
Short-Term and Long-Term Carcinogenic Effects of Food Contaminants (4-Hydroxynonenal and Pesticides) on Colorectal Human Cells: Involvement of Genotoxic and Non-Genomic Mechanisms

 , , and
, , and
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Chemicals and Supplements for Cell-Culture-Media
2.2. Antibodies
2.3. Cell Lines and Maintenance
2.4. Treatments
2.5. Viability Assay
2.6. Immunofluorescence
2.7. Comet Assay
2.8. Western Blot Analysis
2.9. Soft Agar Clonogenicity Assay
2.10. Seahorse
2.11. Statistical Analysis
3. Results
3.1. Acute Viability and DNA Damage of Contaminants in HCECs
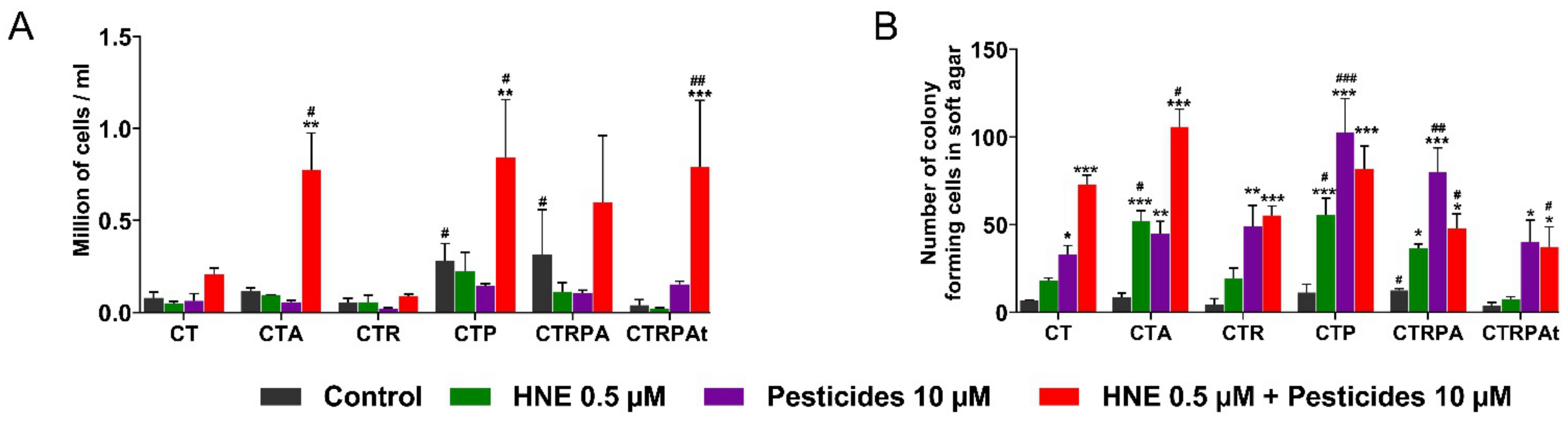
3.2. Cell Growth and Clonogenicity after a Long-Term Exposure to Contaminants in HCECs
3.3. Long-Term DNA Damage of Contaminants in HCECs
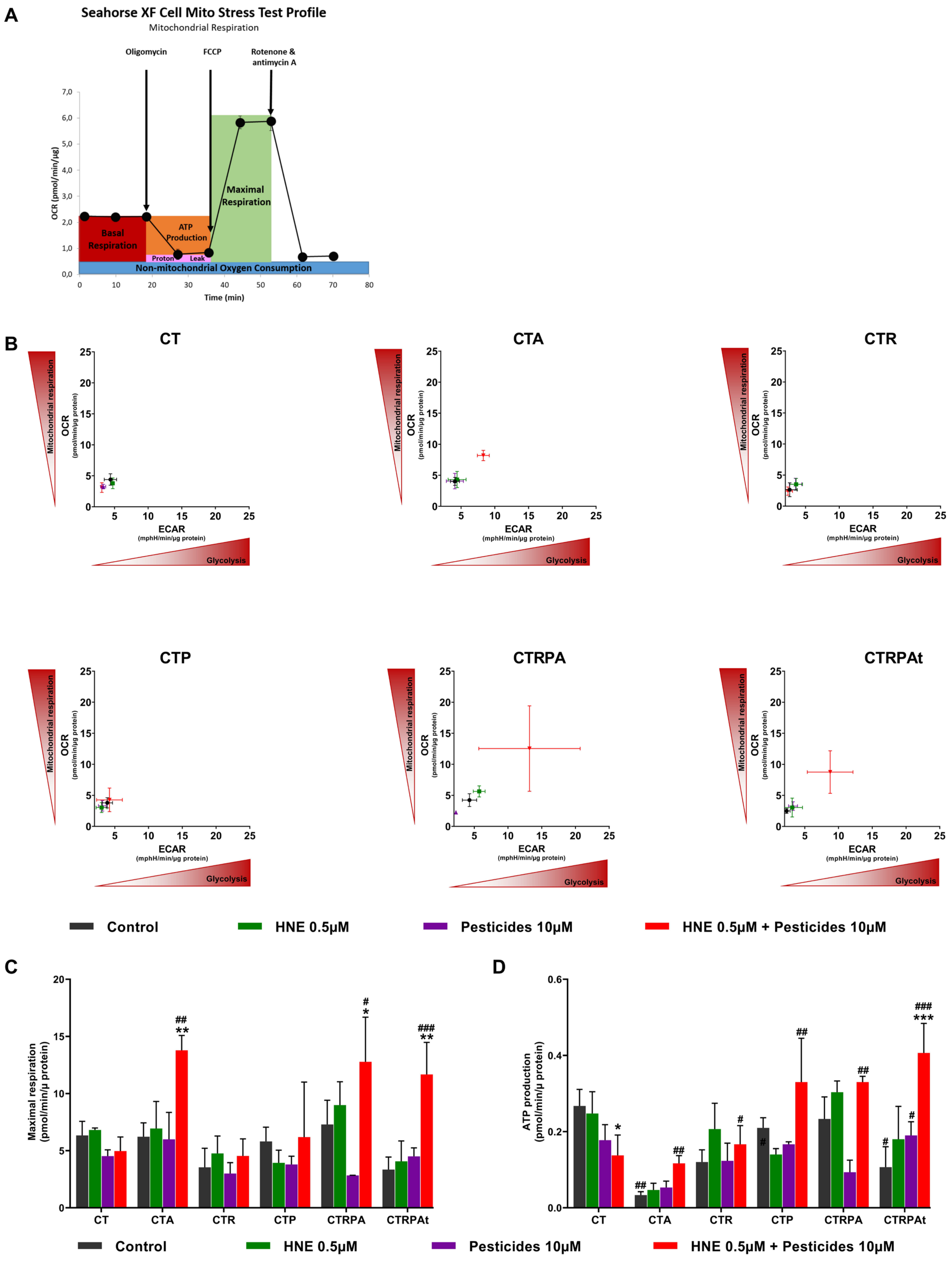
3.4. Metabolic Reprogramming after Long-Term Exposure to Contaminants in HCECs
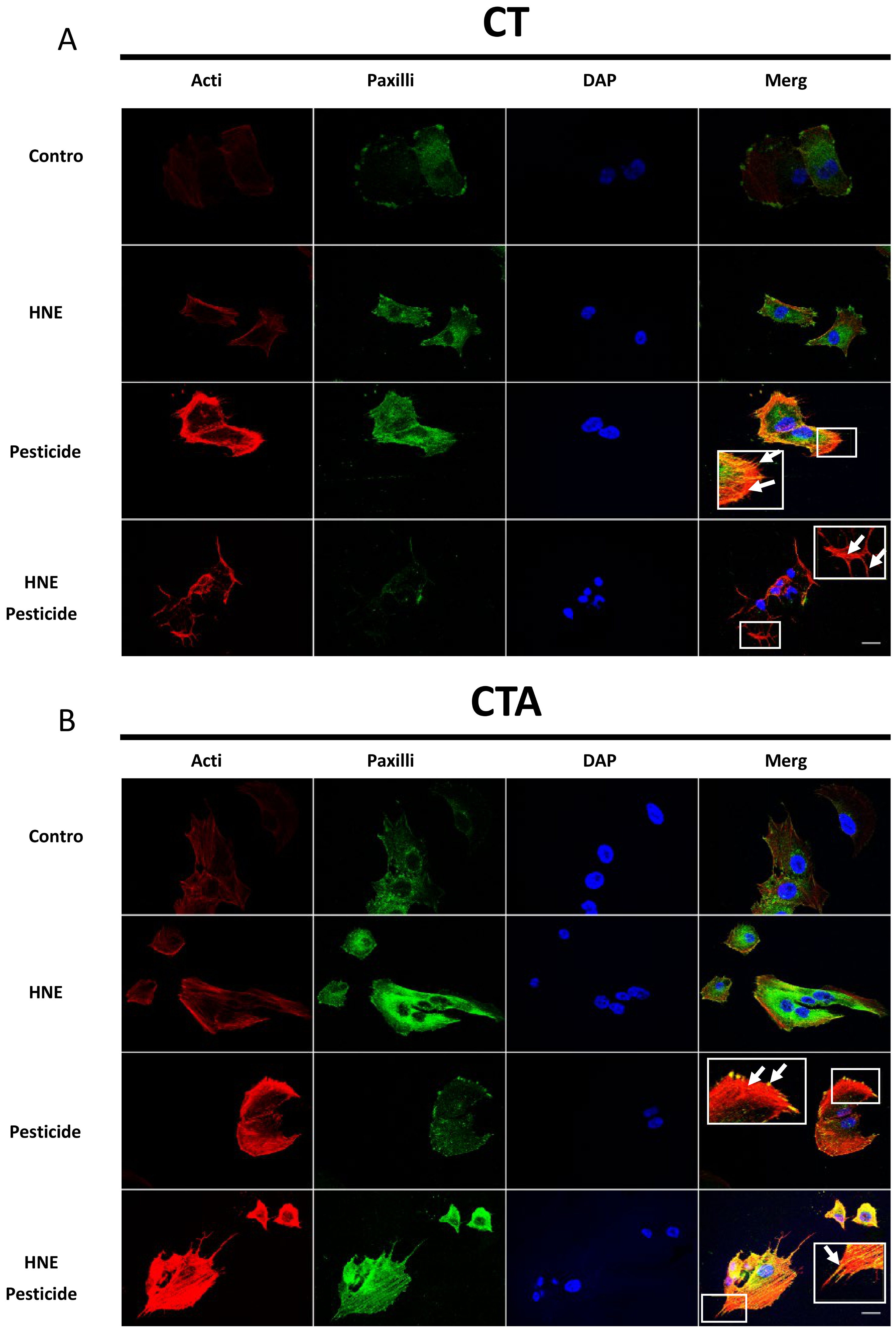
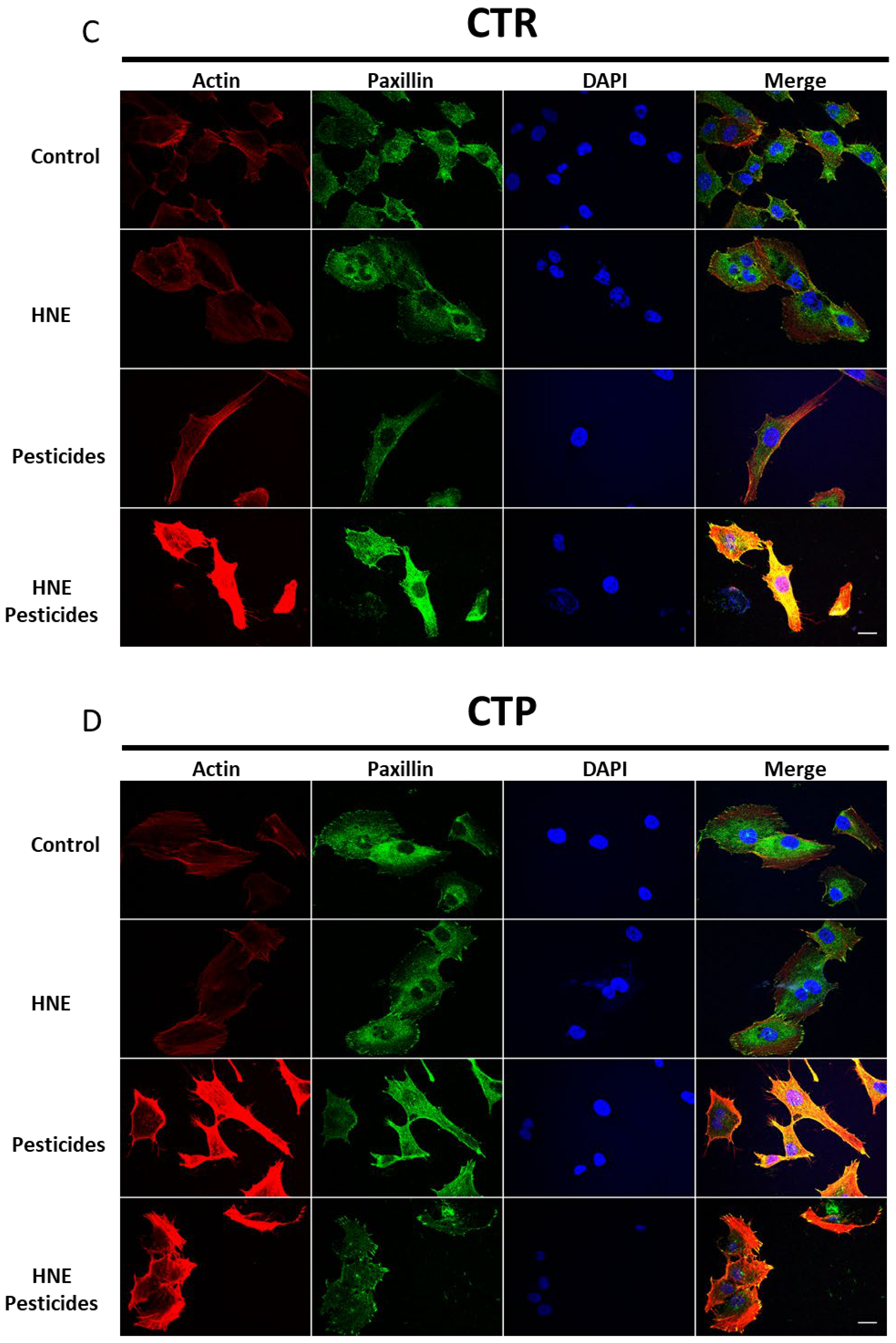
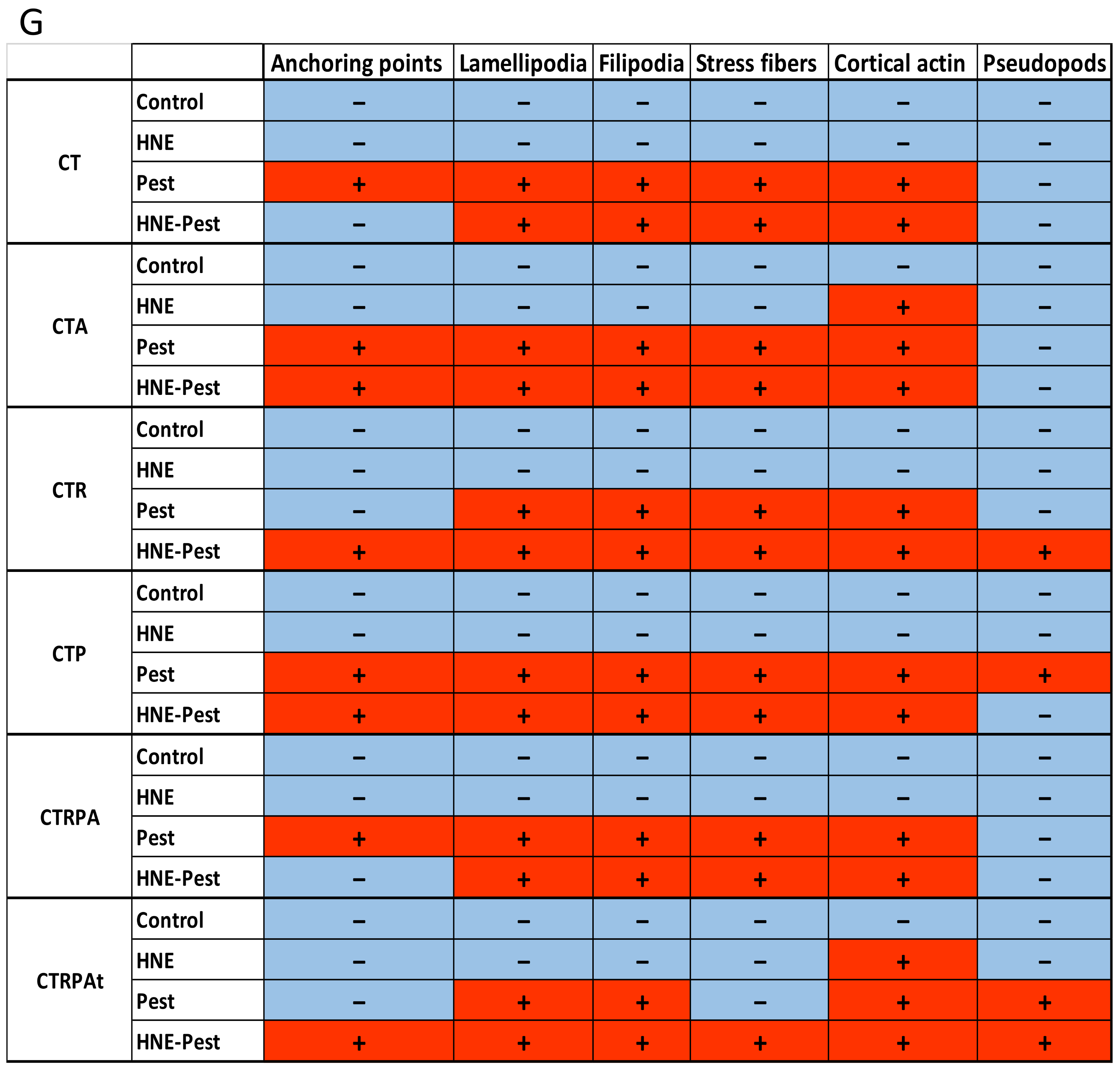
3.5. Cell Morphology after Long-Term Exposure to Contaminants in HCECs
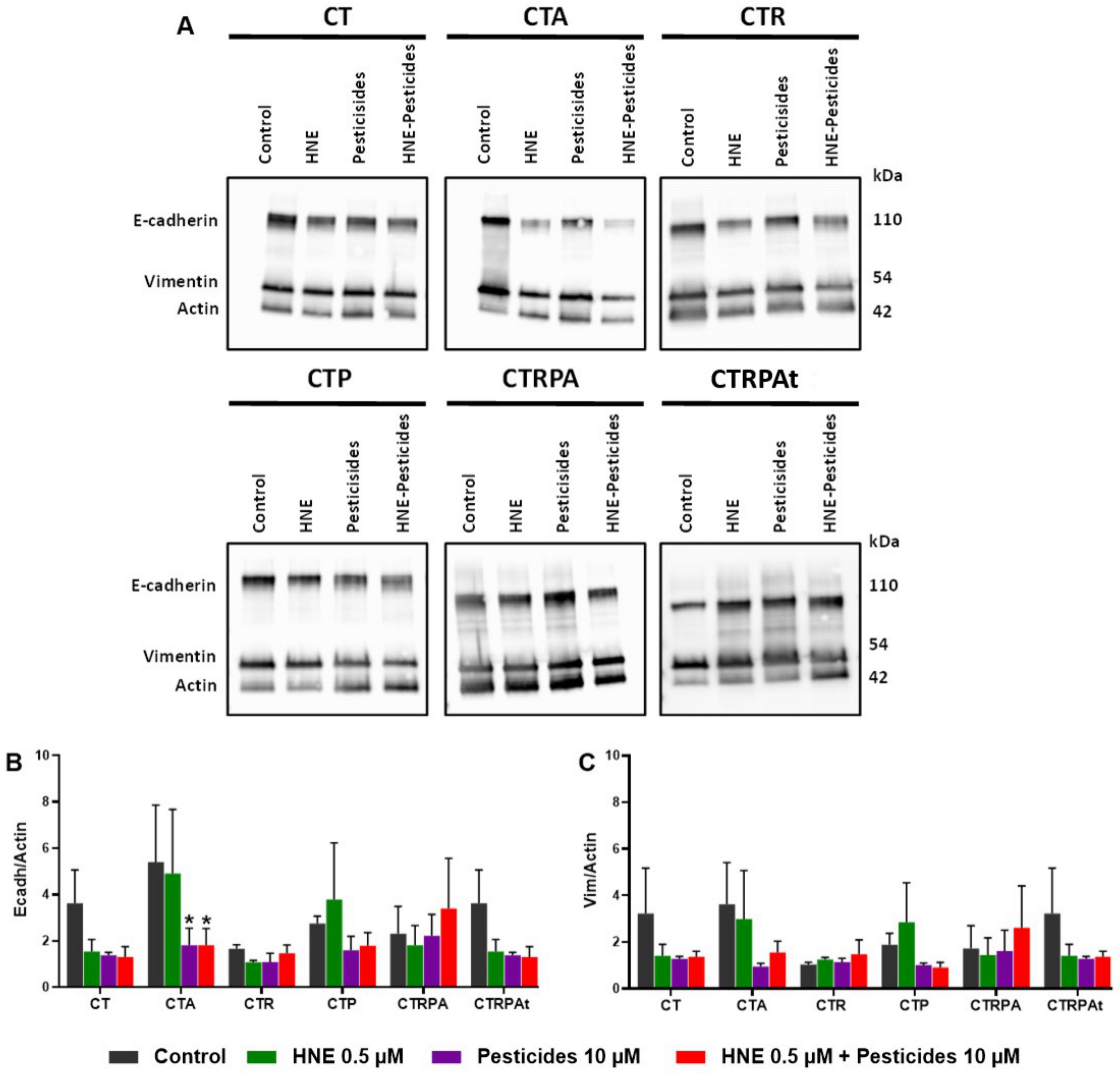
3.6. Epithelial-Mesenchymal Transition (EMT) after Long-Term Exposure to Contaminants in HCECs
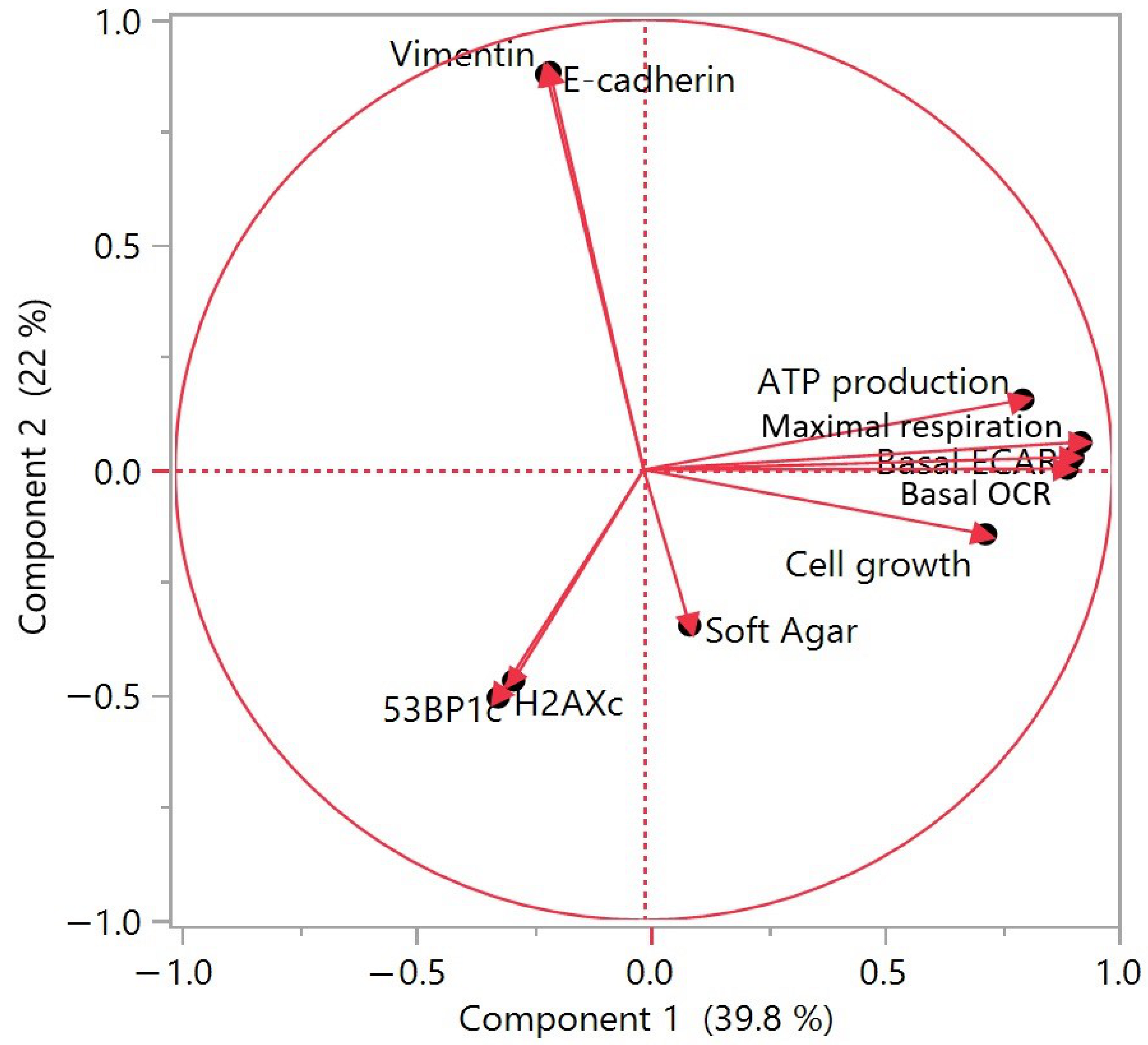
3.7. Principal Component Analysis of Long-Term Exposure Results to Contaminants in HCECs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Pancione, M.; Remo, A.; Colantuoni, V. Genetic and Epigenetic Events Generate Multiple Pathways in Colorectal Cancer Progression. Pathol. Res. Int. 2012, 2012, 509348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, G.; Carey, F.A.; Beattie, J.; Wilkie, M.J.V.; Lightfoot, T.J.; Coxhead, J.; Garner, R.C.; Steele, R.J.C.; Wolf, C.R. Mutations in APC, Kirsten-Ras, and P53—Alternative Genetic Pathways to Colorectal Cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 9433–9438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, H.; Chen, C. The Colorectal Cancer Epidemic: Challenges and Opportunities for Primary, Secondary and Tertiary Prevention. Br. J. Cancer 2018, 119, 785–792. [Google Scholar] [CrossRef] [Green Version]
- Murphy, N.; Moreno, V.; Hughes, D.J.; Vodicka, L.; Vodicka, P.; Aglago, E.K.; Gunter, M.J.; Jenab, M. Lifestyle and Dietary Environmental Factors in Colorectal Cancer Susceptibility. Mol. Asp. Med. 2019, 69, 2–9. [Google Scholar] [CrossRef]
- Parkin, D.M. 5. Cancers Attributable to Dietary Factors in the UK in 2010. II. Meat Consumption. Br. J. Cancer 2011, 105, S24–S26. [Google Scholar] [CrossRef]
- Feng, Y.-L.; Shu, L.; Zheng, P.-F.; Zhang, X.-Y.; Si, C.-J.; Yu, X.-L.; Gao, W.; Zhang, L. Dietary Patterns and Colorectal Cancer Risk: A Meta-Analysis. Eur. J. Cancer Prev. Off. J. Eur. Cancer Prev. Organ. ECP 2017, 26, 201–211. [Google Scholar] [CrossRef]
- Wiseman, M. The Second World Cancer Research Fund/American Institute for Cancer Research Expert Report. Food, Nutrition, Physical Activity, and the Prevention of Cancer: A Global Perspective. Proc. Nutr. Soc. 2008, 67, 253–256. [Google Scholar] [CrossRef] [Green Version]
- Dalleau, S.; Baradat, M.; Guéraud, F.; Huc, L. Cell Death and Diseases Related to Oxidative Stress: 4-Hydroxynonenal (HNE) in the Balance. Cell Death Differ. 2013, 20, 1615–1630. [Google Scholar] [CrossRef] [Green Version]
- Surya, R.; Héliès-Toussaint, C.; Martin, O.C.; Gauthier, T.; Guéraud, F.; Taché, S.; Naud, N.; Jouanin, I.; Chantelauze, C.; Durand, D.; et al. Red Meat and Colorectal Cancer: Nrf2-Dependent Antioxidant Response Contributes to the Resistance of Preneoplastic Colon Cells to Fecal Water of Hemoglobin- and Beef-Fed Rats. Carcinogenesis 2016, 37, 635–645. [Google Scholar] [CrossRef] [Green Version]
- Kortenkamp, A.; Faust, M. Regulate to Reduce Chemical Mixture Risk. Science 2018, 361, 224–226. [Google Scholar] [CrossRef] [Green Version]
- Crépet, A.; Héraud, F.; Béchaux, C.; Gouze, M.E.; Pierlot, S.; Fastier, A.; Leblanc, J.C.; Le Hégarat, L.; Takakura, N.; Fessard, V.; et al. The PERICLES Research Program: An Integrated Approach to Characterize the Combined Effects of Mixtures of Pesticide Residues to Which the French Population Is Exposed. Toxicology 2013, 313, 83–93. [Google Scholar] [CrossRef]
- Crépet, A.; Tressou, J.; Graillot, V.; Béchaux, C.; Pierlot, S.; Héraud, F.; Leblanc, J.C. Identification of the Main Pesticide Residue Mixtures to Which the French Population Is Exposed. Environ. Res. 2013, 126, 125–133. [Google Scholar] [CrossRef]
- Graillot, V.; Takakura, N.; Hegarat, L.L.; Fessard, V.; Audebert, M.; Cravedi, J.-P. Genotoxicity of Pesticide Mixtures Present in the Diet of the French Population. Environ. Mol. Mutagen. 2012, 53, 173–184. [Google Scholar] [CrossRef]
- Edlich, W.; Lyr, H.; Lyr, H. Target Sites of Fungicides with Primary Effects on Lipid Peroxidation. Available online: https://www.taylorfrancis.com/ (accessed on 7 August 2019).
- Radice, S.; Marabini, L.; Gervasoni, M.; Ferraris, M.; Chiesara, E. Adaptation to Oxidative Stress: Effects of Vinclozolin and Iprodione on the HepG2 Cell Line. Toxicology 1998, 129, 183–191. [Google Scholar] [CrossRef]
- Radice, S.; Ferraris, M.; Marabini, L.; Grande, S.; Chiesara, E. Effect of Iprodione, a Dicarboximide Fungicide, on Primary Cultured Rainbow Trout (Oncorhynchus Mykiss) Hepatocytes. Aquat. Toxicol. 2001, 54, 51–58. [Google Scholar] [CrossRef]
- Canal-Raffin, M.; l’Azou, B.; Jorly, J.; Hurtier, A.; Cambar, J.; Brochard, P. Cytotoxicity of Folpet Fungicide on Human Bronchial Epithelial Cells. Toxicology 2008, 249, 160–166. [Google Scholar] [CrossRef]
- Blystone, C.R.; Lambright, C.S.; Furr, J.; Wilson, V.S.; Gray, L.E. Iprodione Delays Male Rat Pubertal Development, Reduces Serum Testosterone Levels, and Decreases Ex Vivo Testicular Testosterone Production. Toxicol. Lett. 2007, 174, 74–81. [Google Scholar] [CrossRef]
- Ostby, J.; Kelce, W.R.; Lambright, C.; Wolf, C.J.; Mann, P.; Gray, L.E. The Fungicide Procymidone Alters Sexual Differentiation in the Male Rat by Acting as an Androgen-Receptor Antagonist in Vivo and in Vitro. Toxicol. Ind. Health 1999, 15, 80–93. [Google Scholar] [CrossRef]
- Go, R.-E.; Kim, C.-W.; Choi, K.-C. Effect of Fenhexamid and Cyprodinil on the Expression of Cell Cycle- and Metastasis-Related Genes via an Estrogen Receptor-Dependent Pathway in Cellular and Xenografted Ovarian Cancer Models. Toxicol. Appl. Pharmacol. 2015, 289, 48–57. [Google Scholar] [CrossRef]
- Huang, X.; Jin, Y.; Zhou, D.; Xu, G.; Huang, J.; Shen, L. IQGAP1 Modulates the Proliferation and Migration of Vascular Smooth Muscle Cells in Response to Estrogen. Int. J. Mol. Med. 2015, 35, 1460–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, C.-C.; Chen, F.-Y.; Chen, C.-R.; Liu, C.-C.; Wong, L.-C.; Liu, Y.-W.; Su, J.-G.J. Cyprodinil as an Activator of Aryl Hydrocarbon Receptor. Toxicology 2013, 304, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Medjakovic, S.; Zoechling, A.; Gerster, P.; Ivanova, M.M.; Teng, Y.; Klinge, C.M.; Schildberger, B.; Gartner, M.; Jungbauer, A. Effect of Nonpersistent Pesticides on Estrogen Receptor, Androgen Receptor, and Aryl Hydrocarbon Receptor. Environ. Toxicol. 2014, 29, 1201–1216. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.D.; O’Neil, J.D.; Woehrling, E.K.; Ndunge, O.B.A.; Hill, E.J.; Menache, A.; Reiss, C.J. A Preliminary Investigation into the Impact of a Pesticide Combination on Human Neuronal and Glial Cell Lines in Vitro. PLoS ONE 2012, 7, e42768. [Google Scholar] [CrossRef] [PubMed]
- Ankley, G.T.; Bencic, D.C.; Cavallin, J.E.; Jensen, K.M.; Kahl, M.D.; Makynen, E.A.; Martinović, D.; Mueller, N.D.; Wehmas, L.C.; Villeneuve, D.L. Dynamic Nature of Alterations in the Endocrine System of Fathead Minnows Exposed to the Fungicide Prochloraz. Toxicol. Sci. 2009, 112, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, T.; Tsutsumi, O.; Ikezuki, Y.; Takai, Y.; Taketani, Y. Positive Relationship between Androgen and the Endocrine Disruptor, Bisphenol A, in Normal Women and Women with Ovarian Dysfunction. Endocr. J. 2004, 51, 165–169. [Google Scholar] [CrossRef] [Green Version]
- Viel, J.-F.; Warembourg, C.; Le Maner-Idrissi, G.; Lacroix, A.; Limon, G.; Rouget, F.; Monfort, C.; Durand, G.; Cordier, S.; Chevrier, C. Pyrethroid Insecticide Exposure and Cognitive Developmental Disabilities in Children: The PELAGIE Mother–Child Cohort. Environ. Int. 2015, 82, 69–75. [Google Scholar] [CrossRef]
- Lukowicz, C.; Ellero-Simatos, S.; Régnier, M.; Polizzi, A.; Lasserre, F.; Montagner, A.; Lippi, Y.; Jamin, E.L.; Martin, J.-F.; Naylies, C.; et al. Metabolic Effects of a Chronic Dietary Exposure to a Low-Dose Pesticide Cocktail in Mice: Sexual Dimorphism and Role of the Constitutive Androstane Receptor. Environ. Health Perspect. 2018, 126, 067007. [Google Scholar] [CrossRef]
- Smith, L.; Klément, W.; Dopavogui, L.; de Bock, F.; Lasserre, F.; Barretto, S.; Lukowicz, C.; Fougerat, A.; Polizzi, A.; Schaal, B.; et al. Perinatal Exposure to a Dietary Pesticide Cocktail Does Not Increase Susceptibility to High-Fat Diet-Induced Metabolic Perturbations at Adulthood but Modifies Urinary and Fecal Metabolic Fingerprints in C57Bl6/J Mice. Environ. Int. 2020, 144, 106010. [Google Scholar] [CrossRef]
- Vodicka, P.; Vodenkova, S.; Buchler, T.; Vodickova, L. DNA Repair Capacity and Response to Treatment of Colon Cancer. Pharmacogenomics 2019, 20, 1225–1233. [Google Scholar] [CrossRef]
- Roig, A.I.; Eskiocak, U.; Hight, S.K.; Kim, S.B.; Delgado, O.; Souza, R.F.; Spechler, S.J.; Wright, W.E.; Shay, J.W. Immortalized Epithelial Cells Derived From Human Colon Biopsies Express Stem Cell Markers and Differentiate in Vitro. Gastroenterology 2010, 138, 1012–1021.e5. [Google Scholar] [CrossRef]
- Graillot, V.; Dormoy, I.; Dupuy, J.; Shay, J.W.; Huc, L.; Mirey, G.; Vignard, J. Genotoxicity of Cytolethal Distending Toxin (CDT) on Isogenic Human Colorectal Cell Lines: Potential Promoting Effects for Colorectal Carcinogenesis. Front. Cell. Infect. Microbiol. 2016, 6, 34. [Google Scholar] [CrossRef]
- Perdry, H.; Gutzkow, K.B.; Chevalier, M.; Huc, L.; Brunborg, G.; Boutet-Robinet, E. Validation of Gelbond® High-Throughput Alkaline and Fpg-Modified Comet Assay Using a Linear Mixed Model. Environ. Mol. Mutagen. 2018, 59, 595–602. [Google Scholar] [CrossRef]
- Collins, A.R. Measuring Oxidative Damage to DNA and Its Repair with the Comet Assay. Biochim. Biophys. Acta 2014, 1840, 794–800. [Google Scholar] [CrossRef]
- European Standards Committee on Oxidative DNA Damage (ESCODD) Measurement of DNA Oxidation in Human Cells by Chromatographic and Enzymic Methods. Free Radic. Biol. Med. 2003, 34, 1089–1099. [CrossRef]
- Gedik, C.M.; Collins, A.; ESCODD (European Standards Committee on Oxidative DNA Damage). Establishing the Background Level of Base Oxidation in Human Lymphocyte DNA: Results of an Interlaboratory Validation Study. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 82–84. [Google Scholar] [CrossRef]
- Fernandez-Vidal, A.; Vignard, J.; Mirey, G. Around and beyond 53BP1 Nuclear Bodies. Int. J. Mol. Sci. 2017, 18, 2611. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Hunt, C.R.; Chakraborty, S.; Pandita, R.K.; Yordy, J.; Ramnarain, D.B.; Horikoshi, N.; Pandita, T.K. Role of 53BP1 in the Regulation of DNA Double-Strand Break Repair Pathway Choice. Radiat. Res. 2014, 181, 1–8. [Google Scholar] [CrossRef] [Green Version]
- De Feraudy, S.; Revet, I.; Bezrookove, V.; Feeney, L.; Cleaver, J.E. A Minority of Foci or Pan-Nuclear Apoptotic Staining of GammaH2AX in the S Phase after UV Damage Contain DNA Double-Strand Breaks. Proc. Natl. Acad. Sci. USA 2010, 107, 6870–6875. [Google Scholar] [CrossRef] [Green Version]
- Vignard, J.; Mirey, G.; Salles, B. Ionizing-Radiation Induced DNA Double-Strand Breaks: A Direct and Indirect Lighting Up. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2013, 108, 362–369. [Google Scholar] [CrossRef] [Green Version]
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem Cell Divisions, Somatic Mutations, Cancer Etiology, and Cancer Prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef] [Green Version]
- Tomasetti, C.; Vogelstein, B. Cancer Etiology. Variation in Cancer Risk among Tissues Can Be Explained by the Number of Stem Cell Divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Powers, S.; Zhu, W.; Hannun, Y.A. Substantial Contribution of Extrinsic Risk Factors to Cancer Development. Nature 2016, 529, 43–47. [Google Scholar] [CrossRef]
- Plutynski, A. Is Cancer a Matter of Luck? Biol. Philos. 2021, 36, 3. [Google Scholar] [CrossRef]
- Bastide, N.M.; Chenni, F.; Audebert, M.; Santarelli, R.L.; Taché, S.; Naud, N.; Baradat, M.; Jouanin, I.; Surya, R.; Hobbs, D.A.; et al. A Central Role for Heme Iron in Colon Carcinogenesis Associated with Red Meat Intake. Cancer Res. 2015, 75, 870–879. [Google Scholar] [CrossRef] [Green Version]
- Martin, O.; Lin, C.; Naud, N.; Tache, S.; Raymond-Letron, I.; Corpet, D.; Pierre, F. Antibiotic Suppression of Intestinal Microbiota Reduces Heme-Induced Lipoperoxidation Associated with Colon Carcinogenesis in Rats. Nutr. Cancer 2014, 67, 119–125. [Google Scholar] [CrossRef]
- Martin, O.C.B.; Olier, M.; Ellero-Simatos, S.; Naud, N.; Dupuy, J.; Huc, L.; Taché, S.; Graillot, V.; Levêque, M.; Bézirard, V.; et al. Haem Iron Reshapes Colonic Luminal Environment: Impact on Mucosal Homeostasis and Microbiome through Aldehyde Formation. Microbiome 2019, 7, 72. [Google Scholar] [CrossRef] [Green Version]
- Karlhuber, G.M.; Bauer, H.C.; Eckl, P.M. Cytotoxic and Genotoxic Effects of 4-Hydroxynonenal in Cerebral Endothelial Cells. Mutat Res. 1997, 381, 209–216. [Google Scholar] [CrossRef]
- Boutet-Robinet, E.; Bortoli, S.; Huc, L. DNA Damage Response upon Environmental Contaminants: An Exhausting Work for Genomic Integrity. Curr. Opin. Toxicol. 2018, 8, 28–33. [Google Scholar] [CrossRef] [Green Version]
- Vodicka, P.; Urbanova, M.; Makovicky, P.; Tomasova, K.; Kroupa, M.; Stetina, R.; Opattova, A.; Kostovcikova, K.; Siskova, A.; Schneiderova, M.; et al. Oxidative Damage in Sporadic Colorectal Cancer: Molecular Mapping of Base Excision Repair Glycosylases in Colorectal Cancer Patients. Int. J. Mol. Sci. 2020, 21, 2473. [Google Scholar] [CrossRef] [Green Version]
- De Azevedo Mello, F.; Magalhaes Silva, B.B.; Barreiro, E.B.V.; Franco, I.B.; Nogueira, I.M.; Nahas Chagas, P.H.; Santos Parizi, J.L.; Pereira, D.R.; Rossi, R.C.; Nai, G.A. Evaluation of Genotoxicity after Acute and Chronic Exposure to 2,4-Dichlorophenoxyacetic Acid Herbicide (2,4-D) in Rodents Using Machine Learning Algorithms. J. Toxicol. Sci. 2020, 45, 737–750. [Google Scholar] [CrossRef]
- Kaur, M.; Sandhir, R. Comparative Effects of Acute and Chronic Carbofuran Exposure on Oxidative Stress and Drug-Metabolizing Enzymes in Liver. Drug Chem. Toxicol. 2006, 29, 415–421. [Google Scholar] [CrossRef]
- Barretto, S.A.; Lasserre, F.; Huillet, M.; Régnier, M.; Polizzi, A.; Lippi, Y.; Fougerat, A.; Person, E.; Bruel, S.; Bétoulières, C.; et al. The Pregnane X Receptor Drives Sexually Dimorphic Hepatic Changes in Lipid and Xenobiotic Metabolism in Response to Gut Microbiota in Mice. Microbiome 2021, 9, 93. [Google Scholar] [CrossRef]
- Spitsbergen, J.M.; Tsai, H.W.; Reddy, A.; Miller, T.; Arbogast, D.; Hendricks, J.D.; Bailey, G.S. Neoplasia in Zebrafish (Danio Rerio) Treated with 7,12-Dimethylbenz[a]Anthracene by Two Exposure Routes at Different Developmental Stages. Toxicol. Pathol. 2000, 28, 705–715. [Google Scholar] [CrossRef]
- Bermejo, J.L.; Hemminki, K. Gene-Environment Studies: Any Advantage over Environmental Studies? Carcinogenesis 2007, 28, 1526–1532. [Google Scholar] [CrossRef] [Green Version]
- Gamage, S.M.K.; Lee, K.T.W.; Dissabandara, D.L.O.; Lam, A.K.-Y.; Gopalan, V. Dual Role of Heme Iron in Cancer; Promotor of Carcinogenesis and an Inducer of Tumour Suppression. Exp. Mol. Pathol. 2021, 120, 104642. [Google Scholar] [CrossRef]
- Zárate, L.V.; Pontillo, C.A.; Español, A.; Miret, N.V.; Chiappini, F.; Cocca, C.; Álvarez, L.; de Pisarev, D.K.; Sales, M.E.; Randi, A.S. Angiogenesis Signaling in Breast Cancer Models Is Induced by Hexachlorobenzene and Chlorpyrifos, Pesticide Ligands of the Aryl Hydrocarbon Receptor. Toxicol. Appl. Pharmacol. 2020, 401, 115093. [Google Scholar] [CrossRef]
- Li, X.-L.; Zhou, J.; Chen, Z.-R.; Chng, W.-J. P53 Mutations in Colorectal Cancer- Molecular Pathogenesis and Pharmacological Reactivation. World J. Gastroenterol. WJG 2015, 21, 84–93. [Google Scholar] [CrossRef]
- Liebl, M.C.; Hofmann, T.G. The Role of P53 Signaling in Colorectal Cancer. Cancers 2021, 13, 2125. [Google Scholar] [CrossRef]
- Cha, P.-H.; Hwang, J.-H.; Kwak, D.-K.; Koh, E.; Kim, K.-S.; Choi, K.-Y. APC Loss Induces Warburg Effect via Increased PKM2 Transcription in Colorectal Cancer. Br. J. Cancer 2021, 124, 634–644. [Google Scholar] [CrossRef]
- Lee, S.Y.; Jeon, H.M.; Ju, M.K.; Kim, C.H.; Yoon, G.; Han, S.I.; Park, H.G.; Kang, H.S. Wnt/Snail Signaling Regulates Cytochrome C Oxidase and Glucose Metabolism. Cancer Res. 2012, 72, 3607–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melnik, S.; Dvornikov, D.; Müller-Decker, K.; Depner, S.; Stannek, P.; Meister, M.; Warth, A.; Thomas, M.; Muley, T.; Risch, A.; et al. Cancer Cell Specific Inhibition of Wnt/β-Catenin Signaling by Forced Intracellular Acidification. Cell Discov. 2018, 4, 37. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.R.; Wu, R.; Kardia, S.L.R.; Levin, A.M.; Huang, C.-C.; Shedden, K.A.; Kuick, R.; Misek, D.E.; Hanash, S.M.; Taylor, J.M.G.; et al. Novel Candidate Targets of Beta-Catenin/T-Cell Factor Signaling Identified by Gene Expression Profiling of Ovarian Endometrioid Adenocarcinomas. Cancer Res. 2003, 63, 2913–2922. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pesticide | Family | Chemical Family | Proportion | Final Concentration |
|---|---|---|---|---|
| Procymidone | Fungicide | Dicarboximide | 42% | 4.2 µM |
| Iprodione | Fungicide | Dicarboximide | 33% | 3.3 µM |
| Cyprodinil | Fungicide | Pyrimidiamine | 15% | 1.5 µM |
| Fludioxonil | Fungicide | Phenylpyrrole | 9% | 0.9 µM |
| Lambda-Cyhalothrin | Insecticide | Synthetic Pyrethroids | 1% | 0.1 µM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnaud, L.C.; Gauthier, T.; Le Naour, A.; Hashim, S.; Naud, N.; Shay, J.W.; Pierre, F.H.; Boutet-Robinet, E.; Huc, L. Short-Term and Long-Term Carcinogenic Effects of Food Contaminants (4-Hydroxynonenal and Pesticides) on Colorectal Human Cells: Involvement of Genotoxic and Non-Genomic Mechanisms. Cancers 2021, 13, 4337. https://doi.org/10.3390/cancers13174337
Arnaud LC, Gauthier T, Le Naour A, Hashim S, Naud N, Shay JW, Pierre FH, Boutet-Robinet E, Huc L. Short-Term and Long-Term Carcinogenic Effects of Food Contaminants (4-Hydroxynonenal and Pesticides) on Colorectal Human Cells: Involvement of Genotoxic and Non-Genomic Mechanisms. Cancers. 2021; 13(17):4337. https://doi.org/10.3390/cancers13174337
Chicago/Turabian StyleArnaud, Liana C., Thierry Gauthier, Augustin Le Naour, Saleha Hashim, Nathalie Naud, Jerry W. Shay, Fabrice H. Pierre, Elisa Boutet-Robinet, and Laurence Huc. 2021. "Short-Term and Long-Term Carcinogenic Effects of Food Contaminants (4-Hydroxynonenal and Pesticides) on Colorectal Human Cells: Involvement of Genotoxic and Non-Genomic Mechanisms" Cancers 13, no. 17: 4337. https://doi.org/10.3390/cancers13174337