
The Multiple Faces of MNT and Its Role as a MYC Modulator



Abstract
:Simple Summary
Abstract
1. Introduction
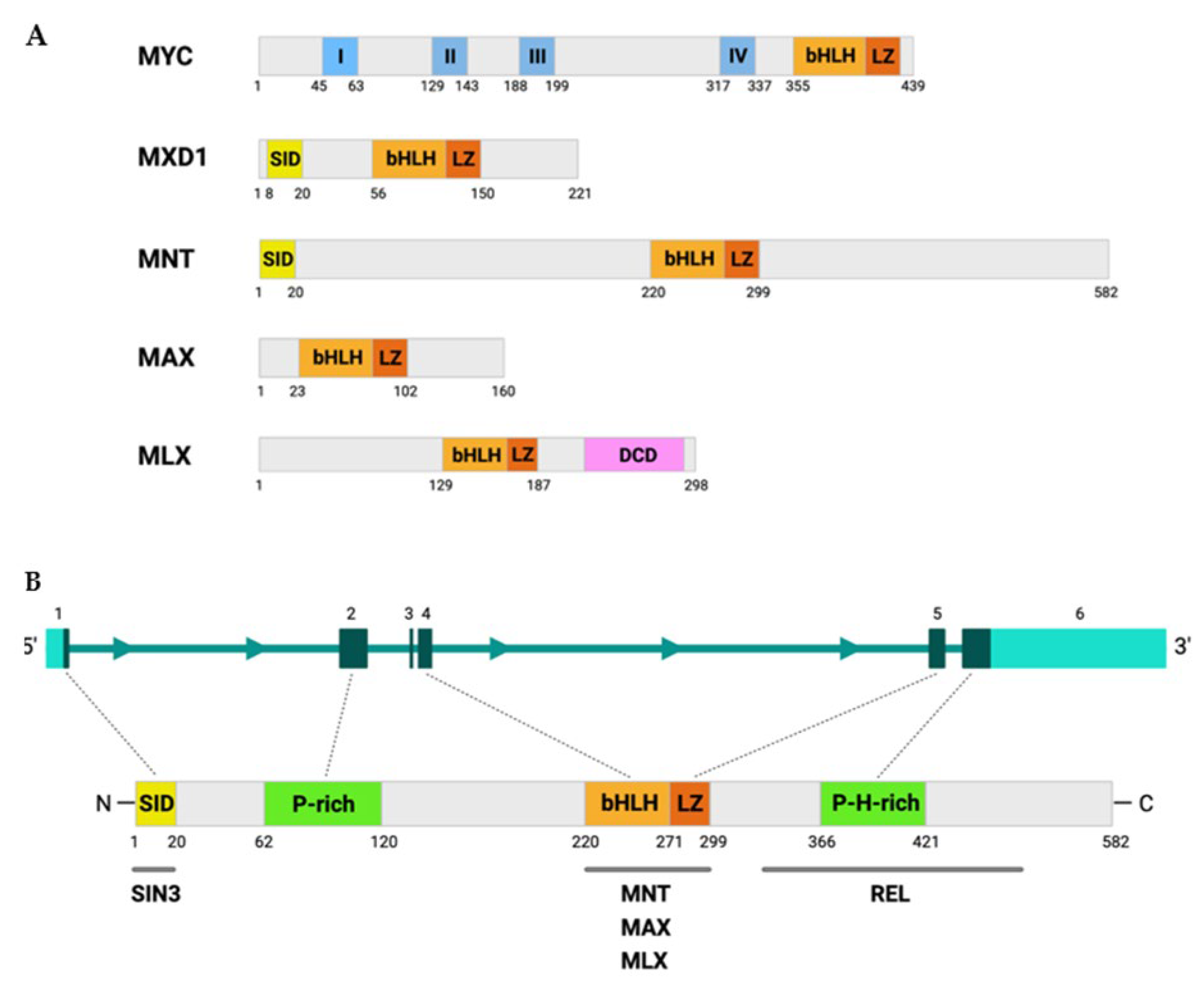
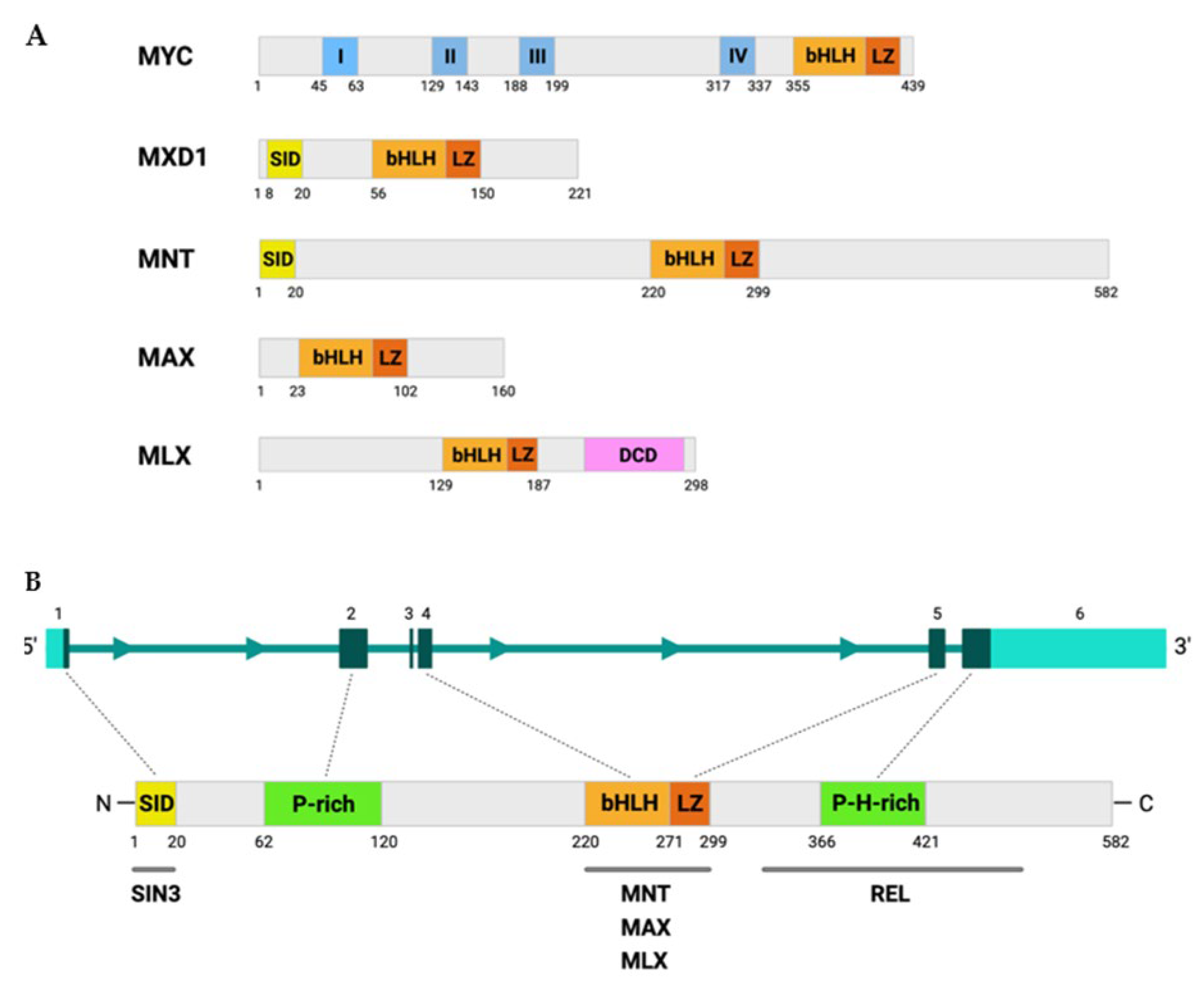
2. MNT Structure and Regulation
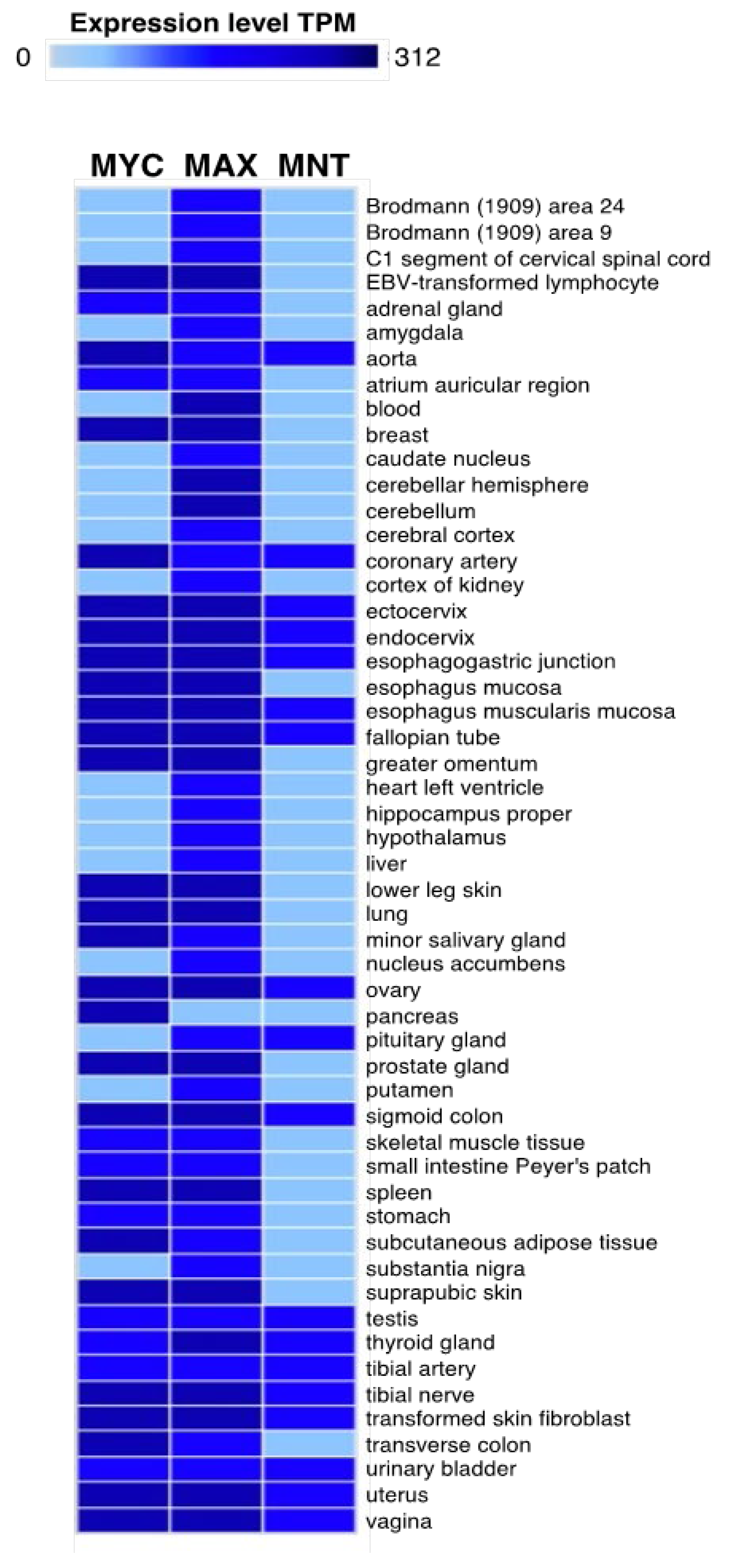
3. MNT Expression
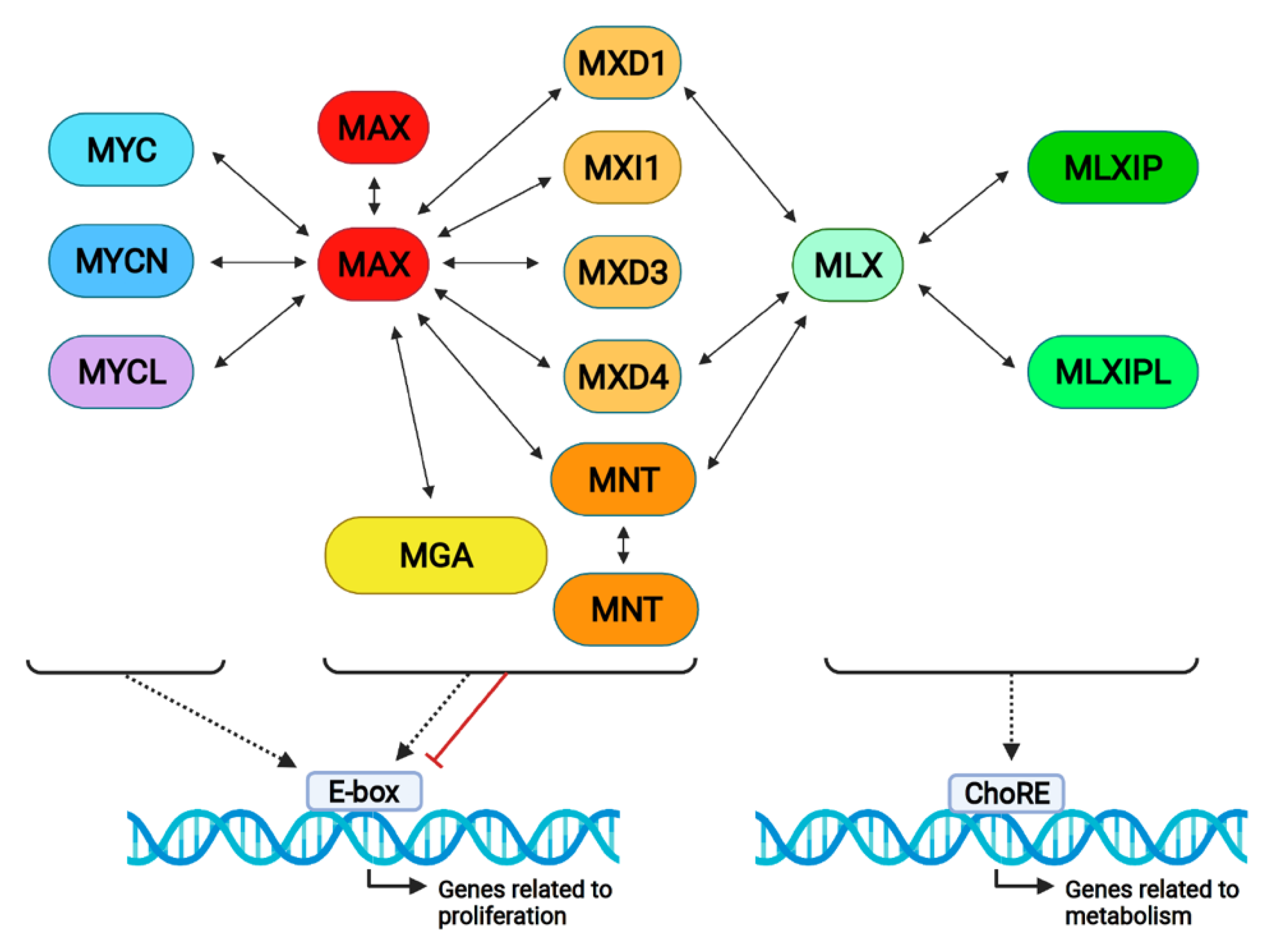
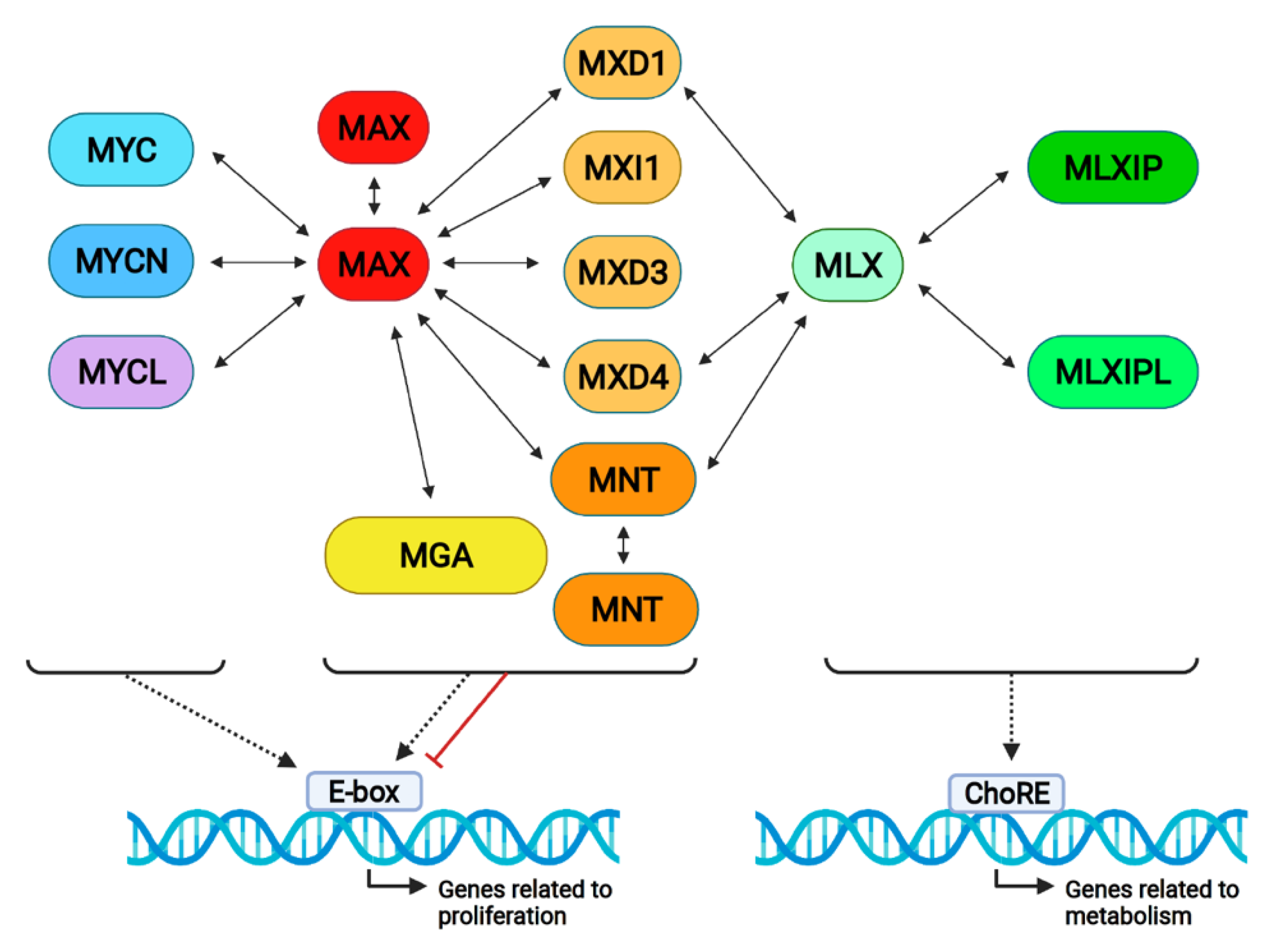
4. MNT Interactions in the Proximal MYC Network
5. MNT Interactions Outside the Proximal MYC Network
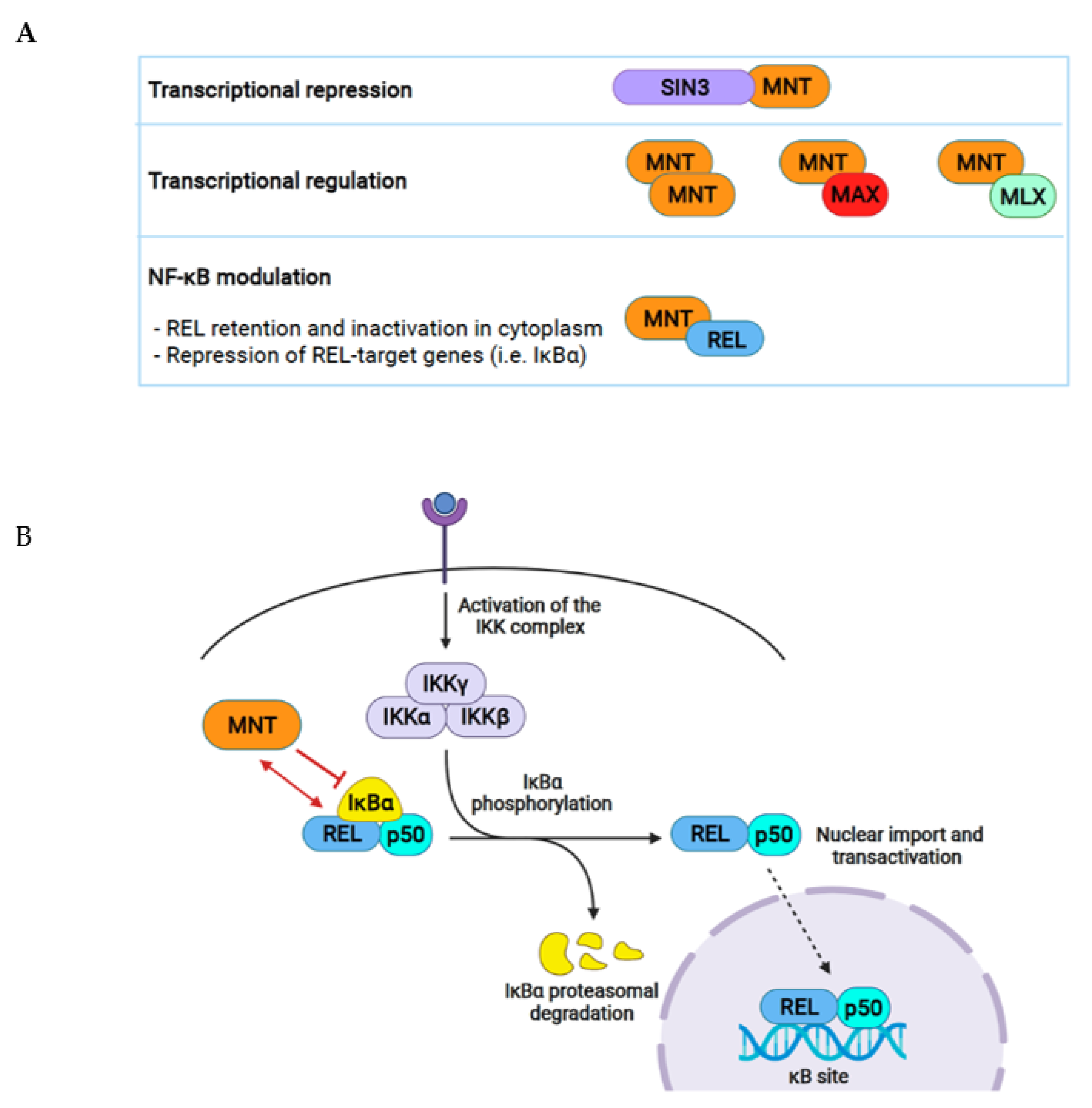
5.1. SIN3 Proteins
5.2. REL
5.3. MATα-1
5.4. Other Proteins
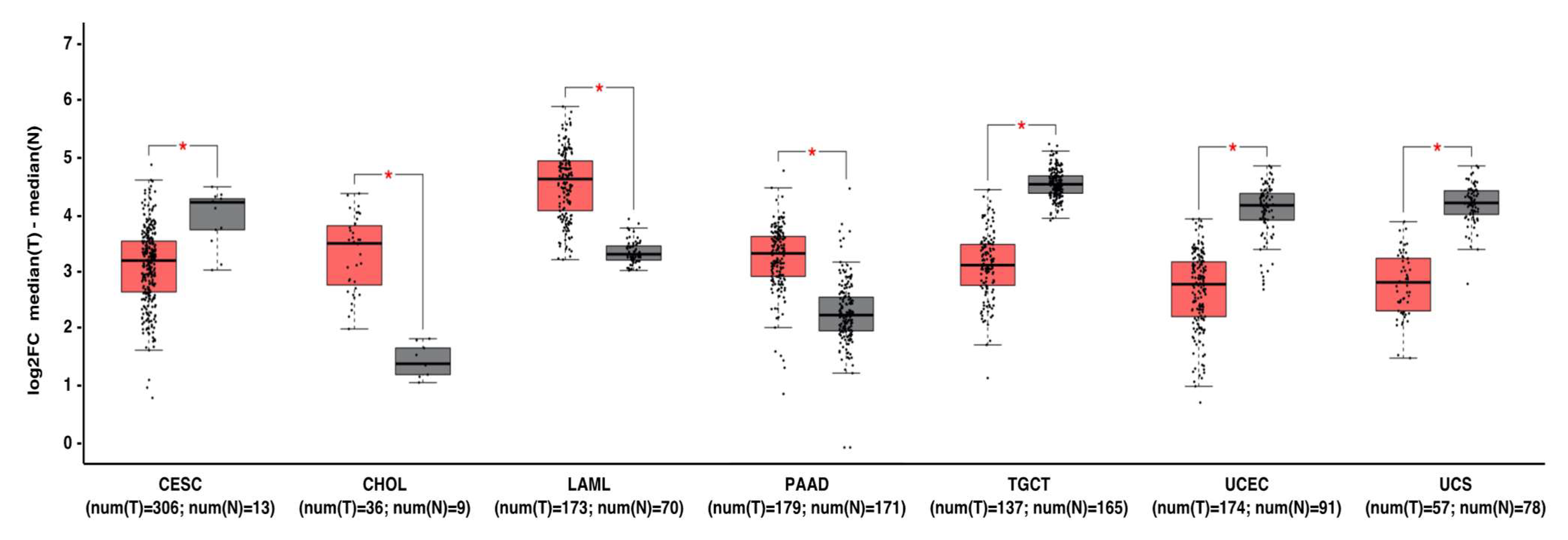
6. MNT Alterations in Cancer
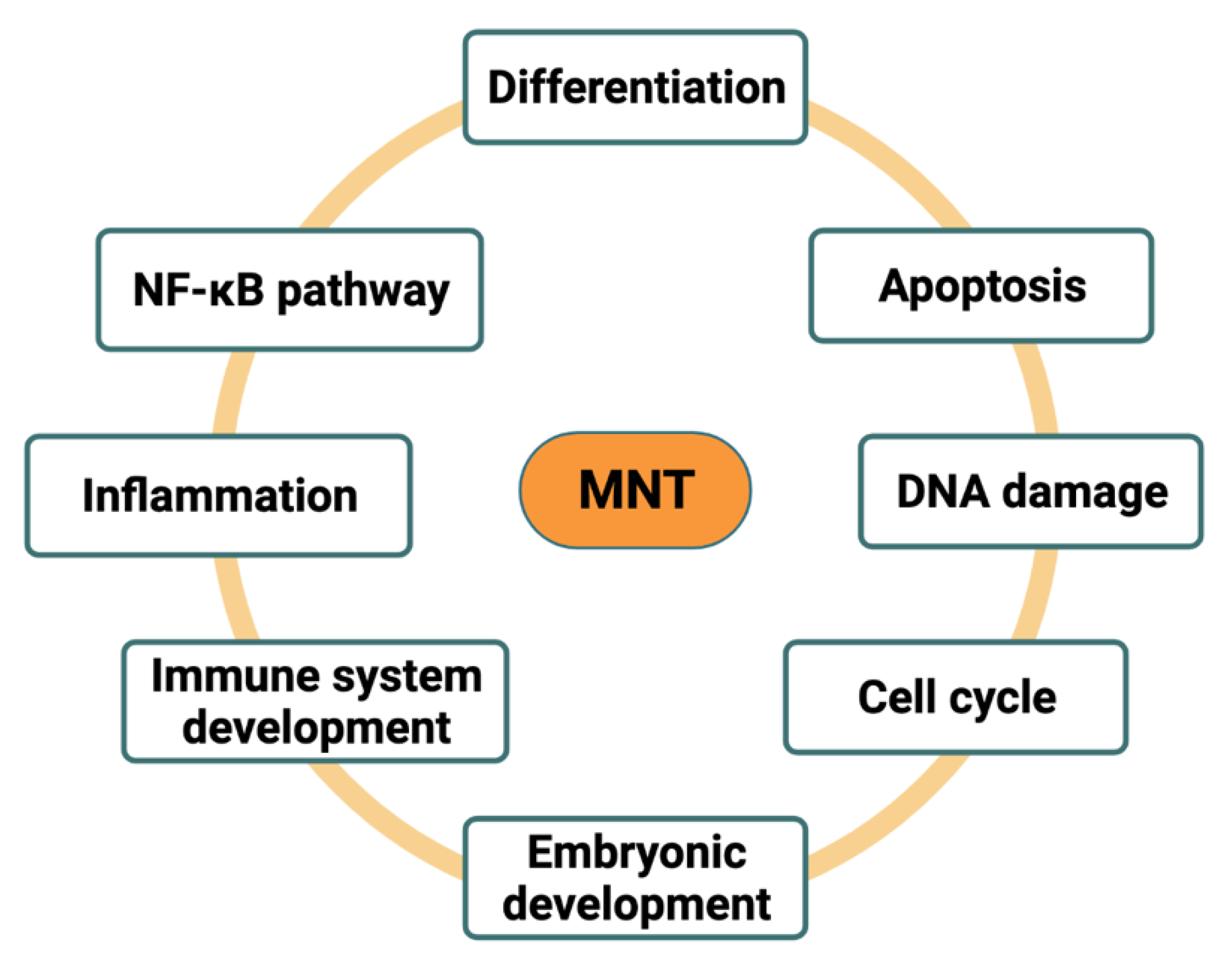
7. Functions of MNT in the Cell
7.1. MNT as MYC Antagonist
7.2. MNT as MYC Cooperator
8. MAX-Independent Roles of MNT
9. Concluding Remarks and Therapeutical Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Hurlin, P.J.; Quéva, C.; Eisenman, R.N. Mnt, a novel Max-interacting protein is coexpressed with Myc in proliferating cells and mediates repression at Myc binding sites. Genes Dev. 1997, 11, 44–58. [Google Scholar] [CrossRef] [Green Version]
- Meroni, G.; Reymond, A.; Alcalay, M.; Borsani, G.; Tanigami, A.; Tonlorenzi, R.; Lo Nigro, C.; Messali, S.; Zollo, M.; Ledbetter, D.H.; et al. Rox, a novel bHLHZip protein expressed in quiescent cells that heterodimerizes with Max, binds a non-canonical E box and acts as a transcriptional repressor. EMBO J. 1997, 16, 2892–2906. [Google Scholar] [CrossRef]
- Ayer, D.E.; Lawrence, Q.A.; Eisenman, R.N. Mad-max transcriptional repression is mediated by ternary complex formation with mammalian homologs of yeast repressor Sin3. Cell 1995, 80, 767–776. [Google Scholar] [CrossRef] [Green Version]
- Grzenda, A.; Lomberk, G.; Zhang, J.S.; Urrutia, R. Sin3: Master scaffold and transcriptional corepressor. Biochim. Biophys. Acta Gene Regul. Mech. 2009, 1789, 443–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raffeiner, P.; Hart, J.R.; García-Caballero, D.; Bar-Peled, L.; Weinberg, M.S.; Vogt, P.K. An MXD1-derived repressor peptide identifies noncoding mediators of MYC-driven cell proliferation. Proc. Natl. Acad. Sci. USA 2020, 117, 6571–6579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurlin, P.J.; Steingrimsson, E.; Copeland, N.G.; Jenkins, N.A.; Eisenman, R.N. Mga, a dual-specificity transcription factor that interacts with Max and contains a T-domain DNA- binding motif. EMBO J. 1999, 18, 7019–7028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rikin, A.; Evans, T. The Tbx/bHlH Transcription Factor mga regulates gata4 and Organogenesis. Dev. Dyn. 2010, 239, 535–547. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, H.; Ishiguro, K.; Gaubatz, S.; Livingston, D.M.; Nakatani, Y. A Complex with Chromatin Modifiers That Occupies E2F- and Myc-Responsive Genes in G0 Cells. Science 2002, 296, 1132–1136. [Google Scholar] [CrossRef]
- Mathsyaraja, H.; Catchpole, J.; Eastwood, E.; Babaeva, E.; Geuenich, M.; Cheng, P.F.; Freie, B.; Ayers, J.; Yu, M.; Wu, N.; et al. Loss of MGA mediated Polycomb repression promotes tumor progression and invasiveness. Elife 2021, 28, e64212. [Google Scholar] [CrossRef]
- Yang, G.; Hurlin, P.J. MNT and Emerging Concepts of MNT-MYC Antagonism. Genes 2017, 8, 83. [Google Scholar] [CrossRef]
- Toyo-oka, K.; Hirotsune, S.; Gambello, M.J.; Zhou, Z.; Olson, L.; Rosenfeld, M.G.; Eisenman, R.; Hurlin, P.; Wynshaw-Boris, A. Loss of the Max-interacting protein Mnt in mice results in decreased viability, defective embryonic growth and craniofacial defects: Relevance to Miller-Dieker syndrome. Hum. Mol. Genet. 2004, 13, 1057–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, K.P.; McArthur, G.A.; Quéva, C.; Hurlin, P.J.; Soriano, P.; Eisenman, R.N. Targeted disruption of the MYC antagonist MAD1 inhibits cell cycle exit during granulocyte differentiation. EMBO J. 1998, 17, 774–785. [Google Scholar] [CrossRef] [Green Version]
- Schreiber-Agus, N.; Meng, Y.; Hoang, T.; Hou, H.; Chen, K.; Greenberg, R.; Cordon-Cardo, C.; Lee, H.-W.; DePinho, R.A. Role of Mxi1 in ageing organ systems and the regulation of normal and neoplastic growth. Nature 1998, 393, 483–487. [Google Scholar] [CrossRef]
- Queva, C.; McArthur, G.A.; Iritani, B.M.; Eisenman, R.N. Targeted Deletion of the S-Phase-Specific Myc Antagonist Mad3 Sensitizes Neuronal and Lymphoid Cells to radiation-induced apoptosis. Mol. Cell. Biol. 2001, 21, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Diolaiti, D.; McFerrin, L.; Carroll, P.A.; Eisenman, R.N. Functional interactions among members of the MAX and MLX transcriptional network during oncogenesis. Biochim. Biophys. Acta 2015, 1849, 484–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018, 6, 282–300.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boonk, S.E.; Zoutman, W.H.; Marie-Cardine, A.; van der Fits, L.; Out-Luiting, J.J.; Mitchell, T.J.; Tosi, I.; Morris, S.L.; Moriarty, B.; Booken, N.; et al. Evaluation of Immunophenotypic and Molecular Biomarkers for Sézary Syndrome Using Standard Operating Procedures: A Multicenter Study of 59 Patients. J. Investig. Dermatol. 2016, 136, 1364–1372. [Google Scholar] [CrossRef] [Green Version]
- Van Doorn, R.; Dijkman, R.; Vermeer, M.H.; Out-Luiting, J.J.; Van Der Raaij-Helmer, E.M.H.; Willemze, R.; Tensen, C.P. Aberrant expression of the tyrosine kinase receptor EphA4 and the transcription factor twist in Sézary syndrome identified by gene expression analysis. Cancer Res. 2004, 64, 5578–5586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeer, M.H.; Van Doorn, R.; Dijkman, R.; Mao, X.; Whittaker, S.; Van Voorst Vader, P.C.; Gerritsen, M.J.P.; Geerts, M.L.; Gellrich, S.; Söderberg, O.; et al. Novel and highly recurrent chromosomal alterations in Sézary Syndrome. Cancer Res. 2008, 68, 2689–2698. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Pan, L.; Zhang, X.; Suo, X.; Niu, Z.; Zhang, J.; Wang, F.; Dong, Z.; Da, W.; Ohno, R. Expression and mutation analysis of genes that encode the Myc antagonists Mad1, Mxi1 and Rox in acute leukaemia. Leuk. Lymphoma 2007, 48, 1200–1207. [Google Scholar] [CrossRef]
- Cvekl, A.; Zavadil, J.; Birshtein, B.K.; Grotzer, M.A.; Cvekl, A. Analysis of transcripts from 17p13.3 in medulloblastoma suggests ROX/MNT as a potential tumour suppressor gene. Eur. J. Cancer 2004, 40, 2525–2532. [Google Scholar] [CrossRef]
- Wahlström, T.; Henriksson, M. Mnt Takes Control as Key Regulator of the Myc/Max/Mxd Network. Adv. Cancer Res. 2007, 97, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Loo, L.W.M.; Secombe, J.; Little, J.T.; Leni-Sue Carlos, L.-S.; Yost, C.; Cheng, P.-F.; Flynn, E.M.; Edgar, B.A.; Eisenman, R.N. The Transcriptional Repressor dMnt Is a Regulator of Growth in Drosophila melanogaster. Mol. Cell. Biol. 2005, 25, 7078–7091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orian, A.; van Steensel, B.; Delrow, J.; Bussemaker, H.J.; Li, L.; Sawado, T.; Williams, E.; Loo, L.W.M.; Cowley, S.M.; Yost, C.; et al. Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev. 2003, 17, 1101–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFerrin, L.G.; Atchley, W.R. Evolution of the Max and Mlx networks in animals. Genome Biol. Evol. 2011, 3, 915–937. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.W.; Llop, J.R.; Farrell, S.F.; Yuan, J.; Stolzenburg, L.R.; Samuelson, A.V. The Caenorhabditis elegans Myc-Mondo/Mad Complexes Integrate Diverse Longevity Signals. PLoS Genet. 2014, 10, e1004278. [Google Scholar] [CrossRef] [Green Version]
- Hurlin, P.J.; Zhou, Z.; Toyo-oka, K.; Ota, S.; Walker, W.L.; Hirotsune, S.; Wynshaw-boris, A. Deletion of Mnt leads to disrupted cell cycle control and tumorigenesis. EMBO J. 2003, 22, 4584–4596. [Google Scholar] [CrossRef] [Green Version]
- Schreiber-Agus, N.; Chin, L.; Chen, K.; Torres, R.; Rao, G.; Guida, P.; Skoultchi, A.; Depinho, R.A. An Amino-Terminal Domain of Mxi1 Mediates Anti-Myc Oncogenic Activity and Interacts with a Homolog of the Yeast Transcriptional Repressor SIN3. Cell 1995, 80, 777–786. [Google Scholar]
- Kay, B.K.; Williamson, M.P.; Sudol, M. The importance of being proline: The interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000, 14, 231–241. [Google Scholar] [CrossRef]
- Kapoor, I.; Kanaujiya, J.; Kumar, Y.; Thota, J.R.; Bhatt, M.L.B.; Chattopadhyay, N.; Sanyal, S.; Trivedi, A.K. Proteomic discovery of MNT as a novel interacting partner of E3 ubiquitin ligase E6AP and a key mediator of myeloid differentiation. Oncotarget 2016, 7, 7640–7656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popov, N.; Wahlström, T.; Hurlin, P.J.; Henriksson, M. Mnt transcriptional repressor is functionally regulated during cell cycle progression. Oncogene 2005, 24, 8326–8337. [Google Scholar] [CrossRef] [Green Version]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Juergens, K.; Rust, B.; Pieler, T.; Henningfeld, K.A. Isolation and comparative expression analysis of the Myc-regulatory proteins Mad1, Mad3, and Mnt during Xenopus development. Dev. Dyn. 2005, 233, 1554–1559. [Google Scholar] [CrossRef]
- Sadaghiani, B.; Thiébaud, C.H. Neural crest development in the Xenopus laevis embryo, studied by interspecific transplantation and scanning electron microscopy. Dev. Biol. 1987, 124, 91–110. [Google Scholar] [CrossRef]
- Lafita-Navarro, M.C.; Liaño-Pons, J.; Quintanilla, A.; Varela, I.; Blanco, R.; Ourique, F.; Bretones, G.; Aresti, J.; Molina, E.; Carroll, P.; et al. The MNT transcription factor autoregulates its expression and supports proliferation in MYC-associated factor X (MAX)-deficient cells. J. Biol. Chem. 2020, 295, 2001–2017. [Google Scholar] [CrossRef]
- Meroni, G.; Cairo, S.; Merla, G.; Messali, S.; Brent, R.; Ballabio, A.; Reymond, A. Mlx, a new Max-like bHLHZip family member: The center stage of a novel transcription factors regulatory pathway? Oncogene 2000, 19, 3266–3277. [Google Scholar] [CrossRef] [Green Version]
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 2014, 4, a014357. [Google Scholar] [CrossRef]
- Heinzel, T.; Lavinsky, R.M.; Mullen, T.; Soderstrom, M.; Lahertyll, C.D.; Torchia, J.; Yang, W.; Branl, G.; Ngo, S.D.; Davie, J.R.; et al. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature 1997, 387, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Laherty, C.D.; Yang, W.; Sun, J.; Davie, J.R.; Seto, E.; Eisenman, R.N. Histone Deacetylases associated with the mSin3 Corepressor mediate Mad Transcriptional Repression. Cell 1997, 89, 349–356. [Google Scholar] [CrossRef] [Green Version]
- Koskinen, P.J.; Ayer, D.E.; Eisenman, R.N. Ras Cotransformation by Mad Is Mediated by Multiple Interactions. Cell Growth Differ. 1995, 6, 623–629. [Google Scholar] [PubMed]
- Liaño-Pons, J.; Lafita-Navarro, M.C.; García-Gaipo, L.; Colomer, C.; Rodríguez, J.; von Kriegsheim, A.; Hurlin, P.J.; Delgado, M.D.; Bigas, A.; Espinosa, M.L.; et al. A novel role of MNT as a negative regulator of REL and the NF-κB pathway. Oncogenesis 2021, 10, 1–14. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared Principles in NF-κB Signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [Green Version]
- Kanarek, N.; London, N.; Schueler-Furman, O.; Ben-Neriah, Y. Ubiquitination and Degradation of the Inhibitors of NF-kB. Cold Spring Harb. Perspect. Biol. 2010, 2, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.C.; Ganchi, P.A.; Ballard, D.W.; Greene, W.C. NF-kappa B controls expression of inhibitor I kappa B alpha: Evidence for an inducible autoregulatory pathway. Science 1993, 259, 1912–1915. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liu, T.; Wang, J.; Li, T.W.H.; Fan, W.; Peng, H.; Krishnan, A.; Gores, G.J.; Mato, J.M.; Lu, S.C. Deregulated methionine adenosyltransferase α1, c-Myc, and Maf proteins together promote cholangiocarcinoma growth in mice and humans‡. Hepatology 2016, 64, 439–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Li, T.W.H.; Ko, K.S.; Xia, M.; Lu, S.C. Switch from Mnt-Max to Myc-Max induces p53 and cyclin D1 expression and apoptosis during cholestasis in mouse and human hepatocytes. Hepatology 2009, 49, 860–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerrato, A.; Merolla, F.; Morra, F.; Celetti, A. CCDC6: The identity of a protein known to be partner in fusion. Int. J. Cancer 2018, 142, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Van den Bergh, F.; Sabina, R.L. Characterization of human AMP deaminase 2 (AMPD2) gene expression reveals alternative transcripts encoding variable N-terminal extensions of isoform L. Biochem. J. 1995, 312, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Dixon, G.; Pan, H.; Yang, D.; Rosen, B.P.; Jashari, T.; Verma, N.; Pulecio, J.; Caspi, I.; Lee, K.; Stransky, S.; et al. QSER1 protects DNA methylation valleys from de novo methylation. Science 2021, 372, eabd0875. [Google Scholar] [CrossRef]
- Rockel, B.; Kopec, K.O.; Lupas, A.N.; Baumeister, W. Structure and function of tripeptidyl peptidase II, a giant cytosolic protease. Biochim. Biophys. Acta-Proteins Proteom. 2012, 1824, 237–245. [Google Scholar] [CrossRef]
- Dezfouli, S.; Bakke, A.; Huang, J.; Wynshaw-Boris, A.; Hurlin, P.J. Inflammatory Disease and Lymphomagenesis Caused by Deletion of the Myc Antagonist Mnt in T Cells. Mol. Cell. Biol. 2006, 26, 2080–2092. [Google Scholar] [CrossRef] [Green Version]
- Toyo-oka, K.; Bowen, T.J.; Hirotsune, S.; Li, Z.; Jain, S.; Ota, S.; Lozach, L.E.; Bassett, I.G.; Lozach, J.; Rosenfeld, M.G.; et al. Mnt-Deficient Mammary Glands Exhibit Impaired Involution and Tumors with Characteristics of Myc Overexpression. Cancer Res. 2006, 66, 5565–6894. [Google Scholar] [CrossRef] [Green Version]
- Edelmann, J.; Holzmann, K.; Miller, F.; Winkler, D.; Buhler, A.; Zenz, T.; Bullinger, L.; Kuhn, M.W.M.; Gerhardinger, A.; Bloehdorn, J.; et al. High-resolution genomic profiling of chronic lymphocytic leukemia reveals new recurrent genomic alterations. Blood 2012, 6, 4783–4794. [Google Scholar] [CrossRef] [Green Version]
- Lo Nigro, C.; Venesio, T.; Reymond, A.; Meroni, G.; Alberici, P.; Cainarca, S.; Enrico, F.; Stack, M.; Ledbetter, D.H.; Liscia, D.S.; et al. The Human ROX Gene: Genomic Structure and Mutation Analysis in Human Breast Tumors. Genomics 1998, 282, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Sommer, A.; Waha, A.; Tonn, J.; Sörensen, N.; Hurlin, P.J.; Eisenman, R.N.; Lüscher, B.; Pietsch, T. Analysis of the MAX-binding protein MNT in human medulloblastomas. Int. J. Cancer 1999, 82, 810–816. [Google Scholar] [CrossRef]
- Phillips, N.; Ziegler, M.; Saha, B.; Xynos, F. Allelic loss on chromosome 17 in human ovarian cancer. Int. J. Cancer 1993, 54, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Saxena, A.; Clark, W.C.; Robertson, J.T.; Ikejiri, B.; Oldfield, E.H.; Ali, I.U. Erratum: Evidence for the involvement of a potential second tumor suppressor gene on chromosome 17 distinct from p53 in malignant astrocytomas (Cancer Research (December 1, 1992) (6716–6721)). Cancer Res. 1993, 53, 1472. [Google Scholar]
- Williamson, M.P.; Elder, P.A.; Knowles, M.A. The spectrum of TP53 mutations in bladder carcinoma. Genes Chromosom. Cancer 1994, 9, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, Å.; Oyjord, T.; Hovig, E.; Holm, R.; Florenes, V.A.; Nesland, J.M.; Myklebost, O.; Hoie, J.; Bruland, S.; Borresen, A.-L.; et al. p53 Abnormalities in Different Subtypes of Human Sarcomas. Cancer Res. 1993, 53, 468–471. [Google Scholar] [PubMed]
- Hurlin, P.J.; Zhou, Z.Q.; Toyo-Oka, K.; Ota, S.; Walker, W.L.; Hirotsune, S.; Wynshaw-Boris, A. Evidence of Mnt-Myc antagonism revealed by Mnt gene deletion. Cell Cycle 2004, 3, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.A.; Maclean, K.H.; Keller, U.B.; Pendeville, H.; Baudino, T.A.; Cleveland, J.L. Mnt loss triggers Myc transcription targets, proliferation, apoptosis, and transformation. Mol. Cell. Biol. 2004, 24, 1560–1569. [Google Scholar] [CrossRef] [Green Version]
- Walker, W.; Zhou, Z.Q.; Ota, S.; Wynshaw-Boris, A.; Hurlin, P.J. Mnt-Max to Myc-Max complex switching regulates cell cycle entry. J. Cell Biol. 2005, 169, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Link, J.M.; Hurlin, P.J. The activities of MYC, MNT and the MAX-interactome in lymphocyte proliferation and oncogenesis. Biochim. Biophys. Acta-Gene Regul. Mech. 2015, 1849, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Pierce, S.B.; Yost, C.; Anderson, S.A.R.; Flynn, E.M.; Delrow, J.; Eisenman, R.N. Drosophila growth and development in the absence of dMyc and dMnt. Dev. Biol. 2008, 315, 303–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, S.B.; Yost, C.; Britton, J.S.; Loo, L.W.M.; Flynn, E.M.; Edgar, B.A.; Eisenman, R.N. dMyc is required for larval growth and endoreplication in Drosophila. Development 2004, 131, 2317–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V.; Kim, J.; Gao, P.; Yustein, J. The Interplay Between MYC and HIF in the Warburg Effect. Nat. Rev. Cancer 2008, 8, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Wei, J.; Guo, T.; Shen, Y.; Liu, F. Knockdown of miR-210 decreases hypoxic glioma stem cells stemness and radioresistance. Exp. Cell Res. 2014, 326, 22–35. [Google Scholar] [CrossRef]
- Zhang, Z.; Sun, H.; Dai, H.; Walsh, R.M.; Imakura, M.; Schelter, J.; Burchard, J.; Dai, X.; Chang, A.N.; Diaz, R.L.; et al. MicroRNA miR-210 modulates cellular response to hypoxia through the MYC antagonist MNT. Cell Cycle 2009, 8, 2756–2768. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Yang, S.L.; Fang, X.; Jiang, J.X.; Sun, C.Y.; Huang, T. Hypoxia disrupts the expression levels of circadian rhythm genes in hepatocellular carcinoma. Mol. Med. Rep. 2015, 11, 4002–4008. [Google Scholar] [CrossRef]
- Blaževitš, O.; Bolshette, N.; Vecchio, D.; Guijarro, A.; Croci, O.; Campaner, S.; Grimaldi, B. MYC-associated factor max is a regulator of the circadian clock. Int. J. Mol. Sci. 2020, 21, 2294. [Google Scholar] [CrossRef] [Green Version]
- Niu, F.; Dzikiewicz-Krawczyk, A.; Koerts, J.; de Jong, D.; Wijenberg, L.; Hernandez, M.F.; Slezak-Prochazka, I.; Winkle, M.; Kooistra, W.; van der Sluis, T.; et al. Mir-378a-3p is critical for burkitt lymphoma cell growth. Cancers 2020, 12, 3546. [Google Scholar] [CrossRef]
- Link, J.M.; Ota, S.; Zhou, Z.; Daniel, C.J.; Sears, R.C.; Hurlin, P.J. A critical role for Mnt in Myc-driven T-cell proliferation and oncogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 19685–19690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, D.J.; Junttila, M.R.; Pouyet, L.; Karnezis, A.; Shchors, K.; Bui, D.A.; Brown-Swigart, L.; Johnson, L.; Evan, G.I. Distinct Thresholds Govern Myc’s Biological Output In Vivo. Cancer Cell 2008, 14, 447–457. [Google Scholar] [CrossRef] [Green Version]
- Vasilevsky, N.A.; Ruby, C.E.; Hurlin, P.J.; Weinberg, A.D. OX40 engagement stabilizes Mxd4 and Mnt protein levels in antigen-stimulated T cells leading to an increase in cell survival. Eur. J. Immunol. 2011, 41, 1024–1034. [Google Scholar] [CrossRef] [Green Version]
- Campbell, K.J.; Vandenberg, C.J.; Anstee, N.S.; Hurlin, P.J.; Cory, S. Mnt modulates Myc-driven lymphomagenesis. Cell Death Differ. 2017, 24, 2117–2126. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.V.; Vandenberg, C.J.; Ng, A.P.; Robati, M.R.; Anstee, N.S.; Rimes, J.; Hawkins, E.D.; Cory, S. Development and survival of MYC-driven lymphomas require the MYC antagonist MNT to curb MYC-induced apoptosis. Blood 2020, 135, 1019–1031. [Google Scholar] [CrossRef]
- Burnichon, N.; Cascoń, A.; Schiavi, F.; Morales, N.P.; Comino-Méndez, I.; Abermil, N.; Inglada-Pérez, L.; De Cubas, A.A.; Amar, L.; Barontini, M.; et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin. Cancer Res. 2012, 18, 2828–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comino-Méndez, I.; Gracia-Aznárez, F.J.; Schiavi, F.; Landa, I.; Leandro-García, L.J.; Letón, R.; Honrado, E.; Ramos-Medina, R.; Caronia, D.; Pita, G.; et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat. Genet. 2011, 43, 663–667. [Google Scholar] [CrossRef]
- Pantaleo, M.A.; Urbini, M.; Indio, V.; Ravegnini, G.; Nannini, M.; De Luca, M.; Tarantino, G.; Angelini, S.; Gronchi, A.; Vincenzi, B.; et al. Genome-Wide Analysis Identifies MEN1 and MAX Mutations and a Neuroendocrine-Like Molecular Heterogeneity in Quadruple WT GIST. Mol. Cancer Res. 2017, 15, 553–562. [Google Scholar] [CrossRef] [Green Version]
- Robledo, M. MAX and MYC: A Heritable Breakup. Cancer Res. 2012, 72, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Romero, O.A.; Torres-Diz, M.; Pros, E.; Savola, S.; Gomez, A.; Moran, S.; Saez, C.; Iwakawa, R.; Villanueva, A.; Montuenga, L.M.; et al. MAX inactivation in small cell lung cancer disrupts MYC-SWI/SNF programs and is synthetic lethal with BRG1. Cancer Discov. 2014, 4, 292–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopewell, R.; Ziff, E.B. The nerve growth factor-responsive PC12 cell line does not express the Myc dimerization partner Max. Mol. Cell. Biol. 1995, 15, 3470–3478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terragni, J.; Nayak, G.; Banerjee, S.; Medrano, J.; Graham, J.R.; Brennan, J.F.; Sepulveda, S.; Cooper, G.M. The E-box Binding Factors Max/Mnt, MITF, and USF1 Act Coordinately with FoxO to Regulate Expression of Proapoptotic and Cell Cycle Control Genes by Phosphatidylinositol 3-Kinase/ Akt/Glycogen Synthase Kinase 3 Signaling. J. Biol. Chem. 2011, 286, 36215–36227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathsyaraja, H.; Freie, B.; Cheng, P.F.; Babaeva, E.; Catchpole, J.T.; Janssens, D.; Henikoff, S.; Eisenman, R.N. Max deletion destabilizes MYC protein and abrogates Eµ-Myc lymphomagenesis. Genes Dev. 2019, 1252–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, P.A.; Diolaiti, D.; McFerrin, L.; Gu, H.; Djukovic, D.; Du, J.; Cheng, P.F.; Anderson, S.; Ulrich, M.; Hurley, J.B.; et al. Deregulated Myc Requires MondoA/Mlx for Metabolic Reprogramming and Tumorigenesis. Cancer Cell 2015, 27, 271–285. [Google Scholar] [CrossRef] [Green Version]
- Sheiness, D.; Fanshier, L.; Bishop, J.M. Identification of nucleotide sequences which may encode the oncogenic capacity of avian retrovirus MC29. J. Virol. 1978, 28, 600–610. [Google Scholar] [CrossRef] [Green Version]
- Sheiness, D.; Bishop, J.M. DNA and RNA from uninfected vertebrate cells contain nucleotide sequences related to the putative transforming gene of avian myelocytomatosis virus. J. Virol. 1979, 31, 514–521. [Google Scholar] [CrossRef] [Green Version]
- Duesberg, P.H.; Bister, K.; Vogt, P.K. The RNA of avian acute leukemia virus MC29. Proc. Natl. Acad. Sci. USA 1977, 74, 4320–4324. [Google Scholar] [CrossRef] [Green Version]
- Massó-Vallés, D.; Soucek, L. Blocking Myc to Treat Cancer: Reflecting on Two Decades of Omomyc. Cells 2020, 9, 883. [Google Scholar] [CrossRef] [Green Version]
- Vita, M.; Henriksson, M. The Myc oncoprotein as a therapeutic target for human cancer. Semin. Cancer Biol. 2006, 16, 318–330. [Google Scholar] [CrossRef]
- Castell, A.; Yan, Q.; Fawkner, K.; Hydbring, P.; Zhang, F.; Verschut, V.; Franco, M.; Zakaria, S.M.; Bazzar, W.; Goodwin, J.; et al. A selective high affinity MYC-binding compound inhibits MYC:MAX interaction and MYC-dependent tumor cell proliferation. Sci. Rep. 2018, 8, 10064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demma, M.J.; Hohn, M.J.; Sun, A.; Mapelli, C.; Hall, B.; Walji, A.; O’Neil, J. Inhibition of Myc transcriptional activity by a mini-protein based upon Mxd1. FEBS Lett. 2020, 594, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Demma, M.J.; Mapelli, C.; Sun, A.; Bodea, S.; Ruprecht, B.; Javaid, S.; Wiswell, D.; Muise, E.; Chen, S.; Zelina, J.; et al. Omomyc Reveals New Mechanisms To Inhibit the MYC Oncogene. Mol. Cell. Biol. 2019, 39, 1–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MNT | MXD1 | MXI1 (MXD2) | MXD3 | MXD4 | MYC | References | |
|---|---|---|---|---|---|---|---|
| Protein size | 582 aa | 221 aa | 228 aa | 206 aa | 209 aa | 439 aa | Uniprot database |
| P-rich sequences | Yes | No | No | No | No | Yes | [2] |
| Phenotype of KO mice | Perinatally lethal/Craniofacial abnormalities | Viable/Increased immature granulocyte progenitors | Viable/Hyperplasia | Viable/Enhanced sensitivity to apoptotic stimuli | ND | Embryonic lethal E9.5–E10.5 | [11,12,13,14] |
| Proximal MYC Network interactors | MNT, MAX, MLX | MAX, MLX | MAX | MAX | MAX, MLX | MAX | Reviewed in [15] |
| Expression in cells | Quiescent and proliferating | Quiescent | Quiescent and proliferating * | Proliferating (S-phase) | Quiescent | Proliferating | Reviewed in [15] |
| Pan-cancer copy number alterations (%) | DEL 10 AMP 3 MUT 1 | DEL 2 AMP 6 MUT < 0.5 | DEL 8 AMP 4 MUT < 0.5 | DEL 7 AMP 8 MUT < 0.5 | DEL 6 AMP 5 MUT < 0.5 | DEL 2 AMP 21 MUT 1 | [16] |
| Involvement in human cancer | DEL in CTCL-SS and ALL. Reduced expression in MB | None or weak | None or weak | None or weak | None or weak | Strongly, 70% of tumors have deregulation | [17,18,19,20,21,22] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liaño-Pons, J.; Arsenian-Henriksson, M.; León, J. The Multiple Faces of MNT and Its Role as a MYC Modulator. Cancers 2021, 13, 4682. https://doi.org/10.3390/cancers13184682
Liaño-Pons J, Arsenian-Henriksson M, León J. The Multiple Faces of MNT and Its Role as a MYC Modulator. Cancers. 2021; 13(18):4682. https://doi.org/10.3390/cancers13184682
Chicago/Turabian StyleLiaño-Pons, Judit, Marie Arsenian-Henriksson, and Javier León. 2021. "The Multiple Faces of MNT and Its Role as a MYC Modulator" Cancers 13, no. 18: 4682. https://doi.org/10.3390/cancers13184682
APA StyleLiaño-Pons, J., Arsenian-Henriksson, M., & León, J. (2021). The Multiple Faces of MNT and Its Role as a MYC Modulator. Cancers, 13(18), 4682. https://doi.org/10.3390/cancers13184682