
Serum Levels of the Cytokine TWEAK Are Associated with Metabolic Status in Patients with Prostate Cancer and Modulate Cancer Cell Lipid Metabolism In Vitro

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Biochemical Data
2.3. Cell Culture and Treatments
2.4. siRNA Experiments
2.5. Inhibitor Treatments
2.6. Determination of Fatty Acid Uptake and Lipid Accumulation
2.7. Gene Expression Analysis
2.8. Western Blot Analysis
2.9. Statistical Analysis
3. Results
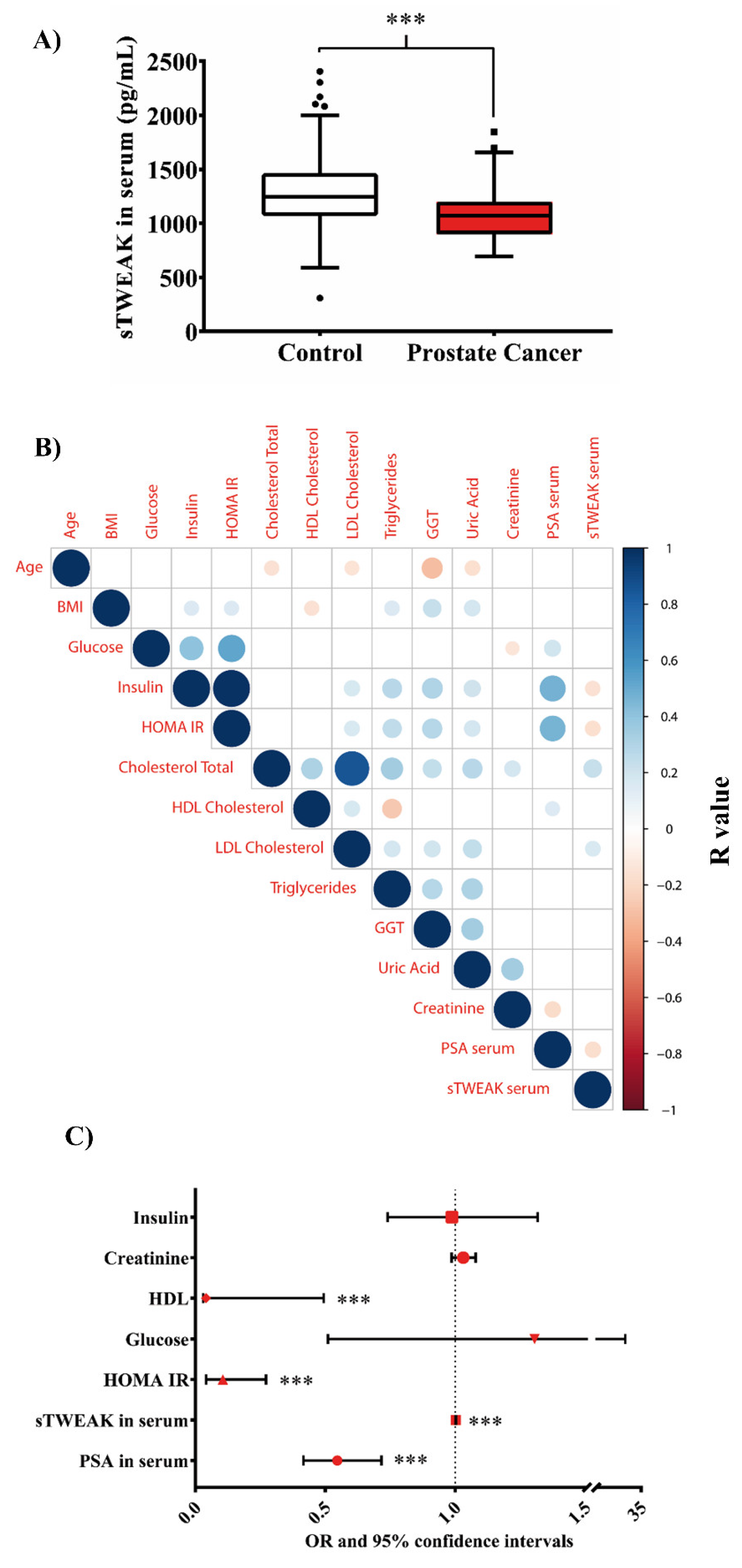
3.1. Low Concentrations of sTWEAK Are Associated with PCa
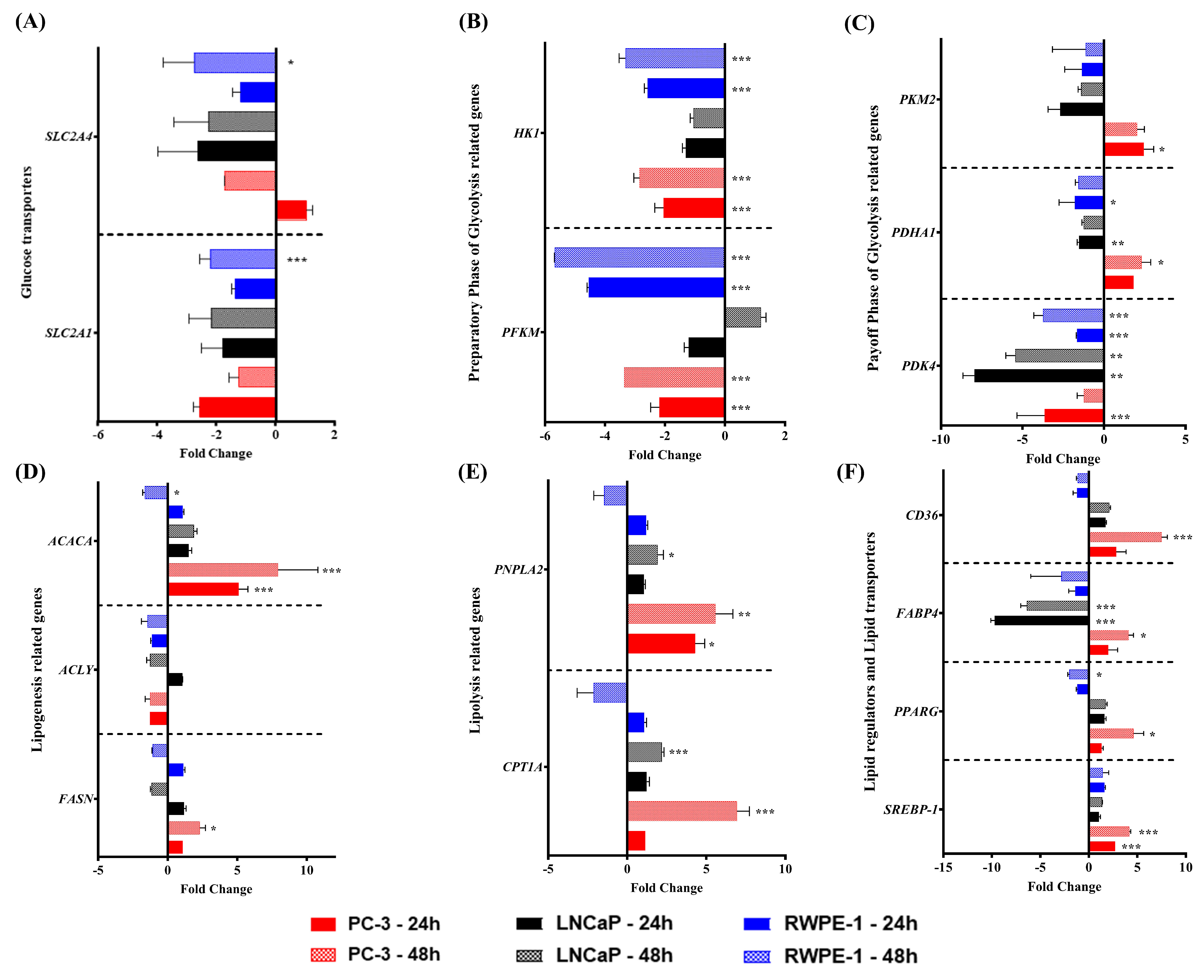
3.2. sTWEAK Treatment of PCa Cell Lines Alters the Expression of Genes Related to Lipid Metabolism but Not to Glucose Metabolism
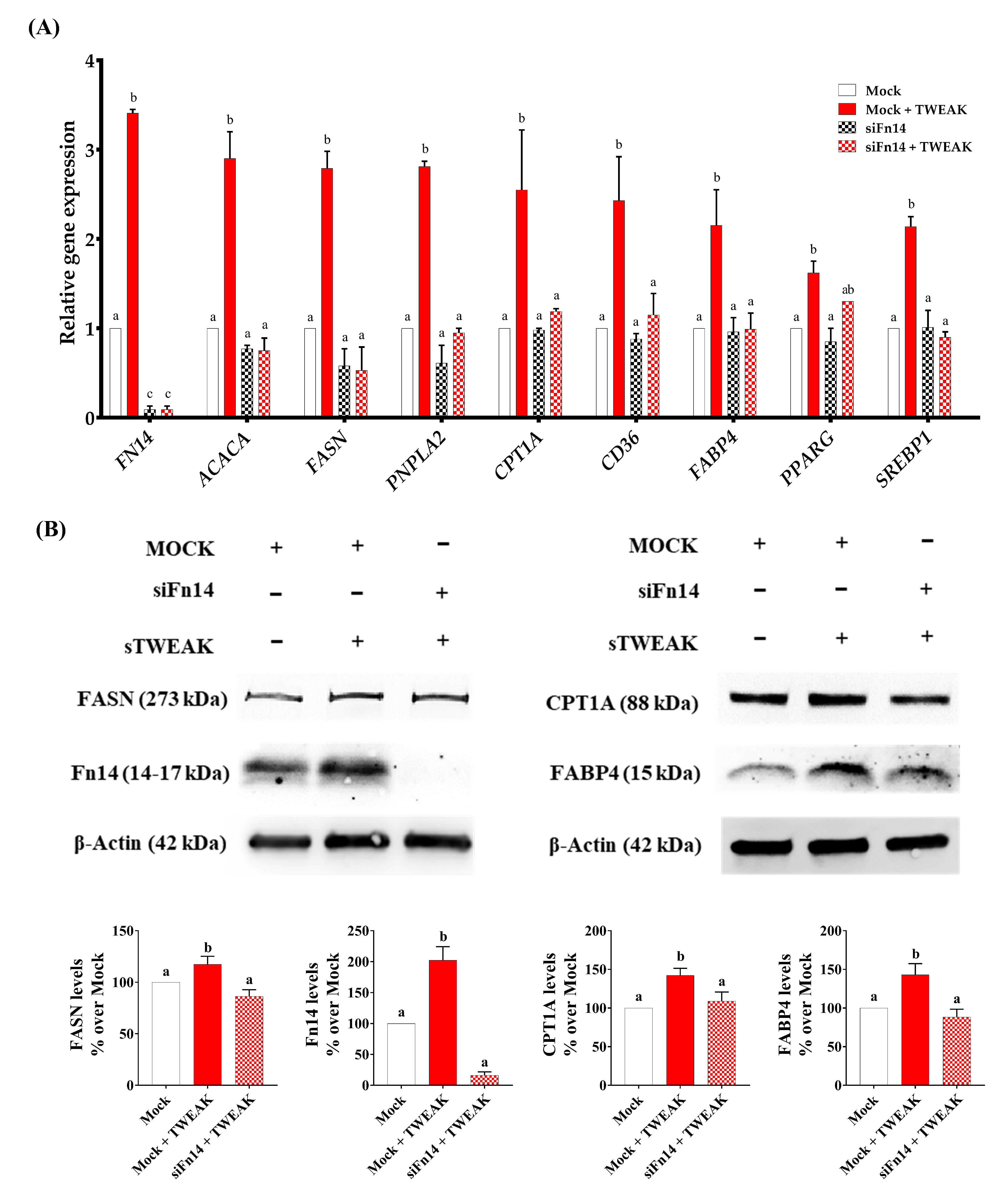
3.3. Silencing of Fn14 (TNFRSF12A) Abrogates the Ability of sTWEAK to Modulate Lipid Metabolism Genes in PC-3 Cells
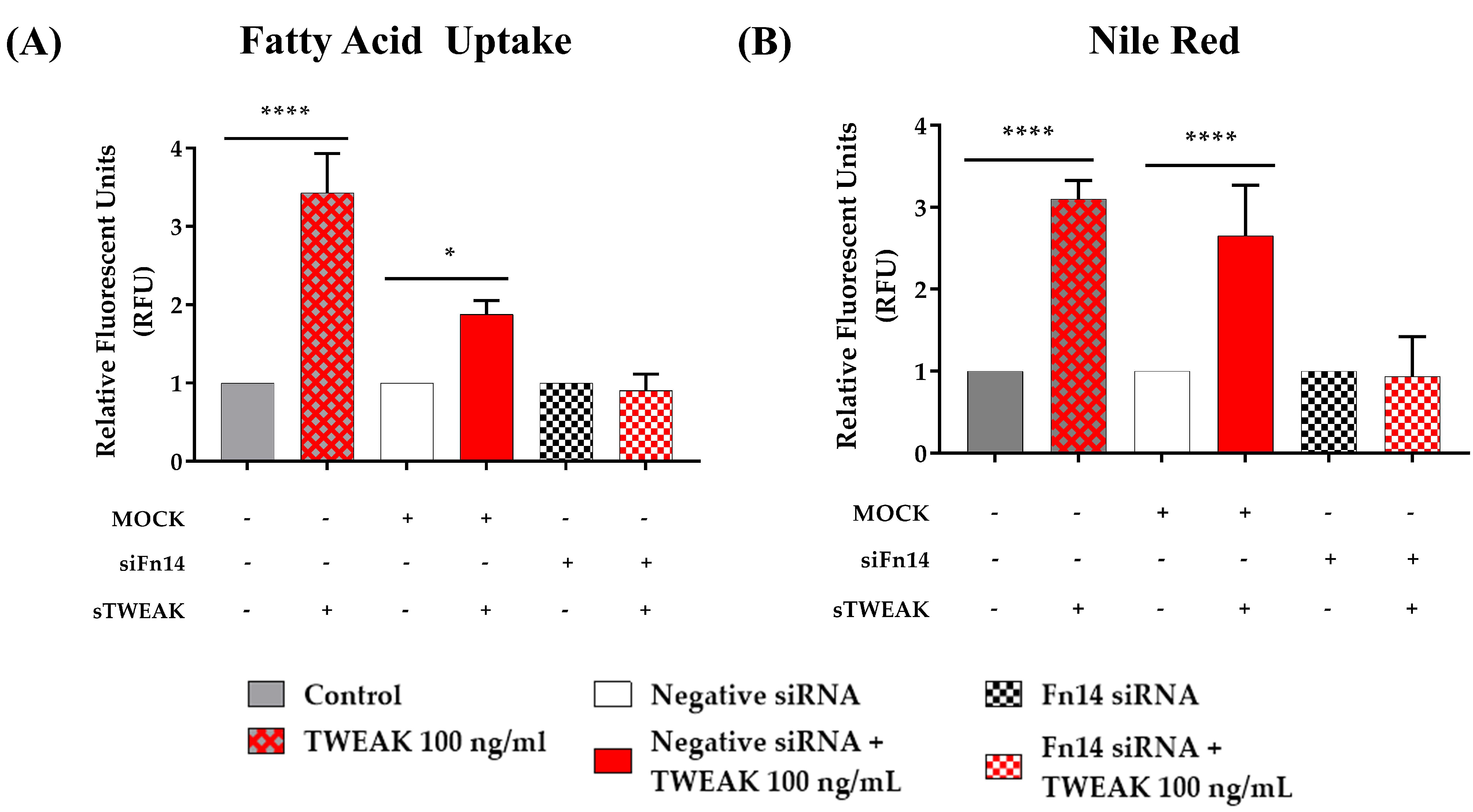
3.4. sTWEAK Stimulates Fatty Acid Uptake and Lipid Accumulation in PC-3 Cells
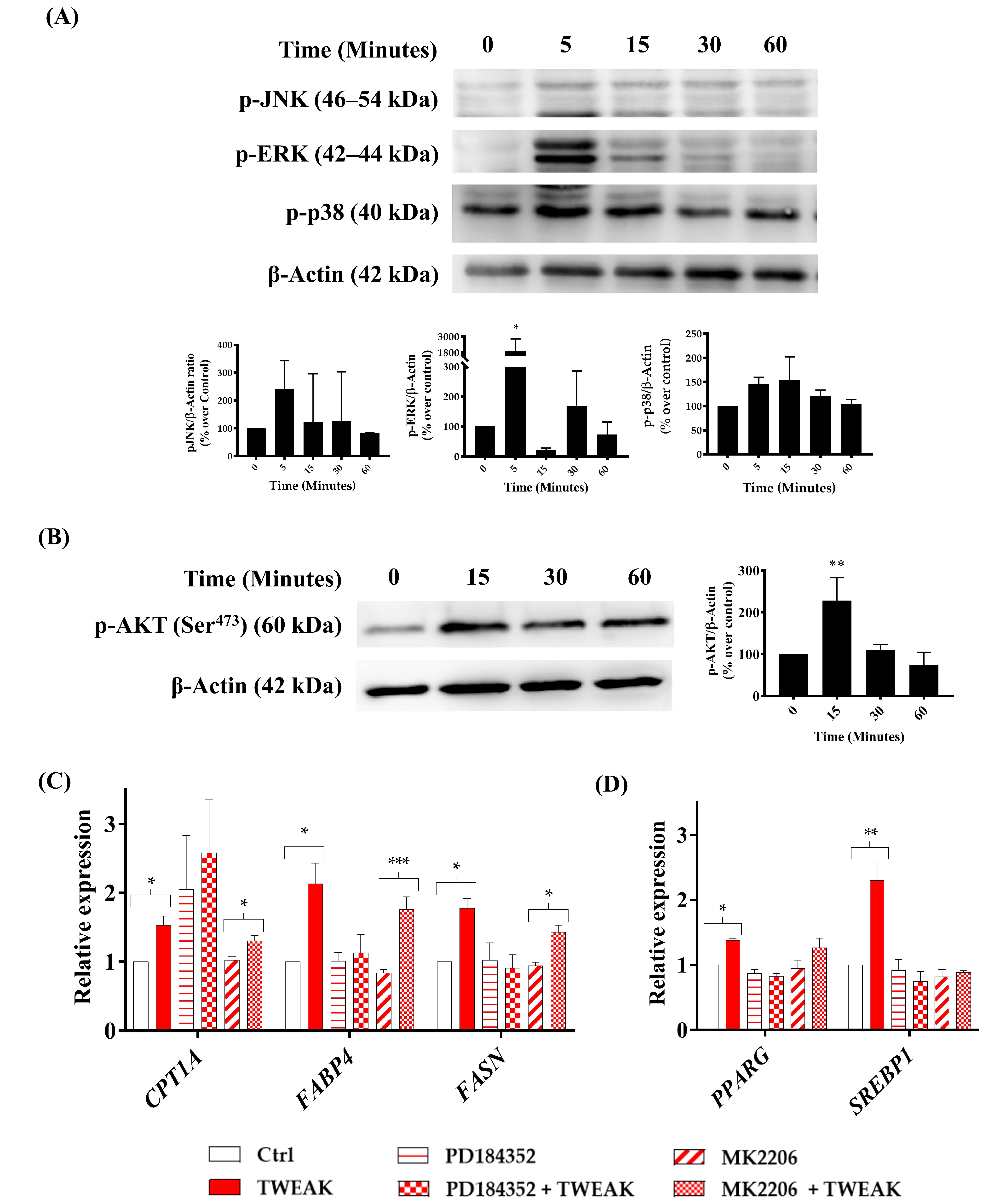
3.5. sTWEAK Stimulates Lipid Metabolism Genes in PC-3 Cells by Phosphorylating ERK1/2 and AKT(ser473)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.I.; Egevad, L.; Amin, M.B.; Delahunt, B.; Srigley, J.R.; Humphrey, P.A. The 2014 International Society of Urological Pathology (ISUP) consensus conference on Gleason grading of prostatic carcinoma: Definition of grading patterns and proposal for a new grading system. Am. J. Surg. Pathol. 2016, 40, 244–252. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, L.P.; Gómez de Cedrón, M.; Ramírez de Molina, A. Alterations of Lipid Metabolism in Cancer: Implications in Prognosis and Treatment. Front. Oncol. 2020, 10, 577420. [Google Scholar] [CrossRef] [PubMed]
- Bader, D.A.; McGuire, S.E. Tumour metabolism and its unique properties in prostate adenocarcinoma. Nat. Rev. Urol. 2020, 17, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Zadra, G.; Photopoulos, C.; Loda, M. The fat side of prostate cancer. Biochim. Biophys. Acta 2013, 1831, 1518–1532. [Google Scholar] [CrossRef] [Green Version]
- Saboori, S.; Rad, E.Y.; Birjandi, M.; Mohiti, S.; Falahi, E. Serum insulin level, HOMA-IR and prostate cancer risk: A systematic review and meta-analysis. Diabetes Metab. Syndr. Clin. Res. Rev. 2019, 13, 110–115. [Google Scholar] [CrossRef]
- Nandeesha, H. Insulin: A novel agent in the pathogenesis of prostate cancer. Int. Urol. Nephrol. 2009, 41, 267–272. [Google Scholar] [CrossRef]
- Sfanos, K.S.; De Marzo, A.M. Prostate cancer and inflammation: The evidence Karen. Histopathology 2012, 60, 199–215. [Google Scholar] [CrossRef] [Green Version]
- Maecker, H.; Varfolomeev, E.; Kischkel, F.; Lawrence, D.; LeBlanc, H.; Lee, W.; Hurst, S.; Danilenko, D.; Li, J.; Filvaroff, E.; et al. TWEAK attenuates the transition from innate to adaptive immunity. Cell 2005, 123, 931–944. [Google Scholar] [CrossRef] [Green Version]
- Chicheportiche, Y.; Bourdon, P.R.; Xu, H.; Hsu, Y.M.; Scott, H.; Hession, C.; Garcia, I.; Browning, J.L. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J. Biol. Chem. 1997, 272, 32401–32410. [Google Scholar] [CrossRef] [Green Version]
- Winkles, J.A. The TWEAK-Fn14 cytokine-receptor axis: Discovery, biology and therapeutic targeting. Nat. Rev. Drug Discov. 2008, 7, 411–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, S.R.; Winkles, J.A. TWEAK, a member of the TNF superfamily, is a multifunctional cytokine that binds the TweakR/Fn14 receptor. Cytokine Growth Factor Rev. 2003, 14, 241–249. [Google Scholar] [CrossRef]
- Narita, S.N.; Tsuchiya, N.; Saito, M.; Inoue, T.; Kumazawa, T.; Yuasa, T.; Nakamura, A.; Habuchi, T. CandidateGenes Involved in Enhanced Growth of Human Prostate Cancer Under High Fat Feeding. Identified by Microarray Analysis. Prostate 2008, 68, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Narita, S.; Tsuchiya, N.; Ma, Z.; Numakura, K.; Obara, T.; Tsuruta, H.; Saito, M.; Inoue, T.; Horikawa, Y.; et al. Overexpression of Fn14 promotes androgen-independent prostate cancer progression through MMP-9 and correlates with poor treatment outcome. Carcinogenesis 2011, 32, 1589–1596. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Plazas, X.; Rodríguez-Gallego, E.; Alves, M.; Altuna-Coy, A.; Lozano-Bartolomé, J.; Portero-Otin, M.; García-Fontgivell, J.F.; Martínez-González, S.; Segarra, J.; Chacón, M.R. Biofluid quantification of TWEAK/Fn14 axis in combination with a selected biomarker panel improves assessment of prostate cancer aggressiveness. J. Transl. Med. 2019, 17, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Plazas, X.; Altuna-Coy, A.; Alves-Santiago, M.; Vila-Barja, J.; García-Fontgivell, J.F.; Martínez-González, S.; Segarra-Tomás, J.; Chacón, M.R. Liquid biopsy-based exo-oncomirnas can predict prostate cancer aggressiveness. Cancers 2021, 13, 250. [Google Scholar] [CrossRef] [PubMed]
- Vendrell, J.; Chacón, M.R. TWEAK: A new player in obesity and diabetes. Front. Immunol. 2013, 4, 488. [Google Scholar] [CrossRef]
- Lozano-Bartolomé, J.; Llauradó, G.; Rodriguez, M.M.; Fernandez-Real, J.M.; Garcia-Fontgivell, J.F.; Puig, J.; Maymó-Masip, E.; Vendrell, J.; Chacón, M.R. Reduced circulating levels of sTWEAK are associated with NAFLD and may affect hepatocyte triglyceride accumulation. Int. J. Obes. 2016, 40, 1337–1345. [Google Scholar] [CrossRef]
- Preston, M.A.; Batista, J.L.; Wilson, K.M.; Carlsson, S.V.; Gerke, T.; Sjoberg, D.D.; Dahl, D.M.; Sesso, H.D.; Feldman, A.S.; Gann, P.H.; et al. Baseline prostate-specific antigen levels in midlife predict lethal prostate cancer. J. Clin. Oncol. 2016, 34, 2705–2711. [Google Scholar] [CrossRef]
- Chen, S.; Liu, J.; Yang, M.; Lai, W.; Ye, L.; Chen, J.; Hou, X.; Ding, H.; Zhang, W.; Wu, Y.; et al. Fn14, a downstream target of the TGF-β signaling pathway, regulates fibroblast activation. PLoS ONE 2015, 10, e0143802. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.S.; Monteiro, M.P.; Costa, M.M.; Ferreira, J.; Alves, M.G.; Oliveira, P.F.; Jarak, I.; Pignatelli, D. MAPK/ERK pathway inhibition is a promising treatment target for adrenocortical tumors. J. Cell. Biochem. 2019, 120, 894–906. [Google Scholar] [CrossRef] [Green Version]
- Tee, S.S.; Suster, I.; Truong, S.; Jeong, S.; Eskandari, R.; DiGialleonardo, V.; Alvarez, J.A.; Aldeborgh, H.N.; Keshari, K.R. Targeted AKT inhibition in prostate cancer cells and spheroids reduces aerobic glycolysis and generation of hyperpolarized [1-13C] lactate. Mol. Cancer Res. 2018, 16, 453–460. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; St Clair, D.K.; Xu, Y.; Crooks, P.A.; St Clair, W.H. A NADPH oxidase-dependent redox signaling pathway mediates the selective radiosensitization effect of parthenolide in prostate cancer cells. Cancer Res. 2010, 70, 2880–2890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, K.L.; Kellner, D.; Bajrami, B.; Anderson, J.E.; Beyna, M.; Bhisetti, G.; Cameron, T.; Capacci, A.G.; Bertolotti-Ciarlet, A.; Feng, J.; et al. CDK12-mediated transcriptional regulation of noncanonical NF-kB components is essential for signaling. Sci. Signal. 2018, 11, eaam8216. [Google Scholar] [CrossRef] [Green Version]
- Vendrell, J.; Maymó-Masip, E.; Tinahones, F.; García-España, A.; Megia, A.; Caubet, E.; García-Fuentes, E.; Chacón, M.R. Tumor necrosis-like weak inducer of apoptosis as a proinflammatory cytokine in human adipocyte cells: Up-regulation in severe obesity is mediated by inflammation but not hypoxia. J. Clin. Endocrinol. Metab. 2010, 95, 2983–2992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, S.; Choy, P.M.; Bubici, C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene 2019, 38, 2223–2240. [Google Scholar] [CrossRef] [Green Version]
- Cooperberg, M.R.; Carroll, P.R.; Klotz, L. Active surveillance for prostate cancer: Progress and promise. J. Clin. Oncol. 2011, 29, 3669–3676. [Google Scholar] [CrossRef] [Green Version]
- Di Sebastiano, K.M.; Pinthus, J.H.; Duivenvoorden, W.C.M.; Mourtzakis, M. Glucose impairments and insulin resistance in prostate cancer: The role of obesity, nutrition and exercise. Obes. Rev. 2018, 19, 1008–1016. [Google Scholar] [CrossRef]
- Hammarsten, J.; Högstedt, B. Hyperinsulinaemia: A prospective risk factor for lethal clinical prostate cancer. Eur. J. Cancer 2005, 41, 2887–2895. [Google Scholar] [CrossRef]
- Weinstein, J.E.; Mackrain, K.; Stolzenberg-Solomon, R.-Z.; Selhub, J.; Virtamo, J.; Albanes, D. Serum creatinine and prostate cancer risk in a prospective study. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2643–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, A.L. Development of cancer metabolism as a therapeutic target: New pathways, patient studies, stratification and combination therapy. Br. J. Cancer 2020, 122, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Centenera, M.M.; Swinnen, J.V. Androgen control of lipid metabolism in prostate cancer: Novel insights and future applications. Endocr. Relat. Cancer 2014, 23, R219–R227. [Google Scholar] [CrossRef]
- Jadvar, H. PET of glucose metabolism and cellular proliferation in prostate cancer. J. Nucl. Med. 2016, 57, 25S–29S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauregard, J.M.; Williams, S.G.; Degrado, T.R.; Roselt, P.; Hicks, R.J. Pilot comparison of 18F-fluorocholine and 18F- fluorodeoxyglucose PET/CT with conventional imaging in prostate cancer. J. Med. Imaging Radiat. Oncol. 2010, 54, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Vigneron, D.B.; Kurhanewicz, J.; Francisco, S. Hyperpolarized 1-[13C]-Pyruvate Magnetic Resonance Imaging Detects an Early Metabolic Response to Androgen Ablation Therapy in Prostate Cancer. Eur. Urol. 2017, 72, 1028–1029. [Google Scholar] [CrossRef]
- Pecinová, A.; Alán, L.; Brázdová, A.; Vrbacký, M.; Pecina, P.; Drahota, Z.; Houštěk, J.; Mráček, T. Role of Mitochondrial Glycerol-3-Phosphate Dehydrogenase in Metabolic Adaptations of Prostate Cancer. Cells 2020, 9, 1764. [Google Scholar] [CrossRef]
- Nomura, D.K.; Lombardi, D.P.; Chang, J.W.; Niessen, S.; Ward, A.M.; Long, J.Z.; Hoover, H.H.; Cravatt, B.F. Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem. Biol. 2011, 18, 846–856. [Google Scholar] [CrossRef] [Green Version]
- Diedrich, J.D.; Rajagurubandara, E.; Herroon, M.K.; Mahapatra, G.; Hüttemann, M.; Podgorski, I. Bone marrow adipocytes promote the warburg phenotype in metastatic prostate tumors via HIF-1α activation. Oncotarget 2016, 7, 64854–64877. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zuckier, L.S.; Ghesani, N.V. Dominant uptake of fatty acid over glucose by prostate cells: A potential new diagnostic and therapeutic approach. Anticancer Res. 2010, 30, 369–374. [Google Scholar]
- Dueregger, A.; Schöpf, B.; Eder, T.; Höfer, J.; Gnaiger, E.; Aufinger, A.; Kenner, L.; Perktold, B.; Ramoner, R.; Klocker, H.; et al. Differential utilization of dietary fatty acids in benign and malignant cells of the prostate. PLoS ONE 2015, 10, e0135704. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 2006, 9, 230–234. [Google Scholar] [CrossRef] [Green Version]
- Dang, Q.; Chen, Y.-A.; Hsieh, J.-T. The dysfunctional lipids in prostate cancer. Am. J. Clin. Exp. Urol. 2019, 7, 273–280. [Google Scholar]
- Loda, M.; Migita, T.; Ruiz, S.; Fornari, A.; Fiorentino, M.; Priolo, C.; Zadra, G.; Inazuka, F.; Grisanzio, C.; Palescandolo, E.; et al. Fatty acid synthase: A metabolic enzyme and candidate oncogene in prostate cancer. J. Natl. Cancer Inst. 2009, 101, 519–532. [Google Scholar] [CrossRef]
- Cao, Z.; Xu, Y.; Guo, F.; Chen, X.; Ji, J.; Xu, H.; He, J.; Yu, Y.; Sun, Y.; Lu, X.; et al. FASN Protein Overexpression Indicates Poor Biochemical Recurrence-Free Survival in Prostate Cancer. Dis. Markers 2020, 2020, 3904947. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. ONCOMINE: A Cancer Microarray Database and Integrated Data-Mining Platform. Neoplasia 2004, 6, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijón, M.A.; Colton, T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glodé, L.M.; Robert, H.; et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol. Cancer Ther. 2014, 13, 2361–2371. [Google Scholar] [CrossRef] [Green Version]
- Mitra, R.; Le, T.T.; Gorjala, P.; Goodman, O.B. Positive regulation of prostate cancer cell growth by lipid droplet forming and processing enzymes DGAT1 and ABHD5. BMC Cancer 2017, 17, 631. [Google Scholar] [CrossRef]
- Luo, X.; Cheng, C.; Tan, Z.; Li, N.; Tang, M.; Yang, L.; Cao, Y. Emerging roles of lipid metabolism in cancer metastasis. Mol. Cancer 2017, 16, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Morrissey, C.; Barrett, B.; Corey, E.; Ylaya, K.; Hewitt, S.; Fang, L.; Tillman, H.; Lake, R.; Vessella, R.; et al. AR-Regulated TWEAK-FN14 Pathway Promotes Prostate Cancer Bone Metastasis. Cancer Res. 2014, 74, 4306–4317. [Google Scholar] [CrossRef] [Green Version]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, T.; Su, Q.; Xu, S.; Deng, J.; Zhou, S.; Zhuang, Y.; Huang, Y.; He, C.; He, S.; Peng, M.; et al. Down-regulation of PKM2 decreases FASN expression in bladder cancer cells through AKT/mTOR/SREBP-1c axis. J. Cell. Physiol. 2019, 234, 3088–3104. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Götting, I.; Jendrossek, V.; Matschke, J. A new twist in protein kinase b/akt signaling: Role of altered cancer cell metabolism in akt-mediated therapy resistance. Int. J. Mol. Sci. 2020, 21, 8563. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krycer, J.R.; Sharpe, L.J.; Luu, W.; Brown, A.J. The Akt-SREBP nexus: Cell signaling meets lipid metabolism. Trends Endocrinol. Metab. 2010, 21, 268–276. [Google Scholar] [CrossRef]
- Namekawa, T.; Ikeda, K.; Horie-Inoue, K.; Inoue, S. Application of Prostate Cancer Models for Preclinical Study: Advantages and Limitations of Cell Lines, Patient-Derived Xenografts, and Three-Dimensional Culture of Patient-Derived Cells. Cells 2019, 8, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Patients | Controls | PCa Patients | p-Value |
|---|---|---|---|
| n = 159 | n = 76 | ||
| Mean ± SD | Mean ± SD | ||
| Anthropometric Parameters | |||
| Age (years) | 63.54 ± 7.26 | 63.51 ± 6.66 | 0.953 |
| BMI (kg/m2) | 28.60 ± 2.31 | 28.24 ± 3.97 | 0.067 |
| ISUP GG (n/%) | - | - | - |
| Low Risk (I and II) | - | 48 (63%) | - |
| High Risk (Group III, IV, V) | - | 28 (37%) | - |
| Glycemic profile | |||
| Glucose (mmol/L) | 5.20 ± 0.53 | 6.03 ± 1.35 | <0.001 |
| Insulin (pmol/L) | 30.60 ± 19.93 | 94.53 ± 55.33 | <0.001 |
| HOMA-IR | 1.04 ± 0.74 | 3.75 ± 2.63 | <0.001 |
| Diabetes (n/%) | 0 (0%) | 8 (10.6%) | <0.001 |
| Lipid profile | |||
| Cholesterol (mmol/L) | 5.17 ± 0.99 | 4.96 ± 1.04 | 0.233 |
| HDL cholesterol (mmol/L) | 1.22 ± 0.28 | 1.47 ± 0.66 | 0.001 |
| LDL cholesterol (mmol/L) | 2.87 ± 0.71 | 3.07 ± 1.23 | 0.185 |
| Triglycerides (mmol/L) | 1.50 ± 0.90 | 1.49 ± 0.81 | 0.925 |
| Hepatic profile | |||
| GGT (µkat/L) | 0.64 ± 0.67 | 0.70 ± 0.79 | 0.404 |
| Renal profile | |||
| Uric acid (µmol/L) | 366.46 ± 78.47 | 413.27 ± 379.57 | 0.527 |
| Creatinine (μmol/L) | 76.74 ± 13.31 | 70.98 ± 14.21 | <0.001 |
| Total PSA (μg/L) | 1.64 ± 2.14 | 10.06 ± 7.73 | <0.001 |
| sTWEAK | 1583.13 ± 1348.60 | 1197.20 ± 579.57 | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altuna-Coy, A.; Ruiz-Plazas, X.; Alves-Santiago, M.; Segarra-Tomás, J.; Chacón, M.R. Serum Levels of the Cytokine TWEAK Are Associated with Metabolic Status in Patients with Prostate Cancer and Modulate Cancer Cell Lipid Metabolism In Vitro. Cancers 2021, 13, 4688. https://doi.org/10.3390/cancers13184688
Altuna-Coy A, Ruiz-Plazas X, Alves-Santiago M, Segarra-Tomás J, Chacón MR. Serum Levels of the Cytokine TWEAK Are Associated with Metabolic Status in Patients with Prostate Cancer and Modulate Cancer Cell Lipid Metabolism In Vitro. Cancers. 2021; 13(18):4688. https://doi.org/10.3390/cancers13184688
Chicago/Turabian StyleAltuna-Coy, Antonio, Xavier Ruiz-Plazas, Marta Alves-Santiago, José Segarra-Tomás, and Matilde R. Chacón. 2021. "Serum Levels of the Cytokine TWEAK Are Associated with Metabolic Status in Patients with Prostate Cancer and Modulate Cancer Cell Lipid Metabolism In Vitro" Cancers 13, no. 18: 4688. https://doi.org/10.3390/cancers13184688