, M.; Alansary, D.; Sonner, J.K.; Green, E.; Deumelandt, K.; et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 2018, 24, 1192–1203. [Google Scholar] [CrossRef] [PubMed]
- Kohanbash, G.; Carrera, D.A.; Shrivastav, S.; Ahn, B.J.; Jahan, N.; Mazor, T.; Chheda, Z.S.; Downey, K.M.; Watchmaker, P.B.; Beppler, C.; et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Investig. 2017, 127, 1425–1437. [Google Scholar] [CrossRef]
- Zhang, X.; Rao, A.; Sette, P.; Deibert, C.; Pomerantz, A.; Kim, W.J.; Kohanbash, G.; Chang, Y.; Park, Y.; Engh, J.; et al. IDH mutant gliomas escape natural killer cell immune surveillance by downregulation of NKG2D ligand expression. Neuro-Oncology 2016, 18, 1402–1412. [Google Scholar] [CrossRef]
- Fathi, A.T.; Nahed, B.V.; Wander, S.A.; Iafrate, A.J.; Borger, D.R.; Hu, R.; Thabet, A.; Cahill, D.P.; Perry, A.M.; Joseph, C.P.; et al. Elevation of Urinary 2-Hydroxyglutarate in IDH-Mutant Glioma. Oncologist 2016, 21, 214–219. [Google Scholar] [CrossRef] [Green Version]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1 -Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Böttcher, M.; Renner, K.; Berger, R.; Mentz, K.; Thomas, S.; Cardenas-Conejo, Z.E.; Dettmer, K.; Oefner, P.J.; Mackensen, A.; Kreutz, M.; et al. D-2-hydroxyglutarate interferes with HIF-1α stability skewing T-cell metabolism towards oxidative phosphorylation and impairing Th17 polarization. Oncoimmunology 2018, 7, e1445454. [Google Scholar] [CrossRef] [Green Version]
- Renault, T.T.; Chipuk, J.E. Getting away with murder: How does the BCL-2 family of proteins kill with immunity? Ann. N. Y. Acad. Sci. 2013, 1285, 59–79. [Google Scholar] [CrossRef] [Green Version]
- Bogenberger, J.M.; Kornblau, S.M.; Pierceall, W.E.; Lena, R.; Chow, D.; Shi, C.X.; Mantei, J.; Ahmann, G.; Gonzales, I.M.; Choudhary, A.; et al. BCL-2 family proteins as 5-Azacytidine-sensitizing targets and determinants of response in myeloid malignancies. Leukemia 2014, 28, 1657–1665. [Google Scholar] [CrossRef] [Green Version]
- Juárez-Salcedo, L.M.; Desai, V.; Dalia, S. Venetoclax: Evidence to date and clinical potential. Drugs Context 2019, 8, 212574. [Google Scholar] [CrossRef] [PubMed]
- Carrington, E.M.; Tarlinton, D.M.; Gray, D.H.; Huntington, N.D.; Zhan, Y.; Lew, A.M. The life and death of immune cell types: The role of BCL-2 anti-apoptotic molecules. Immunol. Cell Biol. 2017, 95, 870–877. [Google Scholar] [CrossRef]
- De Weerdt, I.; Hofland, T.; De Boer, R.; Dobber, J.A.; Dubois, J.; Van Nieuwenhuize, D.; Mobasher, M.; De Boer, F.; Hoogendoorn, M.; Velders, G.A.; et al. Distinct immune composition in lymph node and peripheral blood of CLL patients is reshaped during venetoclax treatment. Blood Adv. 2019, 3, 2642–2652. [Google Scholar] [CrossRef]
- Khaw, S.L.; Mérino, D.; Anderson, M.A.; Glaser, S.P.; Bouillet, P.; Roberts, A.W.; Huang, D.C. Both leukaemic and normal peripheral B lymphoid cells are highly sensitive to the selective pharmacological inhibition of prosurvival Bcl-2 with ABT-199. Leukemia 2014, 28, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Kohlhapp, F.J.; Haribhai, D.; Mathew, R.; Duggan, R.; Ellis, P.A.; Wang, R.; Lasater, E.A.; Shi, Y.; Dave, N.; Riehm, J.J.; et al. Venetoclax increases intratumoral effector t cells and antitumor efficacy in combination with immune checkpoint blockade. Cancer Discov. 2021, 11, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.B.; Khan, D.H.; Hurren, R.; Xu, M.; Na, Y.; Kang, H.; Mirali, S.; Wang, X.; Gronda, M.; Jitkova, Y.; et al. Venetoclax enhances T cell–mediated antileukemic activity by increasing ROS production. Blood 2021, 138, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Lainez-González, D.; Serrano-López, J.; Alonso-Domínguez, J.M. Understanding the hedgehog signaling pathway in acute myeloid leukemia stem cells: A necessary step toward a cure. Biology 2021, 10, 255. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Dombret, H.; Merchant, A.; Tauchi, T.; Dirienzo, C.G.; Sleight, B.; Zhang, X.; Leip, E.P.; Shaik, N.; Bell, T.; et al. Glasdegib plus intensive/nonintensive chemotherapy in untreated acute myeloid leukemia: BRIGHT AML 1019 Phase III trials. Futur. Oncol. 2019, 15, 3531–3545. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.A.; Harrison, C.N.; McLornan, D.P. Targeting of the Hedgehog pathway in myeloid malignancies: Still a worthy chase? Br. J. Haematol. 2015, 170, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Sadarangani, A.; Pineda, G.; Lennon, K.M.; Chun, H.J.; Shih, A.; Schairer, A.E.; Court, A.C.; Goff, D.J.; Prashad, S.L.; Geron, I.; et al. GLI2 inhibition abrogates human leukemia stem cell dormancy. J. Transl. Med. 2015, 13, 98. [Google Scholar] [CrossRef] [Green Version]
- Omland, S.H.; Nielsen, P.S.; Gjerdrum, L.M.; Gniadecki, R. Immunosuppressive environment in basal cell carcinoma: The role of regulatory T cells. Acta Derm. Venereol. 2016, 96, 917–921. [Google Scholar] [CrossRef] [Green Version]
- Hanna, A.; Metge, B.J.; Bailey, S.K.; Chen, D.; Chandrashekar, D.S.; Varambally, S.; Samant, R.S.; Shevde, L.A. Inhibition of Hedgehog signaling reprograms the dysfunctional immune microenvironment in breast cancer. Oncoimmunology 2019, 8, 1548241. [Google Scholar] [CrossRef]
- Papaioannou, E.; Yánez, D.C.; Ross, S.; Lau, C.I.; Solanki, A.; Chawda, M.M.; Virasami, A.; Ranz, I.; Ono, M.; O’Shaughnessy, R.F.; et al. Sonic Hedgehog signaling limits atopic dermatitis via Gli2-driven immune regulation. J. Clin. Investig. 2019, 129, 3153–3170. [Google Scholar] [CrossRef]
- Furmanski, A.L.; Saldana, J.I.; Ono, M.; Sahni, H.; Paschalidis, N.; D’Acquisto, F.; Crompton, T. Tissue-Derived Hedgehog Proteins Modulate Th Differentiation and Disease. J. Immunol. 2013, 190, 2641–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, J.; Holokai, L.; Syu, L.J.; Steele, N.G.; Chang, J.; Wang, J.; Ahmed, S.; Dlugosz, A.; Zavros, Y. Hedgehog signaling induces PD-L1 expression and tumor cell proliferation in gastric cancer. Oncotarget 2018, 9, 37439–37457. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.M.; Nirschl, C.J.; Polanczyk, M.J.; Bell, W.R.; Nirschl, T.R.; Harris-Bookman, S.; Phallen, J.; Hicks, J.; Martinez, D.; Ogurtsova, A.; et al. PD-L1 expression in medulloblastoma: An evaluation by subgroup. Oncotarget 2018, 9, 19177–19191. [Google Scholar] [CrossRef] [Green Version]
- Lipson, E.J.; Lilo, M.T.; Ogurtsova, A.; Esandrio, J.; Xu, H.; Brothers, P.; Schollenberger, M.; Sharfman, W.H.; Taube, J.M. Basal cell carcinoma: PD-L1/PD-1 checkpoint expression and tumor regression after PD-1 blockade. J. Immunother. Cancer 2017, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC class I downregulation in cancer: Underlying mechanisms and potential targets for cancer immunotherapy. Cancers 2020, 12, 1760. [Google Scholar] [CrossRef]
- Otsuka, A.; Dreier, J.; Cheng, P.F.; Nägeli, M.; Lehmann, H.; Felderer, L.; Frew, I.J.; Matsushita, S.; Levesque, M.P.; Dummer, R. Hedgehog pathway inhibitors promote adaptive immune responses in basal cell carcinoma. Clin. Cancer Res. 2015, 21, 1289–1297. [Google Scholar] [CrossRef] [Green Version]
- Feroz, W.; Sheikh, A.M. Exploring the multiple roles of guardian of the genome: P53. Egypt. J. Med. Hum. Genet. 2020, 21, 49. [Google Scholar] [CrossRef]
- Cumbo, C.; Tota, G.; Anelli, L.; Zagaria, A.; Specchia, G.; Albano, F. Molecular Sciences Review TP53 in Myelodysplastic Syndromes: Recent Biological and Clinical Findings. Int. J. Mol. Sci. 2020, 21, 3432. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lane, D.P. P53 in Health and Disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.A.; Chou, W.C.; Kuo, Y.Y.; Liu, C.Y.; Lin, L.I.; Tseng, M.H.; Chiang, Y.C.; Liu, M.C.; Liu, C.W.; Tang, J.L.; et al. TP53 mutations in de novo acute myeloid leukemia patients: Longitudinal follow-ups show the mutation is stable during disease evolution. Blood Cancer J. 2015, 5, e331. [Google Scholar] [CrossRef]
- Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012, 119, 2114–2121. [Google Scholar] [CrossRef]
- Seifert, H.; Mohr, B.; Thiede, C.; Oelschlägel, U.; Schäkel, U.; Illmer, T.; Soucek, S.; Ehninger, G.; Schaich, M. The prognostic impact of 17p (p53) deletion in 2272 adults with acute myeloid leukemia. Leukemia 2009, 23, 656–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Fontela, C.; Mandinova, A.; Aaronson, S.A.; Lee, S.W. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat. Rev. Immunol. 2016, 16, 741–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zheng, M.; Kibe, R.; Huang, Y.; Marrero, L.; Warren, S.; Zieske, A.W.; Iwakuma, T.; Kolls, J.K.; Cui, Y. Trp53 negatively regulates autoimmunity via the STAT3-Th17 axis. FASEB J. 2011, 25, 2387–2398. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.Y.; Zhong, W.Z.; Zhang, X.C.; Su, J.; Xie, Z.; Liu, S.Y.; Tu, H.Y.; Chen, H.J.; Sun, Y.L.; Zhou, Q.; et al. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef] [Green Version]
- Blagih, J.; Zani, F.; Chakravarty, P.; Hennequart, M.; Pilley, S.; Hobor, S.; Hock, A.K.; Walton, J.B.; Morton, J.P.; Gronroos, E.; et al. Cancer-Specific Loss of p53 Leads to a Modulation of Myeloid and T Cell Responses. Cell Rep. 2020, 30, 481–496.e6. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, W.; Simeone, I.; Anjum, S.; Mokrab, Y.; Bertucci, F.; Finetti, P.; Curigliano, G.; Seliger, B.; Cerulo, L.; Tomei, S.; et al. Identification of genetic determinants of breast cancer immune phenotypes by integrative genome-scale analysis. Oncoimmunology 2017, 6, e1253654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, Y.J.; Kim, H.R.; Lee, C.Y.; Cho, B.C.; Shim, H.S. Clinicopathological and prognostic significance of programmed cell death ligand-1 expression in lung adenocarcinoma and its relationship with p53 status. Lung Cancer 2016, 97, 73–80. [Google Scholar] [CrossRef]
- Wörmann, S.M.; Song, L.; Ai, J.; Diakopoulos, K.N.; Kurkowski, M.U.; Görgülü, K.; Ruess, D.; Campbell, A.; Doglioni, C.; Jodrell, D.; et al. Loss of P53 Function Activates JAK2–STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and Is Associated with Patient Survival. Gastroenterology 2016, 151, 180–193.e12. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, J.; Dong, K.; Lin, F.; Long, M.; Ouyang, Y.; Wei, J.; Chen, X.; Weng, Y.; He, T.; et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell. Signal. 2015, 27, 443–452. [Google Scholar] [CrossRef]
- Vadakekolathu, J.; Lai, C.; Reeder, S.; Church, S.E.; Hood, T.; Lourdusamy, A.; Rettig, M.P.; Aldoss, I.; Advani, A.S.; Godwin, J.; et al. TP53 abnormalities correlate with immune infiltration and associate with response to flotetuzumab immunotherapy in AML. Blood Adv. 2020, 4, 5011–5024. [Google Scholar] [CrossRef]
- Lehmann, S.; Bykov, V.J.; Ali, D.; Andreń, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef]
- Rangel, L.P.; Ferretti, G.D.; Costa, C.L.; Andrade, S.M.; Carvalho, R.S.; Costa, D.C.; Silva, J.L. P53 reactivation with induction of massive apoptosis-1 (PRIMA-1) inhibits amyloid aggregation of mutant p53 in cancer cells. J. Biol. Chem. 2019, 294, 3670–3682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molica, M.; Mazzone, C.; Niscola, P.; de Fabritiis, P. TP53 Mutations in Acute Myeloid Leukemia: Still a Daunting Challenge? Front. Oncol. 2021, 10, 3368. [Google Scholar] [CrossRef]
- Ghosh, A.; Michel, J.; Dong, L.; Suek, N.; Zhong, H.; Budhu, S.; de Henau, O.; Wolchok, J.; Merghoub, T. Abstract 4843: TP53-stabilization with APR-246 enhances antitumor effects of immune checkpoint blockade in preclinical models. In Proceedings of the Cancer Research, American Association for Cancer Research (AACR), Atlanta, GA, USA, 29 March–3 April 2019; Volume 79, p. 4843. [Google Scholar]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of RG7112: A small-molecule MDM2 inhibitor in clinical development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daver, N.G.; Garcia, J.S.; Jonas, B.A.; Kelly, K.R.; Assouline, S.; Brandwein, J.M.; Fenaux, P.; Olin, R.L.; Martinelli, G.; Paolini, S.; et al. Updated Results from the Venetoclax (Ven) in Combination with Idasanutlin (Idasa) Arm of a Phase 1b Trial in Elderly Patients (Pts) with Relapsed or Refractory (R/R) AML Ineligible for Cytotoxic Chemotherapy. Blood 2019, 134, 229. [Google Scholar] [CrossRef]
- Namgaladze, D.; Brüne, B. Pharmacological Activation of p53 during Human Monocyte to Macrophage Differentiation Attenuates Their Pro-Inflammatory Activation by TLR4, TLR7 and TLR8 Agonists. Cancers 2021, 13, 958. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Goyama, S.; Liu, X.X.; Tamura, M.; Asada, S.; Tanaka, Y.; Fukuyama, T.; Wunderlich, M.; O’Brien, E.; Mizukawa, B.; et al. Antitumor immunity augments the therapeutic effects of p53 activation on acute myeloid leukemia. Nat. Commun. 2019, 10, 4869. [Google Scholar] [CrossRef] [Green Version]
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Dudenhöffer-Pfeifer, M.; Bryder, D. Immunoediting is not a primary transformation event in a murine model of MLL-ENL AML. Life Sci. Alliance 2018, 1, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Guan, W.; Wang, M.; Chen, J.; Zhang, L.; Xiao, Y.; Wang, L.; Li, Y.; Yu, L. AML1-ETO inhibits acute myeloid leukemia immune escape by CD48. Leuk. Lymphoma 2021, 62, 937–943. [Google Scholar] [CrossRef]
- Bleakley, M.; Riddell, S.R. Molecules and mechanisms of the graft-versus-leukaemia effect. Nat. Rev. Cancer 2004, 4, 371–380. [Google Scholar] [CrossRef]
- Nikiforow, S.; Alyea, E.P. Maximizing GVL in allogeneic transplantation: Role of donor lymphocyte infusions. Hematology 2014, 2014, 570–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epperly, R.; Gottschalk, S.; Velasquez, M.P. A Bump in the Road: How the Hostile AML Microenvironment Affects CAR T Cell Therapy. Front. Oncol. 2020, 10, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Therapeutic Approach | Activity | Drug | Immunomodulation | References |
|---|---|---|---|---|
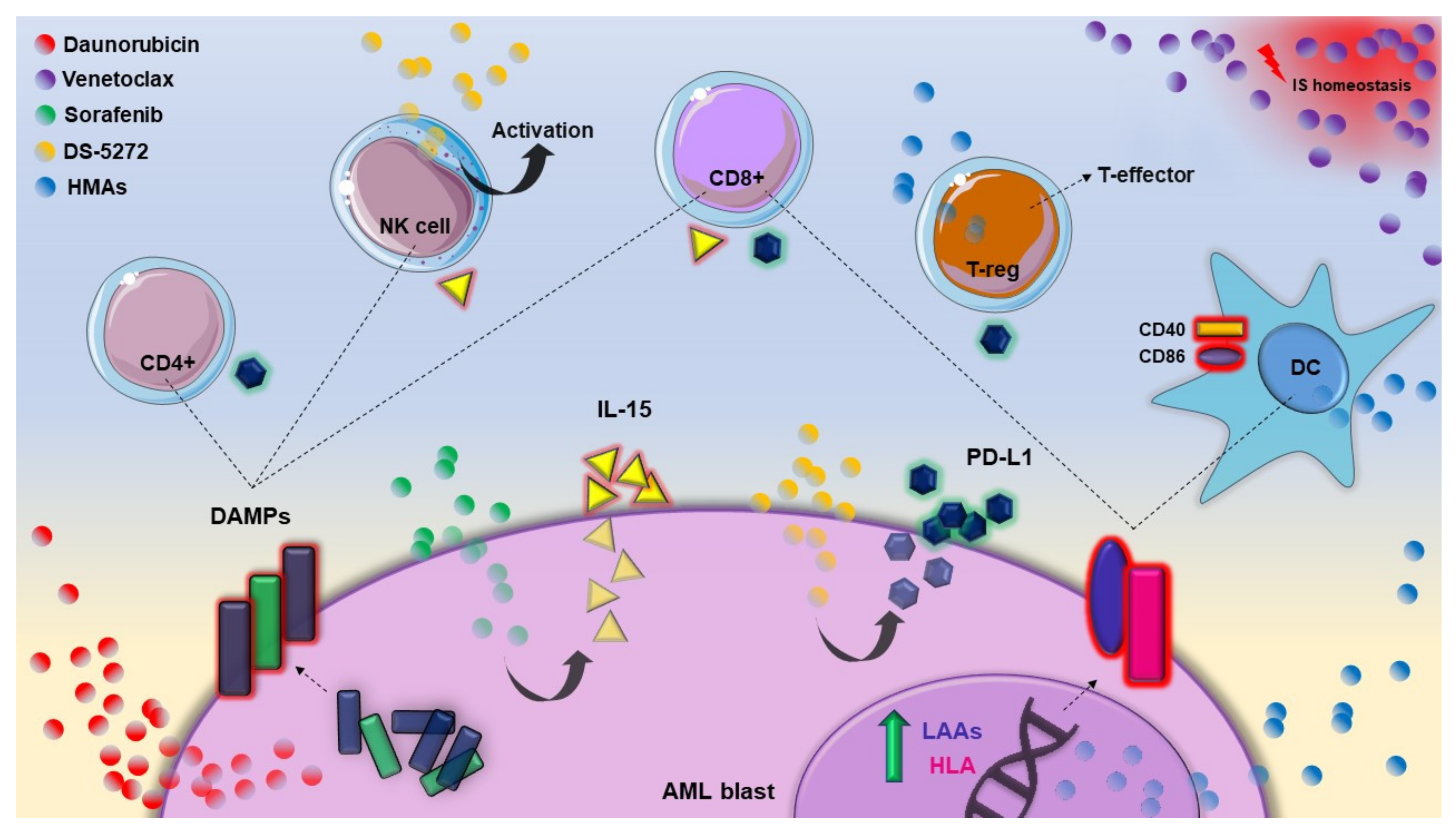
| Conventional chemotherapy | Cytotoxicity | Daunorubicin | IS mediated blast death, through DAMPs translocation on cell surface | [11,12] |
| Cytarabine | T cell reduction and DAMP expression alterations | [13] | ||
| FLT3 inhibitors | Kinase inhibition | Midostaurin | GvL effect reinforced | [14] |
| Sorafenib | CD8+CD279+ T lymphocytes increasing Proinflammatory, IS and angiogenic genes regulation CD8+ T lymphocytes and NK activation | [15,16,17] | ||
| Gilteritinib | NK and CD8+ T cells increased | [18] | ||
| Hypomethylating agents | Gene expression activation | Decitabine | DC activation (CD40 and CD86 overexpression) T cells inhibition (PD-1/PD-l1 axis activation) T helper 1, 2, 17 reduction T-reg shift to T-effector Increasing T cells recognition of CD34+ cells NK activation (NKG2DL and ADCC overexpression) | [19,20,21,22,23] |
| Azacytidine | [24,25,26,27] | |||
| IDH inhibitors | IDH inhibition | Ivosidenib | No available data in AML Target therapies could restore the IS equilibrium altered by R-2-HG accumulation (assumption) | [28,29] |
| Enasidenib | ||||
| BCL-2 inhibitors | BH3 mimetic | Venetoclax | Homeostasis impairment of B, T and myeloid immune cells | [30,31,32] |
| Hedgehog pathway inhibitors | Cell cycle re-entry | Glasdegib | No available data in AML MDSC recruitment, restored MHC-I expression, T cells increase (evidence from solid cancers) | [33,34,35,36,37] |
| p53 targeted therapies | Cell cycle arrest and apoptosis | APR-246 | No available data in AML Increased cytolytic activity of CD8+ T cells (evidence from solid cancers) | [38] |
| MDM2 inhibition | DS-5272 | NK activation and blast PD-L1 overexpression | [39] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarantini, F.; Cumbo, C.; Anelli, L.; Zagaria, A.; Specchia, G.; Musto, P.; Albano, F. Can the New and Old Drugs Exert an Immunomodulatory Effect in Acute Myeloid Leukemia? Cancers 2021, 13, 4121. https://doi.org/10.3390/cancers13164121
Tarantini F, Cumbo C, Anelli L, Zagaria A, Specchia G, Musto P, Albano F. Can the New and Old Drugs Exert an Immunomodulatory Effect in Acute Myeloid Leukemia? Cancers. 2021; 13(16):4121. https://doi.org/10.3390/cancers13164121
Chicago/Turabian StyleTarantini, Francesco, Cosimo Cumbo, Luisa Anelli, Antonella Zagaria, Giorgina Specchia, Pellegrino Musto, and Francesco Albano. 2021. "Can the New and Old Drugs Exert an Immunomodulatory Effect in Acute Myeloid Leukemia?" Cancers 13, no. 16: 4121. https://doi.org/10.3390/cancers13164121