
Implications of Intratumor Heterogeneity on Consensus Molecular Subtype (CMS) in Colorectal Cancer

 ,
,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Collection of Gene Expression Profiles of the Bulk Tumor Samples
2.2. Sample Preparation and Data Processing for Single-Cell RNA Sequencing
2.3. Consensus Molecular Subtyping of the Bulk Tumor Samples
2.4. Single-Sample Gene-Set Enrichment Analyses
2.5. Identification of CMS Specific-Marker Gene Sets
2.6. CMS Assignments to Single Cells
2.7. Software and Tools
3. Results
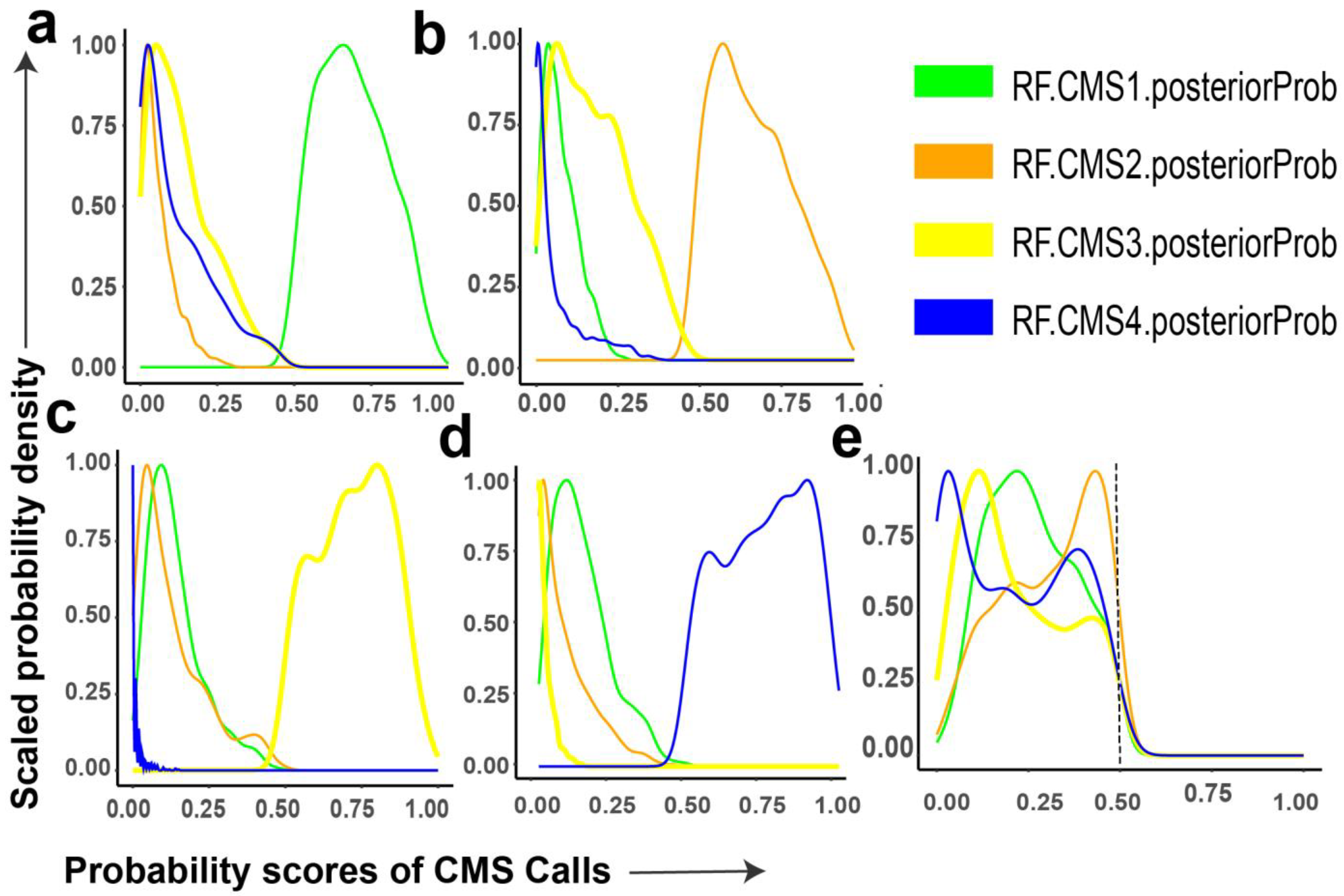
3.1. Probability Distributions of CMS Calls in Bulk Tumor Samples
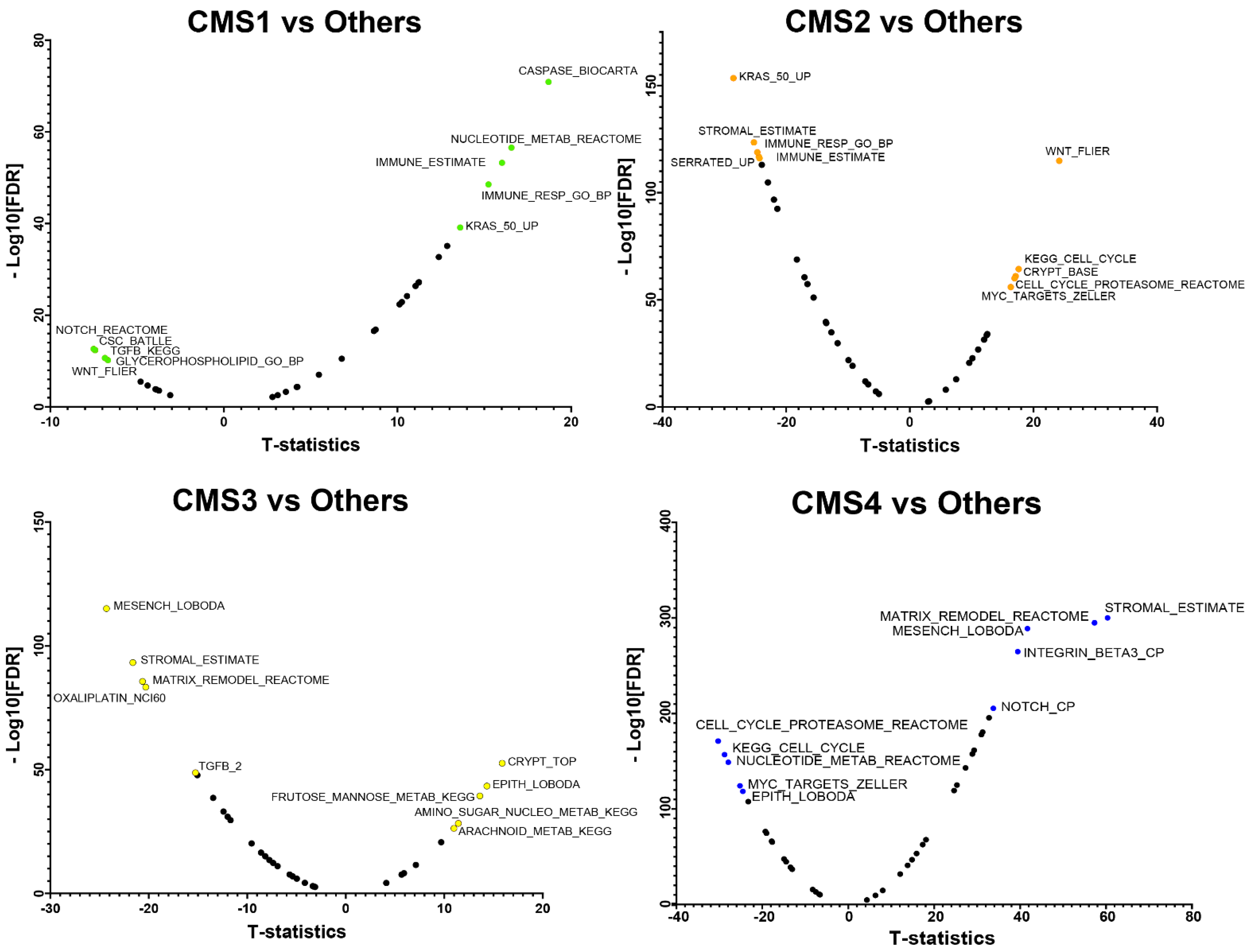
3.2. CMS Specific-Marker Gene Sets
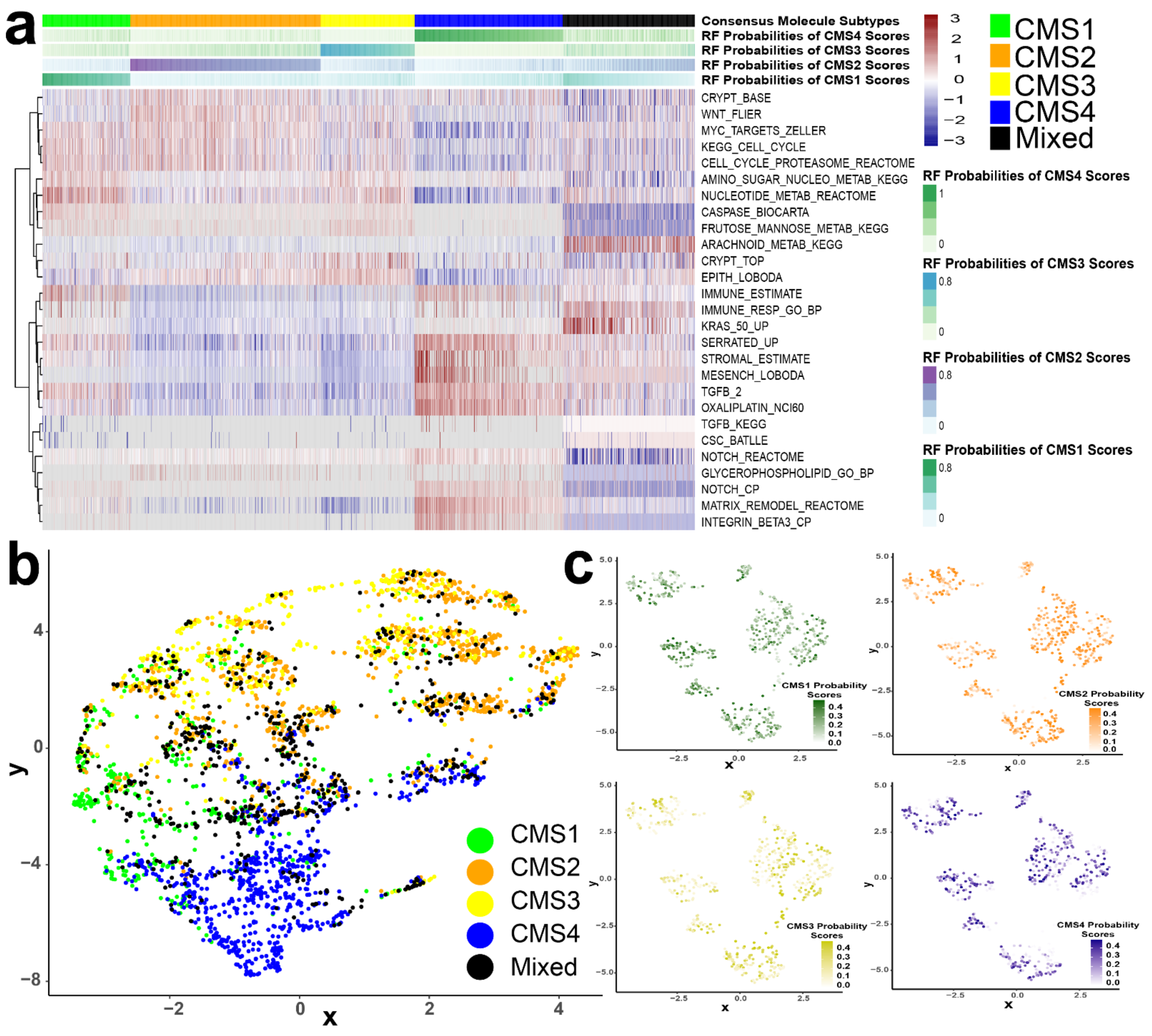
3.3. CMS Represents the Transcriptomic Inter-Tumor Heterogeneity of Colorectal Cancer
3.4. CMS Assignment for Single Cells
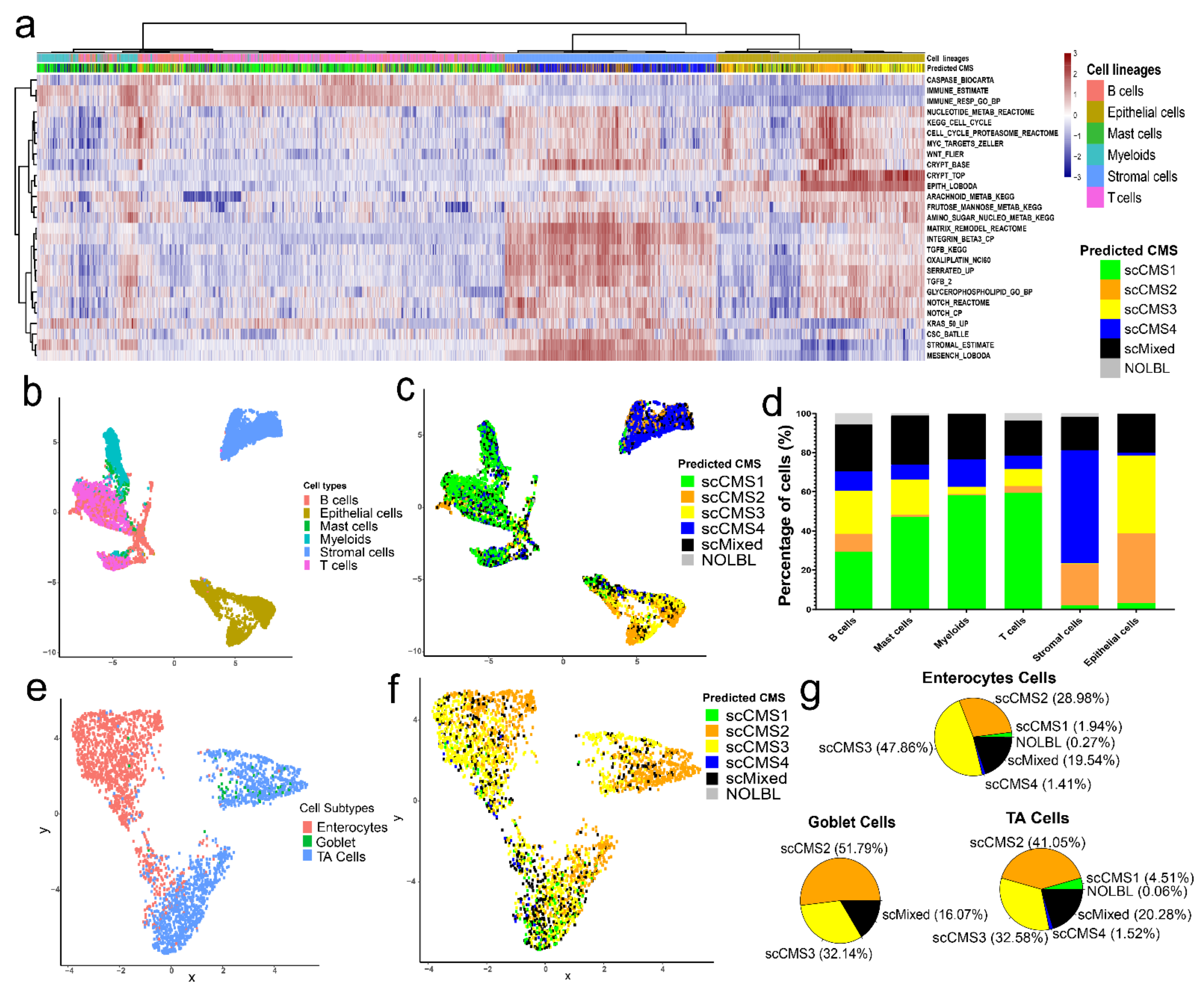
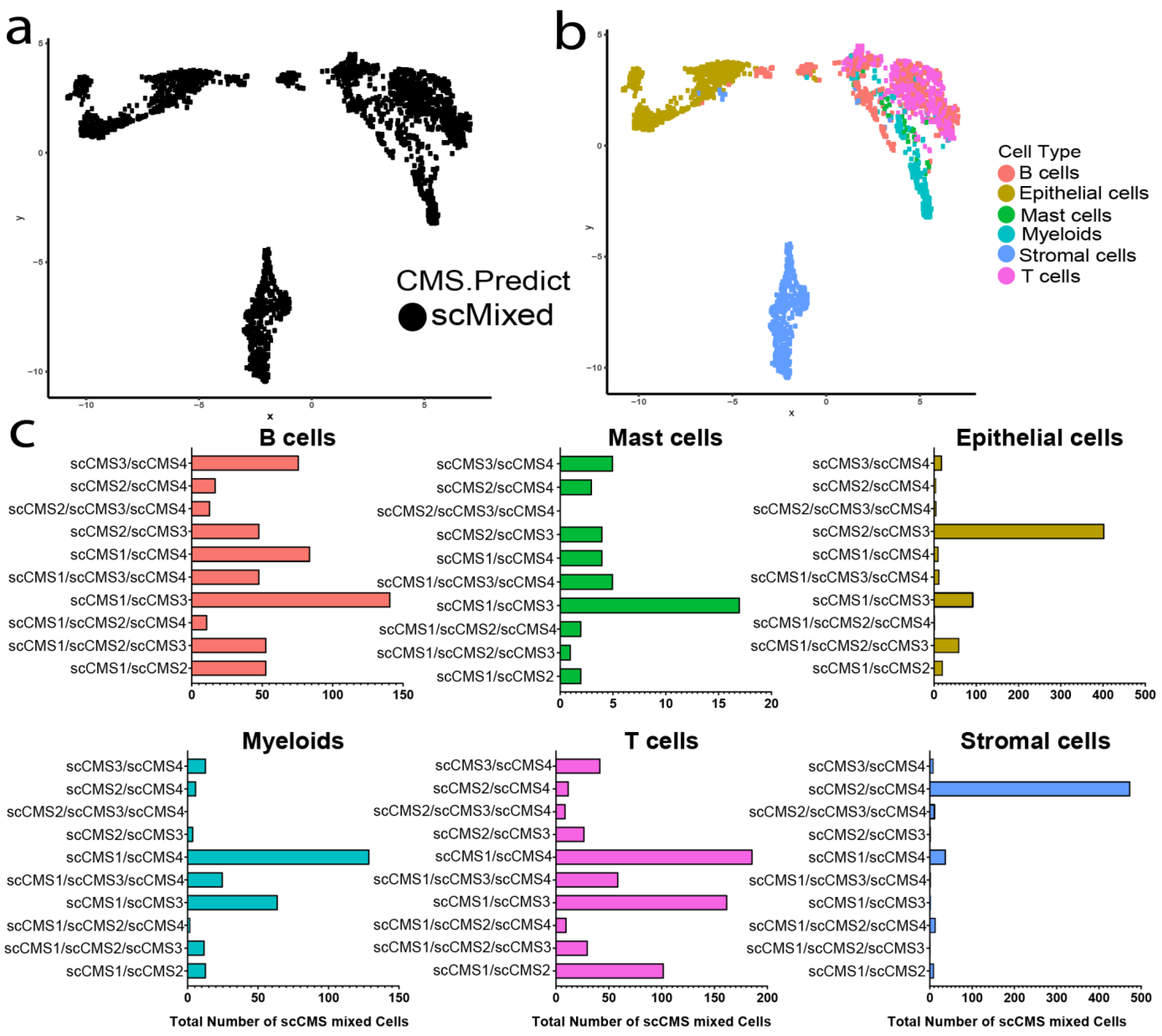
3.5. Distributions of scCMSs Vary among Cell Lineages
3.6. scCMSs Are Associated with Transcriptomic Intra-Tumor Heterogeneity in CRC Tissues
3.7. Frequency Distributions of Single Cells CMSs Do Not Correlate with CMS of Matched Bulk Tumor
3.8. Exploring the Associations of CMS and Intra-Tumor Heterogeneity in an Independent Dataset
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Biller, L.H.; Schrag, D. Diagnosis and Treatment of Metastatic Colorectal Cancer: A Review. JAMA 2021, 325, 669–685. [Google Scholar] [CrossRef]
- Bien, J.; Lin, A. A Review of the Diagnosis and Treatment of Metastatic Colorectal Cancer. JAMA 2021, 325, 2404–2405. [Google Scholar] [CrossRef] [PubMed]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Willauer, A.N.; Liu, Y.; Pereira, A.A.L.; Lam, M.; Morris, J.S.; Raghav, K.P.S.; Morris, V.K.; Menter, D.; Broaddus, R.; Meric-Bernstam, F.; et al. Clinical and molecular characterization of early-onset colorectal cancer. Cancer 2019, 125, 2002–2010. [Google Scholar] [CrossRef] [PubMed]
- Di Nicolantonio, F.; Vitiello, P.P.; Marsoni, S.; Siena, S.; Tabernero, J.; Trusolino, L.; Bernards, R.; Bardelli, A. Precision oncology in metastatic colorectal cancer—From biology to medicine. Nat. Rev. Clin. Oncol. 2021, 18, 506–525. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.C.; Yeh, Y.M.; Lin, B.W.; Lin, S.C.; Chan, R.H.; Chen, P.C.; Shen, M.R. Intratumor Heterogeneity of MYO18A and FBXW7 Variants Impact the Clinical Outcome of Stage III Colorectal Cancer. Front. Oncol. 2020, 10, 588557. [Google Scholar] [CrossRef]
- Feng, M.; Zhao, Z.; Yang, M.; Ji, J.; Zhu, D. T-cell-based immunotherapy in colorectal cancer. Cancer Lett. 2021, 498, 201–209. [Google Scholar] [CrossRef]
- Vitale, I.; Shema, E.; Loi, S.; Galluzzi, L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat. Med. 2021, 27, 212–224. [Google Scholar] [CrossRef]
- Larionova, I.; Tuguzbaeva, G.; Ponomaryova, A.; Stakheyeva, M.; Cherdyntseva, N.; Pavlov, V.; Choinzonov, E.; Kzhyshkowska, J. Tumor-Associated Macrophages in Human Breast, Colorectal, Lung, Ovarian and Prostate Cancers. Front. Oncol. 2020, 10, 566511. [Google Scholar] [CrossRef] [PubMed]
- Fontana, E.; Nyamundanda, G.; Cunningham, D.; Tu, D.; Cheang, M.C.U.; Jonker, D.J.; Siu, L.L.; Sclafani, F.; Eason, K.; Ragulan, C.; et al. Intratumoral Transcriptome Heterogeneity Is Associated with Patient Prognosis and Sidedness in Patients with Colorectal Cancer Treated With Anti-EGFR Therapy From the CO.20 Trial. JCO Precis. Oncol. 2020, 4, 1152–1162. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Yu, T.; Zhao, X.; Gao, X.; Zhao, Y.; Liu, G. Intratumor heterogeneity: A new perspective on colorectal cancer research. Cancer Med. 2020, 9, 7637–7645. [Google Scholar] [CrossRef] [PubMed]
- Adua, D.; Di Fabio, F.; Ercolani, G.; Fiorentino, M.; Gruppioni, E.; Altimari, A.; Rojas Limpe, F.L.; Normanno, N.; Pinna, A.D.; Pinto, C. Heterogeneity in the colorectal primary tumor and the synchronous resected liver metastases prior to and after treatment with an anti-EGFR monoclonal antibody. Mol. Clin. Oncol. 2017, 7, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Joung, J.G.; Oh, B.Y.; Hong, H.K.; Al-Khalidi, H.; Al-Alem, F.; Lee, H.O.; Bae, J.S.; Kim, J.; Cha, H.U.; Alotaibi, M.; et al. Tumor Heterogeneity Predicts Metastatic Potential in Colorectal Cancer. Clin. Cancer Res. 2017, 23, 7209–7216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, H.; Yamanaka, T.; Sakai, D.; Muro, K.; Yamazaki, K.; Nakata, S.; Kimura, H.; Ruff, P.; Kim, T.W.; Peeters, M.; et al. Efficacy of Panitumumab and Cetuximab in Patients with Colorectal Cancer Previously Treated with Bevacizumab; a Combined Analysis of Individual Patient Data from ASPECCT and WJOG6510G. Cancers 2020, 12, 1715. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, S.; Sung, J.S.; Chung, H.J.; Lim, A.R.; Kim, J.W.; Choi, Y.J.; Park, K.H.; Kim, Y.H. Clinical Application of Targeted Deep Sequencing in Metastatic Colorectal Cancer Patients: Actionable Genomic Alteration in K-MASTER project. Cancer Res. Treat. 2020, 53, 123–130. [Google Scholar] [CrossRef]
- Fontana, E.; Eason, K.; Cervantes, A.; Salazar, R.; Sadanandam, A. Context matters-consensus molecular subtypes of colorectal cancer as biomarkers for clinical trials. Ann. Oncol. 2019, 30, 520–527. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 268. [Google Scholar] [CrossRef]
- Lee, M.S.; Menter, D.G.; Kopetz, S. Right Versus Left Colon Cancer Biology: Integrating the Consensus Molecular Subtypes. J. Natl. Compr. Cancer Netw. 2017, 15, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Menter, D.G.; Davis, J.S.; Broom, B.M.; Overman, M.J.; Morris, J.; Kopetz, S. Back to the Colorectal Cancer Consensus Molecular Subtype Future. Curr. Gastroenterol. Rep. 2019, 21, 5. [Google Scholar] [CrossRef]
- Kopetz, S.; Guthrie, K.A.; Morris, V.K.; Lenz, H.J.; Magliocco, A.M.; Maru, D.; Yan, Y.; Lanman, R.; Manyam, G.; Hong, D.S.; et al. Randomized Trial of Irinotecan and Cetuximab with or without Vemurafenib in BRAF-Mutant Metastatic Colorectal Cancer (SWOG S1406). J. Clin. Oncol. 2021, 39, 285–294. [Google Scholar] [CrossRef]
- Dunne, P.D.; McArt, D.G.; Bradley, C.A.; O’Reilly, P.G.; Barrett, H.L.; Cummins, R.; O’Grady, T.; Arthur, K.; Loughrey, M.B.; Allen, W.L.; et al. Challenging the Cancer Molecular Stratification Dogma: Intratumoral Heterogeneity Undermines Consensus Molecular Subtypes and Potential Diagnostic Value in Colorectal Cancer. Clin. Cancer Res. 2016, 22, 4095–4104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becht, E.; de Reynies, A.; Giraldo, N.A.; Pilati, C.; Buttard, B.; Lacroix, L.; Selves, J.; Sautes-Fridman, C.; Laurent-Puig, P.; Fridman, W.H. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clin. Cancer Res. 2016, 22, 4057–4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Tian, X.; Ji, L.; Zhang, X.; Cao, Y.; Shen, C.; Hu, Y.; Wong, J.W.H.; Fang, J.Y.; Hong, J.; et al. A tumor microenvironment-specific gene expression signature predicts chemotherapy resistance in colorectal cancer patients. NPJ Precis. Oncol. 2021, 5, 7. [Google Scholar] [CrossRef]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017, 49, 708–718. [Google Scholar] [CrossRef]
- Katsuta, E.; Rashid, O.M.; Takabe, K. Clinical relevance of tumor microenvironment: Immune cells, vessels, and mouse models. Hum. Cell 2020, 33, 930–937. [Google Scholar] [CrossRef]
- Gallo, G.; Vescio, G.; De Paola, G.; Sammarco, G. Therapeutic Targets and Tumor Microenvironment in Colorectal Cancer. J. Clin. Med. 2021, 10, 2295. [Google Scholar] [CrossRef]
- Thanki, K.; Nicholls, M.E.; Gajjar, A.; Senagore, A.J.; Qiu, S.; Szabo, C.; Hellmich, M.R.; Chao, C. Consensus Molecular Subtypes of Colorectal Cancer and their Clinical Implications. Int. Biol. Biomed. J. 2017, 3, 105–111. [Google Scholar] [PubMed]
- Soldevilla, B.; Carretero-Puche, C.; Gomez-Lopez, G.; Al-Shahrour, F.; Riesco, M.C.; Gil-Calderon, B.; Alvarez-Vallina, L.; Espinosa-Olarte, P.; Gomez-Esteves, G.; Rubio-Cuesta, B.; et al. The correlation between immune subtypes and consensus molecular subtypes in colorectal cancer identifies novel tumour microenvironment profiles, with prognostic and therapeutic implications. Eur. J. Cancer 2019, 123, 118–129. [Google Scholar] [CrossRef] [Green Version]
- Sveen, A.; Bruun, J.; Eide, P.W.; Eilertsen, I.A.; Ramirez, L.; Murumagi, A.; Arjama, M.; Danielsen, S.A.; Kryeziu, K.; Elez, E.; et al. Colorectal Cancer Consensus Molecular Subtypes Translated to Preclinical Models Uncover Potentially Targetable Cancer Cell Dependencies. Clin. Cancer Res. 2018, 24, 794–806. [Google Scholar] [CrossRef] [Green Version]
- Vitiello, P.P.; Martini, G.; Mele, L.; Giunta, E.F.; De Falco, V.; Ciardiello, D.; Belli, V.; Cardone, C.; Matrone, N.; Poliero, L.; et al. Vulnerability to low-dose combination of irinotecan and niraparib in ATM-mutated colorectal cancer. J. Exp. Clin. Cancer Res. 2021, 40, 15. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, T.; Kandimalla, R.; Ishikawa, T.; Takahashi, N.; Yamada, Y.; Yasuno, M.; Kinugasa, Y.; Hansen, T.F.; Fakih, M.; Uetake, H.; et al. A novel mesenchymal-associated transcriptomic signature for risk-stratification and therapeutic response prediction in colorectal cancer. Int. J. Cancer 2020, 147, 3250–3261. [Google Scholar] [CrossRef] [PubMed]
- Koulis, C.; Yap, R.; Engel, R.; Jarde, T.; Wilkins, S.; Solon, G.; Shapiro, J.D.; Abud, H.; McMurrick, P. Personalized Medicine-Current and Emerging Predictive and Prognostic Biomarkers in Colorectal Cancer. Cancers 2020, 12, 812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stintzing, S.; Wirapati, P.; Lenz, H.J.; Neureiter, D.; Fischer von Weikersthal, L.; Decker, T.; Kiani, A.; Kaiser, F.; Al-Batran, S.; Heintges, T.; et al. Consensus molecular subgroups (CMS) of colorectal cancer (CRC) and first-line efficacy of FOLFIRI plus cetuximab or bevacizumab in the FIRE3 (AIO KRK-0306) trial. Ann. Oncol. 2019, 30, 1796–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wlodarczyk, M.; Wlodarczyk, J.; Siwinski, P.; Sobolewska-Wlodarczyk, A.; Fichna, J. Genetic Molecular Subtypes in Optimizing Personalized Therapy for Metastatic Colorectal Cancer. Curr. Drug Targets 2018, 19, 1731–1737. [Google Scholar] [CrossRef]
- Tieng, F.Y.F.; Baharudin, R.; Abu, N.; Mohd Yunos, R.I.; Lee, L.H.; Ab Mutalib, N.S. Single Cell Transcriptome in Colorectal Cancer-Current Updates on Its Application in Metastasis, Chemoresistance and the Roles of Circulating Tumor Cells. Front. Pharmacol. 2020, 11, 135. [Google Scholar] [CrossRef] [Green Version]
- Kyrochristos, I.D.; Roukos, D.H. Comprehensive intra-individual genomic and transcriptional heterogeneity: Evidence-based Colorectal Cancer Precision Medicine. Cancer Treat. Rev. 2019, 80, 101894. [Google Scholar] [CrossRef]
- Eide, P.W.; Bruun, J.; Lothe, R.A.; Sveen, A. CMScaller: An R package for consensus molecular subtyping of colorectal cancer pre-clinical models. Sci. Rep. 2017, 7, 16618. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.O.; Hong, Y.; Etlioglu, H.E.; Cho, Y.B.; Pomella, V.; Van den Bosch, B.; Vanhecke, J.; Verbandt, S.; Hong, H.; Min, J.W.; et al. Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat. Genet. 2020, 52, 594–603. [Google Scholar] [CrossRef]
- Gao, R.; Bai, S.; Henderson, Y.C.; Lin, Y.; Schalck, A.; Yan, Y.; Kumar, T.; Hu, M.; Sei, E.; Davis, A.; et al. Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat. Biotechnol. 2021, 39, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Smillie, C.S.; Biton, M.; Ordovas-Montanes, J.; Sullivan, K.M.; Burgin, G.; Graham, D.B.; Herbst, R.H.; Rogel, N.; Slyper, M.; Waldman, J.; et al. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 2019, 178, 714–730.e22. [Google Scholar] [CrossRef] [PubMed]
- Krug, K.; Mertins, P.; Zhang, B.; Hornbeck, P.; Raju, R.; Ahmad, R.; Szucs, M.; Mundt, F.; Forestier, D.; Jane-Valbuena, J.; et al. A Curated Resource for Phosphosite-specific Signature Analysis. Mol. Cell. Proteom. 2019, 18, 576–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakdash, J.Z.; Marusich, L.R. Repeated Measures Correlation. Front. Psychol. 2017, 8, 456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooi, J.K.; Wirapati, P.; Asher, R.; Lee, C.K.; Savas, P.; Price, T.J.; Townsend, A.; Hardingham, J.; Buchanan, D.; Williams, D.; et al. The prognostic impact of consensus molecular subtypes (CMS) and its predictive effects for bevacizumab benefit in metastatic colorectal cancer: Molecular analysis of the AGITG MAX clinical trial. Ann. Oncol. 2018, 29, 2240–2246. [Google Scholar] [CrossRef]
- Koenitzer, J.R.; Wu, H.; Atkinson, J.J.; Brody, S.L.; Humphreys, B.D. Single-Nucleus RNA-Sequencing Profiling of Mouse Lung. Reduced Dissociation Bias and Improved Rare Cell-Type Detection Compared with Single-Cell RNA Sequencing. Am. J. Respir. Cell. Mol. Biol. 2020, 63, 739–747. [Google Scholar] [CrossRef]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef]
- Denisenko, E.; Guo, B.B.; Jones, M.; Hou, R.; de Kock, L.; Lassmann, T.; Poppe, D.; Clement, O.; Simmons, R.K.; Lister, R.; et al. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol. 2020, 21, 130. [Google Scholar] [CrossRef]
- O’Flanagan, C.H.; Campbell, K.R.; Zhang, A.W.; Kabeer, F.; Lim, J.L.P.; Biele, J.; Eirew, P.; Lai, D.; McPherson, A.; Kong, E.; et al. Dissociation of solid tumor tissues with cold active protease for single-cell RNA-seq minimizes conserved collagenase-associated stress responses. Genome Biol. 2019, 20, 210. [Google Scholar] [CrossRef] [Green Version]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef]
- Buechler, C.; Ritter, M.; Orsó, E.; Langmann, T.; Klucken, J.; Schmitz, G. Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro- and antiinflammatory stimuli. J. Leukoc. Biol. 2000, 67, 97–103. [Google Scholar] [CrossRef]
- Loke, P.n.; Allison, J.P. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc. Natl. Acad. Sci. USA 2003, 100, 5336–5341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slyper, M.; Porter, C.B.M.; Ashenberg, O.; Waldman, J.; Drokhlyansky, E.; Wakiro, I.; Smillie, C.; Smith-Rosario, G.; Wu, J.; Dionne, D.; et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med. 2020, 26, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Mereu, E.; Lafzi, A.; Moutinho, C.; Ziegenhain, C.; McCarthy, D.J.; Alvarez-Varela, A.; Batlle, E.; Sagar; Grun, D.; Lau, J.K.; et al. Benchmarking single-cell RNA-sequencing protocols for cell atlas projects. Nat. Biotechnol. 2020, 38, 747–755. [Google Scholar] [CrossRef]
- Massoni-Badosa, R.; Iacono, G.; Moutinho, C.; Kulis, M.; Palau, N.; Marchese, D.; Rodriguez-Ubreva, J.; Ballestar, E.; Rodriguez-Esteban, G.; Marsal, S.; et al. Sampling time-dependent artifacts in single-cell genomics studies. Genome Biol. 2020, 21, 112. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Wang, J.R.; Ji, S.; Yang, P.; Montierth, M.D.; Guo, S.; Shen, J.P.; Zhao, X.; Chen, J.; Lee, J.J.; et al. Tumor cell total mRNA expression shapes the molecular and clinical phenotype of cancer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Rebersek, M. Consensus molecular subtypes (CMS) in metastatic colorectal cancer—Personalized medicine decision. Radiol. Oncol. 2020, 54, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Marisa, L.; Blum, Y.; Taieb, J.; Ayadi, M.; Pilati, C.; Le Malicot, K.; Lepage, C.; Salazar, R.; Aust, D.; Duval, A.; et al. Intratumor CMS Heterogeneity Impacts Patient Prognosis in Localized Colon Cancer. Clin. Cancer Res. 2021, 27, 4768–4780. [Google Scholar] [CrossRef]
- Sawayama, H.; Miyamoto, Y.; Ogawa, K.; Yoshida, N.; Baba, H. Investigation of colorectal cancer in accordance with consensus molecular subtype classification. Ann. Gastroenterol. Surg. 2020, 4, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Arnadottir, S.S.; Mattesen, T.B.; Vang, S.; Madsen, M.R.; Madsen, A.H.; Birkbak, N.J.; Bramsen, J.B.; Andersen, C.L. Transcriptomic and proteomic intra-tumor heterogeneity of colorectal cancer varies depending on tumor location within the colorectum. PLoS ONE 2020, 15, e0241148. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.; Berral-Gonzalez, A.; Lopez-Cade, I.; Galindo-Pumarino, C.; Bueno-Fortes, S.; Martin-Merino, M.; Carrato, A.; Ocana, A.; De La Pinta, C.; Lopez-Alfonso, A.; et al. Cancer-associated fibroblast-derived gene signatures determine prognosis in colon cancer patients. Mol. Cancer 2021, 20, 73. [Google Scholar] [CrossRef] [PubMed]
- Isella, C.; Terrasi, A.; Bellomo, S.E.; Petti, C.; Galatola, G.; Muratore, A.; Mellano, A.; Senetta, R.; Cassenti, A.; Sonetto, C.; et al. Stromal contribution to the colorectal cancer transcriptome. Nat. Genet. 2015, 47, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Khaliq, A.M.; Kurt, Z.; Grunvald, M.W.; Erdogan, C.; Turgut, S.S.; Rand, T.; Khare, S.; Borgia, J.A.; Hayden, D.M.; Pappas, S.G.; et al. Redefining colorectal cancer classification and clinical stratification through a single-cell atlas. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gieniec, K.A.; Butler, L.M.; Worthley, D.L.; Woods, S.L. Cancer-associated fibroblasts-heroes or villains? Br. J. Cancer 2019, 121, 293–302. [Google Scholar] [CrossRef]
- Trinh, A.; Trumpi, K.; De Sousa, E.M.F.; Wang, X.; de Jong, J.H.; Fessler, E.; Kuppen, P.J.; Reimers, M.S.; Swets, M.; Koopman, M.; et al. Practical and Robust Identification of Molecular Subtypes in Colorectal Cancer by Immunohistochemistry. Clin. Cancer Res. 2017, 23, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Ten Hoorn, S.; Trinh, A.; de Jong, J.; Koens, L.; Vermeulen, L. Classification of Colorectal Cancer in Molecular Subtypes by Immunohistochemistry. Methods Mol. Biol. 2018, 1765, 179–191. [Google Scholar] [CrossRef]
- Buechler, S.A.; Stephens, M.T.; Hummon, A.B.; Ludwig, K.; Cannon, E.; Carter, T.C.; Resnick, J.; Gokmen-Polar, Y.; Badve, S.S. ColoType: A forty gene signature for consensus molecular subtyping of colorectal cancer tumors using whole-genome assay or targeted RNA-sequencing. Sci. Rep. 2020, 10, 12123. [Google Scholar] [CrossRef]
- Chen, K.Y.; Srinivasan, T.; Lin, C.; Tung, K.L.; Gao, Z.; Hsu, D.S.; Lipkin, S.M.; Shen, X. Single-Cell Transcriptomics Reveals Heterogeneity and Drug Response of Human Colorectal Cancer Organoids. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2018, 2018, 2378–2381. [Google Scholar] [CrossRef]
- Angell, H.K.; Bruni, D.; Barrett, J.C.; Herbst, R.; Galon, J. The Immunoscore: Colon Cancer and Beyond. Clin. Cancer Res. 2020, 26, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.S.; Luthra, R.; Liu, Y.; Duose, D.Y.; Lee, W.; Reddy, N.G.; Windham, J.; Chen, H.; Tong, Z.; Zhang, B.; et al. Development and Validation of a Gene Signature Classifier for Consensus Molecular Subtyping of Colorectal Carcinoma in a CLIA-Certified Setting. Clin. Cancer Res. 2021, 27, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.E. The first five years of single-cell cancer genomics and beyond. Genome Res. 2015, 25, 1499–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Li, T.; Liu, F.; Chen, Y.; Yao, J.; Li, Z.; Huang, Y.; Wang, J. Comparative Analysis of Droplet-Based Ultra-High-Throughput Single-Cell RNA-Seq Systems. Mol. Cell 2019, 73, 130–142.e5. [Google Scholar] [CrossRef] [Green Version]
- Ozturk, K.; Dow, M.; Carlin, D.E.; Bejar, R.; Carter, H. The Emerging Potential for Network Analysis to Inform Precision Cancer Medicine. J. Mol. Biol. 2018, 430, 2875–2899. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.P.; Ideker, T. Synthetic Lethal Networks for Precision Oncology: Promises and Pitfalls. J. Mol. Biol. 2018, 430, 2900–2912. [Google Scholar] [CrossRef]
- Jew, B.; Alvarez, M.; Rahmani, E.; Miao, Z.; Ko, A.; Garske, K.M.; Sul, J.H.; Pietilainen, K.H.; Pajukanta, P.; Halperin, E. Accurate estimation of cell composition in bulk expression through robust integration of single-cell information. Nat. Commun. 2020, 11, 1971. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Lenz, H.J.; Ou, F.S.; Venook, A.P.; Hochster, H.S.; Niedzwiecki, D.; Goldberg, R.M.; Mayer, R.J.; Bertagnolli, M.M.; Blanke, C.D.; Zemla, T.; et al. Impact of Consensus Molecular Subtype on Survival in Patients With Metastatic Colorectal Cancer: Results From CALGB/SWOG 80405 (Alliance). J. Clin. Oncol. 2019, 37, 1876–1885. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chowdhury, S.; Hofree, M.; Lin, K.; Maru, D.; Kopetz, S.; Shen, J.P. Implications of Intratumor Heterogeneity on Consensus Molecular Subtype (CMS) in Colorectal Cancer. Cancers 2021, 13, 4923. https://doi.org/10.3390/cancers13194923
Chowdhury S, Hofree M, Lin K, Maru D, Kopetz S, Shen JP. Implications of Intratumor Heterogeneity on Consensus Molecular Subtype (CMS) in Colorectal Cancer. Cancers. 2021; 13(19):4923. https://doi.org/10.3390/cancers13194923
Chicago/Turabian StyleChowdhury, Saikat, Matan Hofree, Kangyu Lin, Dipen Maru, Scott Kopetz, and John Paul Shen. 2021. "Implications of Intratumor Heterogeneity on Consensus Molecular Subtype (CMS) in Colorectal Cancer" Cancers 13, no. 19: 4923. https://doi.org/10.3390/cancers13194923
APA StyleChowdhury, S., Hofree, M., Lin, K., Maru, D., Kopetz, S., & Shen, J. P. (2021). Implications of Intratumor Heterogeneity on Consensus Molecular Subtype (CMS) in Colorectal Cancer. Cancers, 13(19), 4923. https://doi.org/10.3390/cancers13194923