
Challenges and Prospects for Designer T and NK Cells in Glioblastoma Immunotherapy


Abstract
:Simple Summary
Abstract
1. Glioblastoma—The Current Setting
2. T Cell Patrols with a License to Kill
3. Past Experiences and Thinking forward with CAR T Cell Therapy
4. Challenging Solid Brain Tumours with Natural Killers
In Vitro and In Vivo Clinical Studies for Designer NK Cells in GBM
5. Picking a Suitable Target for GBM Immunotherapy
5.1. Growth Factor Receptors as Targets
5.2. Interleukin-13 Receptor Alpha 2
5.3. Co-Stimulator B7H3
5.4. Multitarget Approach with NKG2D
6. Genetic Modification as an Aid to Existing Therapies
7. Experimental Targets for GBM Therapy
7.1. Synergistic Effects Targeting Chondroitin Sulfate Proteoglycan 4 (CSPG4)
7.2. The Pan-Population Antigen MR1
7.3. Targeting Extracellular ATP
7.4. Clinical Value of 2-HG in IDH-Mutant Gliomas
8. Developing Combination Approaches
9. Mode of Delivery—Anti-Tumour Effect vs. Adverse Effects
10. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organisation International Agency for Research on Cancer (IARC). GLOBOCAN 2020: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2020. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/39-All-cancers-fact-sheet.pdf (accessed on 27 September 2021).
- World Health Organisation International Agency for Research on Cancer. World Cancer Report: Cancer Research for Cancer Prevention; World Health Organisation: Regional Office For Europe: Copenhagen, Denmark, 2020. [Google Scholar]
- Dyba, T.; Randi, G.; Bray, F.; Martos, C.; Giusti, F.; Nicholson, N.; Gavin, A.; Flego, M.; Neamtiu, L.; Dimitrova, N.; et al. The European cancer burden in 2020: Incidence and mortality estimates for 40 countries and 25 major cancers. Eur. J. Cancer 2021, 157, 308–347. [Google Scholar] [CrossRef]
- Dolecek, T.A.; Propp, J.M.; Stroup, N.E.; Kruchko, C. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumours Diagnosed in the United States in 2005-2009. Neuro Oncol. 2012, Suppl. 5. [Google Scholar] [CrossRef]
- Tumours, W.H.O.C.o. Tumours of the Nervous System: Pathology and Genetics; IARCpress: Lyon, France, 2000. [Google Scholar]
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.W.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [Green Version]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavare, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanai, N.; Polley, M.Y.; McDermott, M.W.; Parsa, A.T.; Berger, M.S. An extent of resection threshold for newly diagnosed glioblastomas. J. Neurosurg. 2011, 115, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stummer, W.; Pichlmeier, U.; Meinel, T.; Wiestler, O.D.; Zanella, F.; Reulen, H.J. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: A randomised controlled multicentre phase III trial. Lancet Oncol. 2006, 7, 392–401. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.B.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Blakstad, H.J.B.; Rahman, M.A.; Arnesen, V.S.; Brandal, P.; Lie, S.A.; Chekenya, M.; Goplen, D. Survival in a consecutive series of 467 recurrent glioblastoma patients: Impact of prognostic factors and treatment at two independent institutions. Acta Oncologica 2021. submitted. [Google Scholar]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. NeuroOncol. 2013, 15. [Google Scholar] [CrossRef] [Green Version]
- Smoll, N.R.; Schaller, K.; Gautschi, O.P. Long-term survival of patients with glioblastoma multiforme (GBM). J. Clin. Neurosci. 2013, 20, 670–675. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, N.; Delbridge, C.; Gempt, J.; Feuchtinger, A.; Walch, A.; Schirmer, L.; Bunk, W.; Aschenbrenner, T.; Liesche-Starnecker, F.; Schlegel, J. The Intratumoral Heterogeneity Reflects the Intertumoral Subtypes of Glioblastoma Multiforme: A Regional Immunohistochemistry Analysis. Front. Oncol. 2020, 10, 494. [Google Scholar] [CrossRef] [PubMed]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- Culliton, B.J. Fighting cancer with designer cells. Science 1989, 244, 1430–1433. [Google Scholar] [CrossRef]
- Burnet, F.M. The concept of immunological surveillance. Prog. Exp. Tumor Res. 1970, 13, 1–27. [Google Scholar]
- Shi, L.; Kam, C.M.; Powers, J.C.; Aebersold, R.; Greenberg, A.H. Purification of three cytotoxic lymphocyte granule serine proteases that induce apoptosis through distinct substrate and target cell interactions. J. Exp. Med. 1992, 176, 1521–1529. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Kraut, R.P.; Aebersold, R.; Greenberg, A.H. A natural killer cell granule protein that induces DNA fragmentation and, apoptosis. J. Exp. Med. 1992, 175, 553–566. [Google Scholar] [CrossRef] [Green Version]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. CTLA-4-mediated inhibition in regulation of T cell responses: Mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594. [Google Scholar] [CrossRef] [Green Version]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutloff, A.; Dittrich, A.M.; Beier, K.C.; Eljaschewitsch, B.; Kraft, R.; Anagnostopoulos, I.; Kroczek, R.A. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature 1999, 397, 263–266. [Google Scholar] [CrossRef]
- Tindemans, I.; Serafini, N.; Di Santo, J.P.; Hendriks, R.W. GATA-3 function in innate and adaptive immunity. Immunity 2014, 41, 191–206. [Google Scholar] [CrossRef] [Green Version]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef] [Green Version]
- Bahador, M.; Gras Navarro, A.; Rahman, M.A.; Dominguez-Valentin, M.; Sarowar, S.; Ulvestad, E.; Njølstad, G.; Lie, S.A.; Kristoffersen, E.K.; Bratland, E.; et al. Increased infiltration and tolerised antigen-specific CD8(+) T(EM) cells in tumor but not peripheral blood have no impact on survival of HCMV(+) glioblastoma patients. Oncoimmunology 2017, 6, e1336272. [Google Scholar] [CrossRef] [Green Version]
- Kmiecik, J.; Poli, A.; Brons, N.H.; Waha, A.; Eide, G.E.; Enger, P.O.; Zimmer, J.; Chekenya, M. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol. 2013, 264, 71–83. [Google Scholar] [CrossRef] [Green Version]
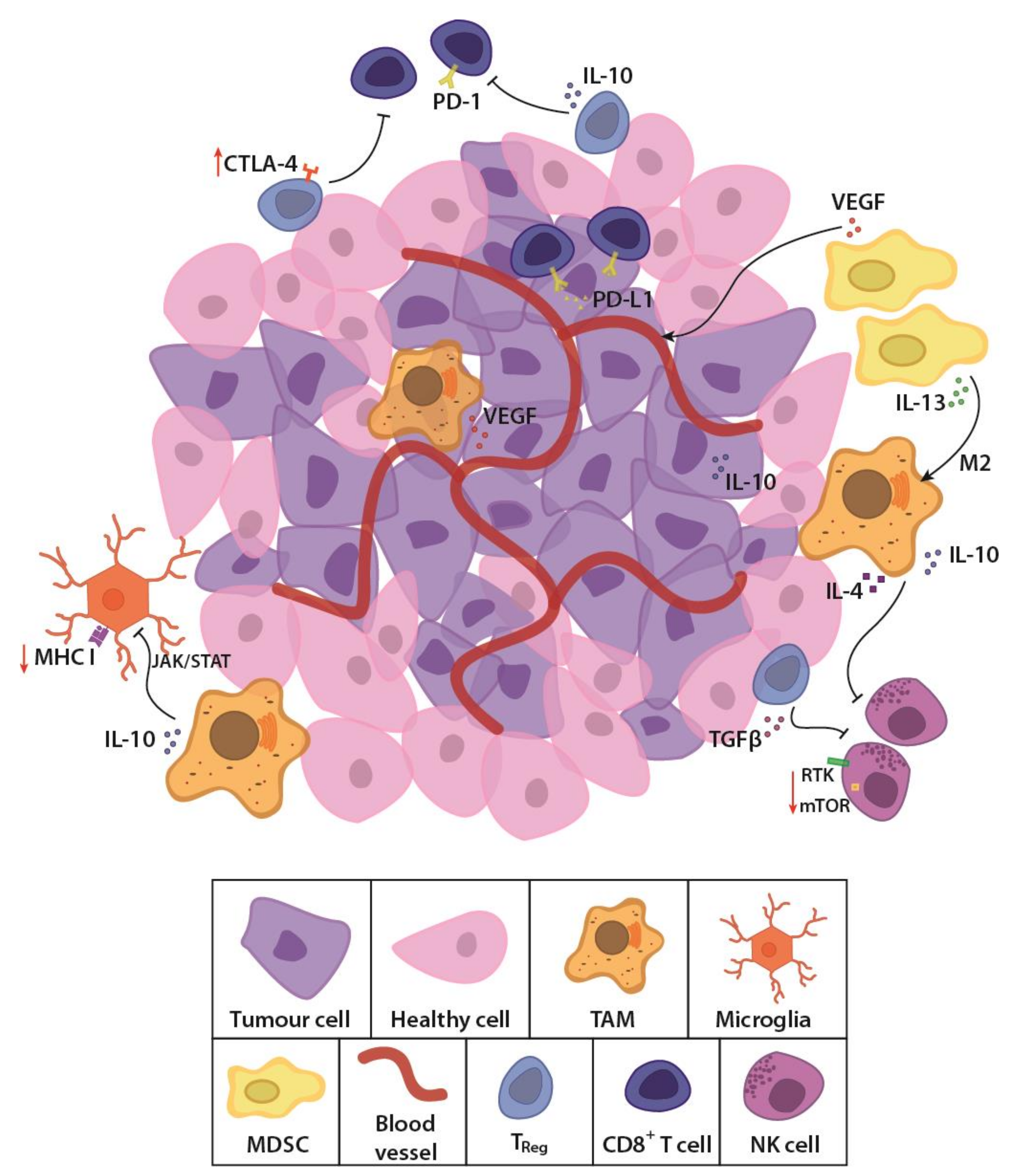
- Grabowski, M.M.; Sankey, E.W.; Ryan, K.J.; Chongsathidkiet, P.; Lorrey, S.J.; Wilkinson, D.S.; Fecci, P.E. Immune suppression in gliomas. J. Neurooncol. 2021, 151, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.R.D.; Cuzzubbo, S.; McArthur, S.; Durrant, L.G.; Adhikaree, J.; Tinsley, C.J.; Pockley, A.G.; McArdle, S.E.B. Immune Escape in Glioblastoma Multiforme and the Adaptation of Immunotherapies for Treatment. Front. Immunol. 2020, 11, 582106. [Google Scholar] [CrossRef]
- Moserle, L.; Casanovas, O. Anti-angiogenesis and metastasis: A tumour and stromal cell alliance. J. Intern. Med. 2013, 273, 128–137. [Google Scholar] [CrossRef]
- Rampling, R.; Cruickshank, G.; Lewis, A.D.; Fitzsimmons, S.A.; Workman, P. Direct measurement of pO2 distribution and bioreductive enzymes in human malignant brain tumors. Int. J. Radiat. Oncol. Biol. Phys. 1994, 29, 427–431. [Google Scholar] [CrossRef]
- Collingridge, D.R.; Piepmeier, J.M.; Rockwell, S.; Knisely, J.P. Polarographic measurements of oxygen tension in human glioma and surrounding peritumoural brain tissue. Radiother. Oncol. 1999, 53, 127–131. [Google Scholar] [CrossRef]
- Evans, S.M.; Jenkins, K.W.; Chen, H.I.; Jenkins, W.T.; Judy, K.D.; Hwang, W.T.; Lustig, R.A.; Judkins, A.R.; Grady, M.S.; Hahn, S.M.; et al. The Relationship among Hypoxia, Proliferation, and Outcome in Patients with De Novo Glioblastoma: A Pilot Study. Transl. Oncol. 2010, 3, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Hutchins, A.P.; Diez, D.; Miranda-Saavedra, D. The IL-10/STAT3-mediated anti-inflammatory response: Recent developments and future challenges. Brief. Funct. Genom. 2013, 12, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, M.; Sankowski, R.; Bunse, L.; Kilian, M.; Green, E.; Ramallo Guevara, C.; Pusch, S.; Poschet, G.; Sanghvi, K.; Hahn, M.; et al. Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH-mutant gliomas. Nat. Cancer 2021, 2, 723–740. [Google Scholar] [CrossRef]
- Han, J.; Alvarez-Breckenridge, C.A.; Wang, Q.-E.; Yu, J. TGF-β signaling and its targeting for glioma treatment. Am. J. Cancer Res. 2015, 5, 945–955. [Google Scholar]
- Hao, C.; Chen, G.; Zhao, H.; Li, Y.; Chen, J.; Zhang, H.; Li, S.; Zhao, Y.; Chen, F.; Li, W.; et al. PD-L1 Expression in Glioblastoma, the Clinical and Prognostic Significance: A Systematic Literature Review and Meta-Analysis. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- McCune, J.S. Rapid Advances in Immunotherapy to Treat Cancer. Clin. Pharmacol. Ther. 2018, 103, 540–544. [Google Scholar] [CrossRef]
- Hunter, P. The fourth pillar. EMBO Rep. 2017, 18, 1889–1892. [Google Scholar] [CrossRef] [PubMed]
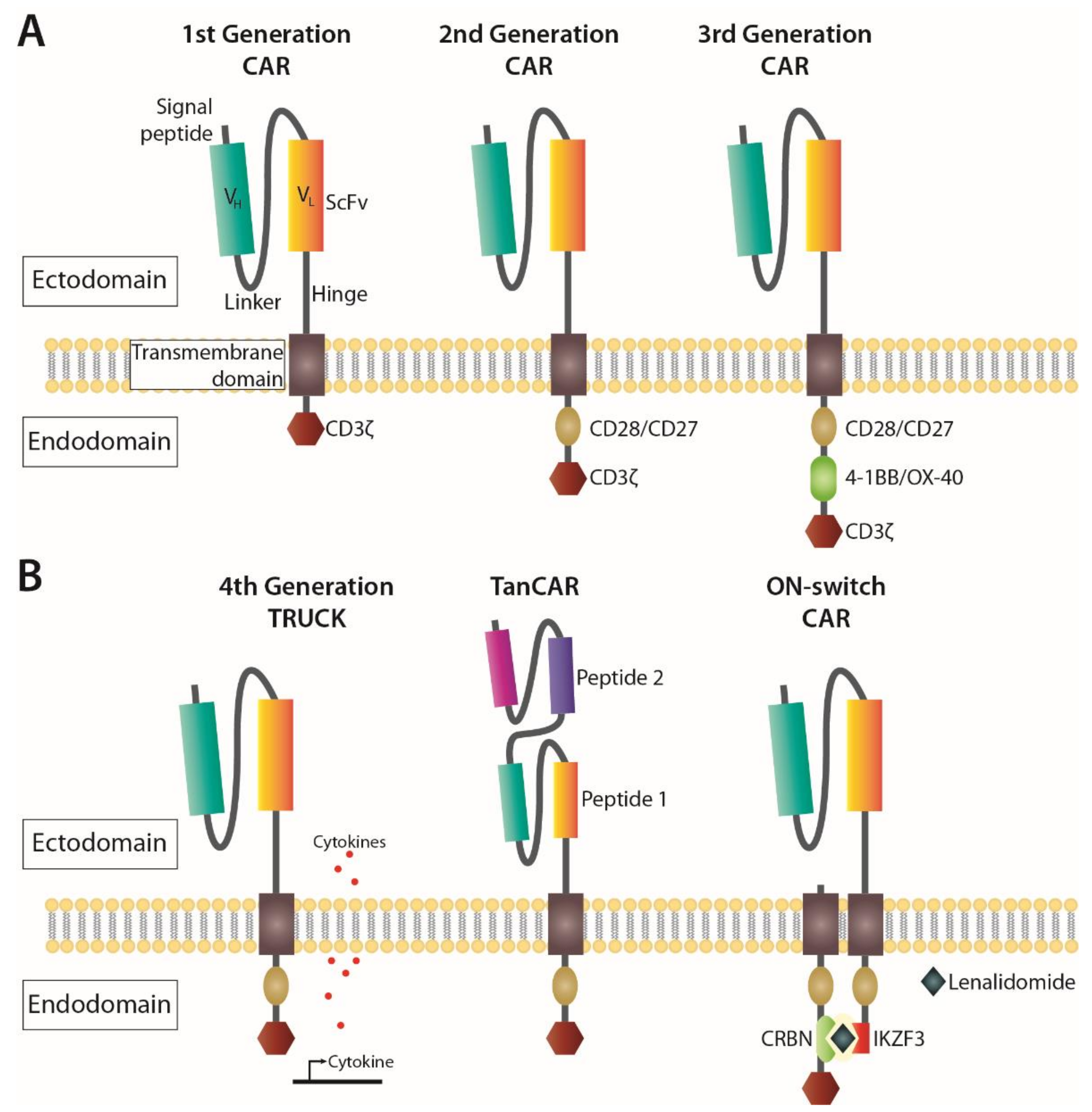
- Kuwana, Y.; Asakura, Y.; Utsunomiya, N.; Nakanishi, M.; Arata, Y.; Itoh, S.; Nagase, F.; Kurosawa, Y. Expression of chimeric receptor composed of immunoglobulin-derived V resions and T-cell receptor-derived C regions. Biochem. Biophys. Res. Commun. 1987, 149, 960–968. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [Green Version]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: Functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef] [Green Version]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 Costimulation: From Mechanism to Therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Ramos, C.A.; Rouce, R.; Robertson, C.S.; Reyna, A.; Narala, N.; Vyas, G.; Mehta, B.; Zhang, H.; Dakhova, O.; Carrum, G.; et al. In Vivo Fate and Activity of Second- versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin’s Lymphomas. Mol. Ther. 2018, 26, 2727–2737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Yeku, O.O.; Purdon, T.J.; Koneru, M.; Spriggs, D.; Brentjens, R.J. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 2017, 7, 10541. [Google Scholar] [CrossRef] [Green Version]
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schönfeld, K.; Koch, J.; Dotti, G.; et al. TanCAR: A novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol. Ther. Nucleic Acids 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Roybal, K.T.; Puchner, E.M.; Onuffer, J.; Lim, W.A. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 2015, 350, 6258. [Google Scholar] [CrossRef] [Green Version]
- Jan, M.; Scarfò, I.; Larson, R.C.; Walker, A.; Schmidts, A.; Guirguis, A.A.; Gasser, J.A.; Słabicki, M.; Bouffard, A.A.; Castano, A.P.; et al. Reversible ON- and OFF-switch chimeric antigen receptors controlled by lenalidomide. Sci. Transl. Med. 2021, 13, 575. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Rivière, I. Clinical manufacturing of CAR T cells: Foundation of a promising therapy. Mol. Ther. Oncolytics 2016, 3, 16015. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, N.V.; Shaw, P.A.; Hexner, E.O.; Pequignot, E.; Gill, S.; Luger, S.M.; Mangan, J.K.; Loren, A.W.; Perl, A.E.; Maude, S.L.; et al. Optimizing Chimeric Antigen Receptor T-Cell Therapy for Adults With Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2020, 38, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Bouchkouj, N.; Kasamon, Y.L.; Claro, R.A.d.; George, B.; Lin, X.; Lee, S.; Blumenthal, G.M.; Bryan, W.; McKee, A.E.; Pazdur, R. FDA approval summary: Axicabtagene Ciloleucel for Relapsed or Refractory Large B-cell Lymphoma. Clin. Cancer Res. 2019, 25, 1702–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, M.C.; Lu, X.; Huang, Y.; Lin, X.; Mahmood, I.; Przepiorka, D.; Gavin, D.; Lee, S.; Liu, K.; George, B.; et al. FDA Approval Summary: Tisagenlecleucel for Treatment of Patients with Relapsed or Refractory B-cell Precursor Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2019, 25, 1142–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef]
- Si, J.; Shi, X.; Sun, S.; Zou, B.; Li, Y.; An, D.; Lin, X.; Gao, Y.; Long, F.; Pang, B.; et al. Hematopoietic Progenitor Kinase1 (HPK1) Mediates T Cell Dysfunction and Is a Druggable Target for T Cell-Based Immunotherapies. Cancer Cell 2020, 38, 551–566. [Google Scholar] [CrossRef]
- Zhang, E.; Gu, J.; Xu, H. Prospects for chimeric antigen receptor-modified T cell therapy for solid tumors. Mol. Cancer 2018, 17, 1–12. [Google Scholar] [CrossRef]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Tallantyre, E.C.; Evans, N.A.; Parry-Jones, J.; Morgan, M.P.G.; Jones, C.H.; Ingram, W. Neurological updates: Neurological complications of CAR-T therapy. J. Neurol. 2021, 268, 1544–1554. [Google Scholar] [CrossRef]
- Fitzgerald, J.C.; Weiss, S.L.; Maude, S.L.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; Shaw, P.; Berg, R.A.; June, C.H.; Porter, D.L.; et al. Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia. Ann. Intensive Care 2017, 45, 2. [Google Scholar] [CrossRef]
- Mehta, R.S.; Rezvani, K. Chimeric Antigen Receptor Expressing Natural Killer Cells for the Immunotherapy of Cancer. Front. Immunol. 2018, 9, 283. [Google Scholar] [CrossRef] [Green Version]
- Imai, C.; Iwamoto, S.; Campana, D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 2005, 106, 376–383. [Google Scholar] [CrossRef] [Green Version]
- Töpfer, K.; Cartellieri, M.; Michen, S.; Wiedemuth, R.; Müller, N.; Lindemann, D.; Bachmann, M.; Füssel, M.; Schackert, G.; Temme, A. DAP12-Based Activating Chimeric Antigen Receptor for NK Cell Tumor Immunotherapy. J. Immunol. 2015, 194, 3201–3212. [Google Scholar] [CrossRef]
- Kiessling, R.; Klein, E.; Pross, H.; Wigzell, H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur. J. Immunol. 1975, 5, 117–121. [Google Scholar] [CrossRef]
- Herberman, R.B.; Nunn, M.E.; Holden, H.T.; Lavrin, D.H. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int. J. Cancer 1975, 16, 230–239. [Google Scholar] [CrossRef]
- Lanier, L.L. Natural killer cell receptor signaling. Curr. Opin. Immunol. 2003, 15, 308–314. [Google Scholar] [CrossRef]
- Pende, D.; Parolini, S.; Pessino, A.; Sivori, S.; Augugliaro, R.; Morelli, L.; Marcenaro, E.; Accame, L.; Malaspina, A.; Biassoni, R.; et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J. Exp. Med. 1999, 190, 1505–1516. [Google Scholar] [CrossRef]
- Vitale, M.; Bottino, C.; Sivori, S.; Sanseverino, L.; Castriconi, R.; Marcenaro, E.; Augugliaro, R.; Moretta, L.; Moretta, A. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J. Exp. Med. 1998, 187, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Pessino, A.; Sivori, S.; Bottino, C.; Malaspina, A.; Morelli, L.; Moretta, L.; Biassoni, R.; Moretta, A. Molecular cloning of NKp46: A novel member of the immunoglobulin superfamily involved in triggering of natural cytotoxicity. J. Exp. Med. 1998, 188, 953–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretta, A.; Biassoni, R.; Bottino, C.; Pende, D.; Vitale, M.; Poggi, A.; Mingari, M.C.; Moretta, L. Major histocompatibility complex class I-specific receptors on human natural killer and T lymphocytes. Immunol. Rev. 1997, 155, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.S.; Purdy, A.K. Structure/function of human killer cell immunoglobulin-like receptors: Lessons from polymorphisms, evolution, crystal structures and mutations. Immunology 2011, 132, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Poggi, A.; Pella, N.; Morelli, L.; Spada, F.; Revello, V.; Sivori, S.; Augugliaro, R.; Moretta, L.; Moretta, A. p40, a novel surface molecule involved in the regulation of the non-major histocompatibility complex-restricted cytolytic activity in humans. Eur. J. Immunol. 1995, 25, 369–376. [Google Scholar] [CrossRef]
- Ljunggren, H.G.; Kärre, K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol. Today 1990, 11, 237–244. [Google Scholar] [CrossRef]
- Lanier, L.L.; Yu, G.; Phillips, J.H. Co-association of CD3 zeta with a receptor (CD16) for IgG Fc on human natural killer cells. Nature 1989, 342, 803–805. [Google Scholar] [CrossRef]
- Haspels, H.N.; Rahman, M.A.; Joseph, J.V.; Navarro, A.G.; Chekenya, M. Glioblastoma stem-like cells are more susceptible than differentiated cells to natural killer cell lysis mediated through killer immunoglobulin-like receptors-human leukocyte antigen ligand mismatch and activation receptor-ligand interactions. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Gras Navarro, A.; Kmiecik, J.; Leiss, L.; Zelkowski, M.; Engelsen, A.; Bruserud, Ø.; Zimmer, J.; Enger, P.Ø.; Chekenya, M. NK Cells with KIR2DS2 Immunogenotype Have a Functional Activation Advantage To Efficiently Kill Glioblastoma and Prolong Animal Survival. J. Immunol. 2014, 193, 6192–6206. [Google Scholar] [CrossRef] [Green Version]
- Shaim, H.; Shanley, M.; Basar, R.; Daher, M.; Gumin, J.; Zamler, D.B.; Uprety, N.; Wang, F.; Huang, Y.; Gabrusiewicz, K.; et al. Targeting the αv integrin/TGF-β axis improves natural killer cell function against glioblastoma stem cells. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. NKG2D Receptor and Its Ligands in Host Defense. Cancer Immunol. Res. 2015, 3, 575–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza-Fonseca-Guimaraes, F.; Cursons, J.; Huntington, N.D. The Emergence of Natural Killer Cells as a Major Target in Cancer Immunotherapy. Trends Immunol. 2019, 40, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Kärre, K. Natural killer cell recognition of missing self. Nat. Immunol. 2008, 9, 477–480. [Google Scholar] [CrossRef]
- Cichocki, F.; Bjordahl, R.; Gaidarova, S.; Mahmood, S.; Abujarour, R.; Wang, H.; Tuininga, K.; Felices, M.; Davis, Z.B.; Bendzick, L.; et al. iPSC-derived NK cells maintain high cytotoxicity and enhance in vivo tumor control in concert with T cells and anti–PD-1 therapy. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Fang, F.; Xiao, W.; Tian, Z. NK cell-based immunotherapy for cancer. Semin. Immunol. 2017, 31, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Santomasso, B.D.; Park, J.H.; Salloum, D.; Riviere, I.; Flynn, J.; Mead, E.; Halton, E.; Wang, X.; Senechal, B.; Purdon, T.; et al. Clinical and biological correlates of neurotoxicity associated with car t-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov. 2018, 8, 958–971. [Google Scholar] [CrossRef] [Green Version]
- Basar, R.; Daher, M.; Rezvani, K. Next-generation cell therapies: The emerging role of CAR-NK cells. Blood Adv. 2020, 4, 5868–5876. [Google Scholar] [CrossRef] [PubMed]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Gobius, I.; Souza-Fonseca-Guimaraes, F. Natural killer cell engineering—A new hope for cancer immunotherapy. Semin. Hematol. 2020, 57, 194–200. [Google Scholar] [CrossRef]
- Rezvani, K.; Rouce, R.; Liu, E.; Shpall, E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol. Ther. 2017, 25, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Rautela, J.; Surgenor, E.; Huntington, N.D. Drug target validation in primary human natural killer cells using CRISPR RNP. J. Leukoc. Biol. 2020, 108, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, T.; Nakazawa, T.; Matsuda, R.; Nishimura, F.; Nakamura, M.; Yamada, S.; Nakagawa, I.; Park, Y.S.; Tsujimura, T.; Nakase, H. CRISPR-Cas9-Mediated TIM3 Knockout in Human Natural Killer Cells Enhances Growth Inhibitory Effects on Human Glioma Cells. Int. J. Mol. Sci. 2021, 22, 3489. [Google Scholar] [CrossRef]
- Teratake, Y.; Takashina, T.; Iijima, K.; Sakuma, T.; Yamamoto, T.; Ishizaka, Y. Development of a protein-based system for transient epigenetic repression of immune checkpoint molecule and enhancement of antitumour activity of natural killer cells. Br. J. Cancer 2020, 122, 823–834. [Google Scholar] [CrossRef]
- Gurney, M.; Stikvoort, A.; Nolan, E.; Kirkham-McCarthy, L.; Khoruzhenko, S.; Shivakumar, R.; Zweegman, S.; Van de Donk, N.; Mutis, T.; Szegezdi, E.; et al. CD38 knockout natural killer cells expressing an affinity optimized CD38 chimeric antigen receptor successfully target acute myeloid leukemia with reduced effector cell fratricide. Haematologica 2020. Available online: https://haematologica.org/article/view/haematol.2020.271908 (accessed on 2 October 2021). [CrossRef]
- Angelo, L.S.; Banerjee, P.P.; Monaco-Shawver, L.; Rosen, J.B.; Makedonas, G.; Forbes, L.R.; Mace, E.M.; Orange, J.S. Practical NK cell phenotyping and variability in healthy adults. Immunol. Res. 2015, 62, 341–356. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wallace, D.L.; De Lara, C.M.; Ghattas, H.; Asquith, B.; Worth, A.; Griffin, G.E.; Taylor, G.P.; Tough, D.F.; Beverley, P.C.L.; et al. In vivo kinetics of human natural killer cells: The effects of ageing and acute and chronic viral infection. Immunology 2007, 121, 258–265. [Google Scholar] [CrossRef]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef]
- Hodgins, J.J.; Khan, S.T.; Park, M.M.; Auer, R.C.; Ardolino, M. Killers 2.0: NK cell therapies at the forefront of cancer control. J. Clin. Investig. 2019, 129, 3499–3510. [Google Scholar] [CrossRef] [Green Version]
- Mo, F.; Mamonkin, M.; Brenner, M.K.; Heslop, H.E. Taking T-Cell Oncotherapy Off-the-Shelf. Trends Immunol. 2021, 42, 261–272. [Google Scholar] [CrossRef]
- Morgan, M.A.; Büning, H.; Sauer, M.; Schambach, A. Use of Cell and Genome Modification Technologies to Generate Improved “Off-the-Shelf” CAR T and CAR NK Cells. Front. Immunol. 2020, 11, 1965. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367. [Google Scholar] [CrossRef] [PubMed]
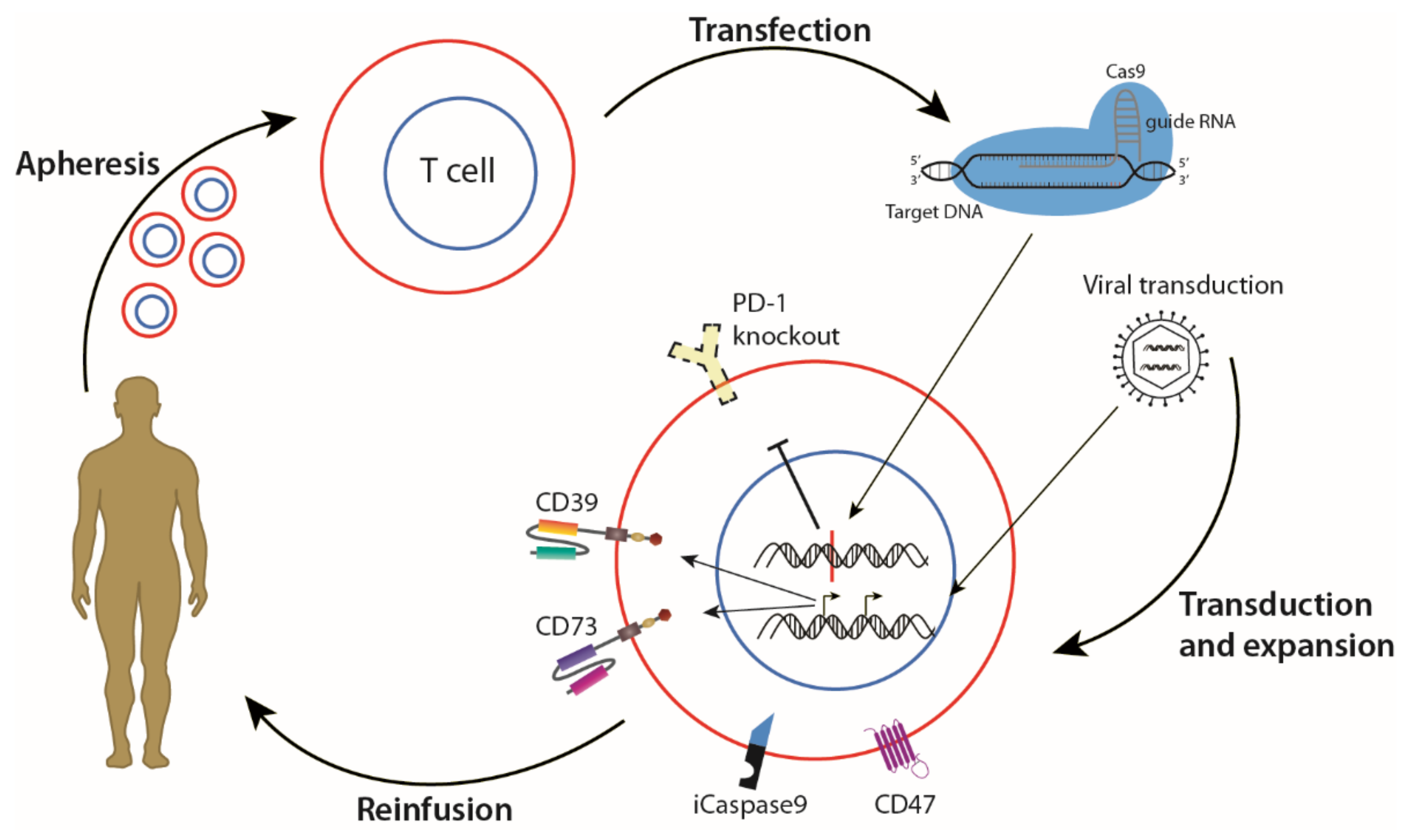
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfò, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torikai, H.; Reik, A.; Soldner, F.; Warren, E.H.; Yuen, C.; Zhou, Y.; Crossland, D.L.; Huls, H.; Littman, N.; Zhang, Z.; et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood 2013, 122, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, R.; Graham, C.; Yallop, D.; Jozwik, A.; Mirci-Danicar, O.C.; Lucchini, G.; Pinner, D.; Jain, N.; Kantarjian, H.; Boissel, N.; et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: Results of two phase 1 studies. Lancet 2020, 396, 1885–1894. [Google Scholar] [CrossRef]
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015, 75, 3853–3864. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, S.; Long, E.O. A human histocompatibility leukocyte antigen (HLA)-G-specific receptor expressed on all natural killer cells. J. Exp. Med. 1999, 189, 1093–1100. [Google Scholar] [CrossRef] [Green Version]
- Faure, M.; Long, E.O. KIR2DL4 (CD158d), an NK cell-activating receptor with inhibitory potential. J. Immunol. 2002, 168, 6208–6214. [Google Scholar] [CrossRef] [Green Version]
- Castriconi, R.; Daga, A.; Dondero, A.; Zona, G.; Poliani, P.L.; Melotti, A.; Griffero, F.; Marubbi, D.; Spaziante, R.; Bellora, F.; et al. NK Cells Recognize and Kill Human Glioblastoma Cells with Stem Cell-Like Properties. J. Immunol. 2009, 182, 3530–3539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostafa, H.; Pala, A.; Hogel, J.; Hlavac, M.; Dietrich, E.; Westhoff, M.A.; Nonnenmacher, L.; Burster, T.; Georgieff, M.; Wirtz, C.R.; et al. Immune phenotypes predict survival in patients with glioblastoma multiforme. J. Hematol. Oncol. 2016, 9, 77. [Google Scholar] [CrossRef] [Green Version]
- Zagzag, D.; Salnikow, K.; Chiriboga, L.; Yee, H.; Lan, L.; Ali, M.A.; Garcia, R.; Demaria, S.; Newcomb, E.W. Downregulation of major histocompatibility complex antigens in invading glioma cells: Stealth invasion of the brain. Lab. Investig. 2005, 85, 328–341. [Google Scholar] [CrossRef]
- Dominguez-Valentin, M.; Navarro, A.G.; Rahman, A.M.; Kumar, S.; Retière, C.; Ulvestad, E.; Kristensen, V.; Lund-Johansen, M.; Lie, B.A.; Enger, P.Ø.; et al. Identification of a natural killer cell receptor allele that prolongs survival of cytomegalovirus-positive glioblastoma patients. Cancer Res. 2016, 76, 5326–5336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef] [PubMed]
- Uherek, C.; Tonn, T.; Uherek, B.; Becker, S.; Schnierle, B.; Klingemann, H.G.; Wels, W. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood 2002, 100, 1265–1273. [Google Scholar] [CrossRef]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. eBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
- Murakami, T.; Nakazawa, T.; Natsume, A.; Nishimura, F.; Nakamura, M.; Matsuda, R.; Omoto, K.; Tanaka, Y.; Shida, Y.; Park, Y.S.; et al. Novel human NK cell line carrying CAR targeting EGFRvIII induces antitumor effects in glioblastoma cells. Anticancer Res. 2018, 38, 5049–5056. [Google Scholar] [CrossRef]
- Nakazawa, T.; Murakami, T.; Natsume, A.; Nishimura, F.; Morimoto, T.; Matsuda, R.; Nakamura, M.; Yamada, S.; Nakagawa, I.; Park, Y.-S.; et al. KHYG-1 Cells With EGFRvIII-specific CAR Induced a Pseudoprogression-like Feature in Subcutaneous Tumours Derived from Glioblastoma-like Cells. Anticancer Res. 2020, 40, 3231–3237. [Google Scholar] [CrossRef]
- Han, J.; Chu, J.; Keung Chan, W.; Zhang, J.; Wang, Y.; Cohen, J.B.; Victor, A.; Meisen, W.H.; Kim, S.H.; Grandi, P.; et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Genßler, S.; Burger, M.C.; Zhang, C.; Oelsner, S.; Mildenberger, I.; Wagner, M.; Steinbach, J.P.; Wels, W.S. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. OncoImmunology 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genßler, S.; Schönfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J. Natl. Cancer Inst. 2015, 108. [Google Scholar] [CrossRef]
- Burger, M.C.; Zhang, C.; Harter, P.N.; Romanski, A.; Strassheimer, F.; Senft, C.; Tonn, T.; Steinbach, J.P.; Wels, W.S. CAR-Engineered NK Cells for the Treatment of Glioblastoma: Turning Innate Effectors Into Precision Tools for Cancer Immunotherapy. Front. Immunol. 2019, 10, 2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; de Carvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef] [Green Version]
- Medikonda, R.; Dunn, G.; Rahman, M.; Fecci, P.; Lim, M. A review of glioblastoma immunotherapy. J. Neurooncol. 2021, 151, 41–53. [Google Scholar] [CrossRef]
- Razavi, S.-M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune Evasion Strategies of Glioblastoma. Front. Surg. 2016, 3. [Google Scholar] [CrossRef]
- Park, S.; Shevlin, E.; Vedvyas, Y.; Zaman, M.; Park, S.; Hsu, Y.M.S.; Min, I.M.; Jin, M.M. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filley, A.C.; Henriquez, M.; Dey, M. Recurrent glioma clinical trial, CheckMate-143: The game is not over yet. Oncotarget 2017, 8, 91779–91794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Saikali, S.; Avril, T.; Collet, B.; Hamlat, A.; Bansard, J.Y.; Drenou, B.; Guegan, Y.; Quillien, V. Expression of nine tumour antigens in a series of human glioblastoma multiforme: Interest of EGFRvIII, IL-13Ralpha2, gp100 and TRP-2 for immunotherapy. J. Neurooncol. 2007, 81, 139–148. [Google Scholar] [CrossRef]
- Ahmed, N.; Salsman, V.S.; Kew, Y.; Shaffer, D.; Powell, S.; Zhang, Y.J.; Grossman, R.G.; Heslop, H.E.; Gottschalk, S. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin. Cancer Res. 2010, 16, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, J.; Marzese, D.M.; Wang, X.; Yang, Z.; Li, C.; Zhang, H.; Zhang, J.; Chen, C.C.; Kelly, D.F.; et al. B7H3 regulates differentiation and serves as a potential biomarker and theranostic target for human glioblastoma. Lab. Investig. 2019, 99, 1117–1129. [Google Scholar] [CrossRef]
- Montano, N.; Cenci, T.; Martini, M.; D’Alessandris, Q.G.; Pelacchi, F.; Ricci-Vitiani, L.; Maira, G.; Maria, R.D.; Larocca, L.M.; Pallini, R. Expression of EGFRvIII in Glioblastoma: Prognostic Significance Revisited. Neoplasia 2011, 13, 1113–1121. [Google Scholar] [CrossRef]
- Tripathy, K.; Das, B.; Singh, A.K.; Misra, A.; Misra, S.; Misra, S.S. Prognostic Significance of Epidermal Growth Factor Receptor in Patients of Glioblastoma Multiforme. J. Clin. Diagn. Res. 2017, 11, EC05–EC08. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Ying, H.; Zeng, G.; Wheeler, C.J.; Black, K.L.; Yu, J.S. HER-2, gp100, and MAGE-1 Are Expressed in Human Glioblastoma and Recognized by Cytotoxic T Cells. Cancer Res. 2004, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347. [Google Scholar] [CrossRef] [PubMed]
- Portillo, A.L.; Hogg, R.; Poznanski, S.M.; Rojas, E.A.; Cashell, N.J.; Hammill, J.A.; Chew, M.V.; Shenouda, M.M.; Ritchie, T.M.; Cao, Q.T.; et al. Expanded human NK cells armed with CAR uncouple potent anti-tumor activity from off-tumor toxicity against solid tumors. iScience 2021, 24, 102619. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.P.; Wang, G.Y.; Arima, K.; Guan, S.P.; Waters, M.R.; Cavenee, W.K.; Pan, E.; Aliwarga, E.; Chong, S.T.; Kok, C.Y.L.; et al. Interleukin-13 receptor alpha 2 cooperates with EGFRvIII signaling to promote glioblastoma multiforme. Nat. Commun. 2017, 8, 1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapoval, A.I.; Ni, J.; Lau, J.S.; Wilcox, R.A.; Flies, D.B.; Liu, D.; Dong, H.; Sica, G.L.; Zhu, G.; Tamada, K.; et al. B7-H3: A costimulatory molecule for T cell activation and IFN-γ production. Nat. Immunol. 2001, 2, 269–274. [Google Scholar] [CrossRef]
- Prasad, D.V.R.; Nguyen, T.; Li, Z.; Yang, Y.; Duong, J.; Wang, Y.; Dong, C. Murine B7-H3 Is a Negative Regulator of T Cells. J. Immunol. 2004, 173, 2500–2506. [Google Scholar] [CrossRef] [Green Version]
- Lemke, D.; Pfenning, P.N.; Sahm, F.; Klein, A.C.; Kempf, T.; Warnken, U.; Schnolzer, M.; Tudoran, R.; Weller, M.; Platten, M.; et al. Costimulatory protein 4IgB7H3 drives the malignant phenotype of glioblastoma by mediating immune escape and invasiveness. Clin. Cancer Res. 2012, 18, 105–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Wang, Y.; Huang, J.; Zhang, Z.; Liu, F.; Xu, J.; Guo, G.; Wang, W.; Tong, A.; Zhou, L. Administration of B7-H3 targeted chimeric antigen receptor-T cells induce regression of glioblastoma. Signal Transduct. Target 2021, 6, 125. [Google Scholar] [CrossRef]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress- inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef]
- Wu, J.; Song, Y.; Bakker, A.B.H.; Bauer, S.; Spies, T.; Lanier, L.L.; Phillips, J.H. An activating immunoreceptor complex formed by NKG2D and DAP10. Science 1999, 285, 730–732. [Google Scholar] [CrossRef]
- Lee, J.; Zhang, T.; Hwang, I.; Kim, A.; Nitschke, L.; Kim, M.; Scott, J.M.; Kamimura, Y.; Lanier, L.L.; Kim, S. Epigenetic Modification and Antibody-Dependent Expansion of Memory-like NK Cells in Human Cytomegalovirus-Infected Individuals. Immunity 2015, 42, 431–442. [Google Scholar] [CrossRef] [Green Version]
- Weiss, T.; Schneider, H.; Silginer, M.; Steinle, A.; Pruschy, M.; Polic, B.; Weller, M.; Roth, P. NKG2D-Dependent antitumor effects of chemotherapy and radiotherapy against glioblastoma. Clin. Cancer Res. 2018, 24, 882–895. [Google Scholar] [CrossRef] [Green Version]
- Weiss, T.; Weller, M.; Guckenberger, M.; Sentman, C.L.; Roth, P. NKG2D-Based CAR T Cells and Radiotherapy Exert Synergistic Efficacy in Glioblastoma. Cancer Res. 2018, 78, 1031–1043. [Google Scholar] [CrossRef] [Green Version]
- The Nobel Prize in Chemistry 2020. Available online: https://www.nobelprize.org/page/2/?np-keyword=press-release (accessed on 2 October 2021).
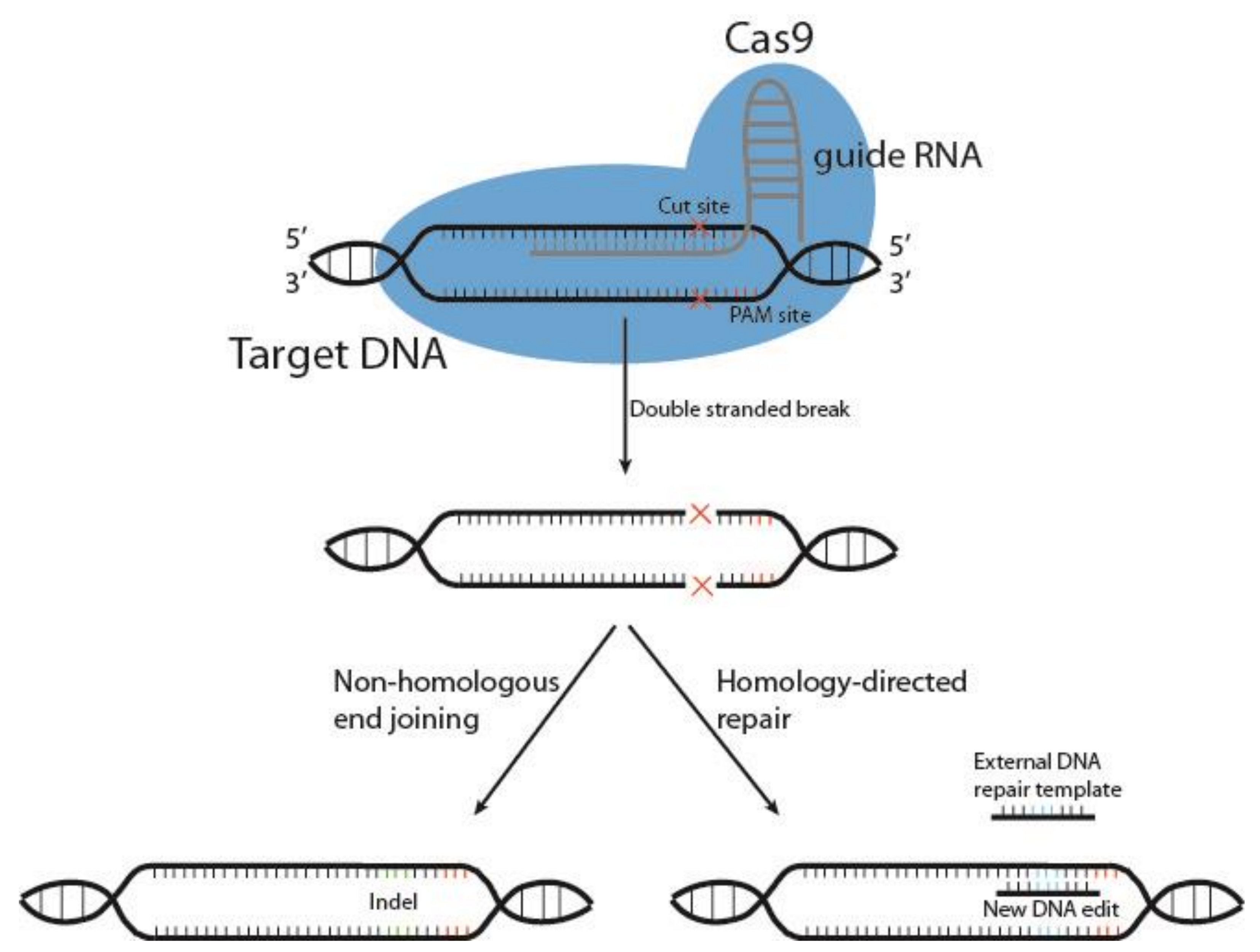
- Garneau, J.E.; Dupuis, M.E.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadan, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef]
- Iliakis, G.; Wang, H.; Perrault, A.R.; Boecker, W.; Rosidi, B.; Windhofer, F.; Wu, W.; Guan, J.; Terzoudi, G.; Panteliasc, G. Mechanisms of DNA double strand break repair and chromosome aberration formation. Cytogenet. Genome Res. 2004, 104, 14–20. [Google Scholar] [CrossRef]
- Heyer, W.D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiel, J.A.; Chou, E.; Mayer, M.; Anderson, E.M.; Van, A.; Smith, B. Homology-Directed Repair with Dharmacon™ Edit-R™ CRISPR-Cas9 Reagents and Single-Stranded DNA Oligos; Dharmacon, GE Healthcare: Buckinghamshire, UK, 2015. [Google Scholar]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Liu, M.; Rehman, S.; Tang, X.; Gu, K.; Fan, Q.; Chen, D.; Ma, W. Methodologies for improving HDR efficiency. Front Genet. 2019, 10, 691. [Google Scholar] [CrossRef]
- Baltimore, D.; Berg, P.; Botchan, M.; Carroll, D.; Charo, R.A.; Church, G.; Corn, J.E.; Daley, G.Q.; Doudna, J.A.; Fenner, M.; et al. A prudent path forward for genomic engineering and germline gene modification. Science 2015, 348, 36–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanphier, E.; Urnov, F. Don’t edit the human germ line. Nature 2015, 519, 410–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Long, L.; Zheng, W.; Dhungana, Y.; Lim, S.A.; Guy, C.; Wang, Y.; Wang, Y.D.; Qian, C.; Xu, B.; et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature 2019, 576, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Mahlakõiv, T.; Ye, Q.; Somanchi, S.; He, S.; Rana, H.; DiFiglia, A.; Gleason, J.; van der Touw, W.; Hariri, R.; et al. CBLB ablation with CRISPR/Cas9 enhances cytotoxicity of human placental stem cell-derived NK cells for cancer immunotherapy. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Tee, L.Y.; Wang, X.G.; Huang, Q.S.; Yang, S.H. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef]
- Schene, I.F.; Joore, I.P.; Oka, R.; Mokry, M.; van Vugt, A.H.M.; van Boxtel, R.; van der Doef, H.P.J.; van der Laan, L.J.W.; Verstegen, M.M.A.; van Hasselt, P.M.; et al. Prime editing for functional repair in patient-derived disease models. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pellegatta, S.; Savoldo, B.; Ianni, N.D.; Corbetta, C.; Chen, Y.; Patané, M.; Sun, C.; Pollo, B.; Ferrone, S.; DiMeco, F.; et al. Constitutive and TNFα-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Sci. Transl. Med. 2018, 10, 430. [Google Scholar] [CrossRef] [Green Version]
- Crowther, M.D.; Dolton, G.; Legut, M.; Caillaud, M.E.; Lloyd, A.; Attaf, M.; Galloway, S.A.E.; Rius, C.; Farrell, C.P.; Szomolay, B.; et al. Genome-wide CRISPR-Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nat. Immunol. 2020, 21, 178–185. [Google Scholar] [CrossRef]
- Li, X.Y.; Moesta, A.K.; Xiao, C.; Nakamura, K.; Casey, M.; Zhang, H.; Madore, J.; Lepletier, A.; Aguilera, A.R.; Sundarrajan, A.; et al. Targeting CD39 in Cancer Reveals an Extracellular ATP- and Inflammasome-Driven Tumor Immunity. Cancer Discov. 2019, 9, 1754–1773. [Google Scholar] [CrossRef] [Green Version]
- Bunse, L.; Pusch, S.; Bunse, T.; Sahm, F.; Sanghvi, K.; Friedrich, M.; Alansary, D.; Sonner, J.K.; Green, E.; Deumelandt, K.; et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 2018, 24, 1192–1203. [Google Scholar] [CrossRef]
- Chekenya, M.; Hjelstuen, M.; Enger, P.Ø.; Thorsen, F.; Jacob, A.L.; Probst, B.; Haraldseth, O.; Pilkington, G.; Butt, A.; Levine, J.M.; et al. NG2 proteoglycan promotes angiogenesis-dependent tumor growth in the central nervous system by sequestering angiostatin. FASEB J. 2002, 16, 586–588. [Google Scholar] [CrossRef]
- Harper, J.R.; Bumol, T.F.; Reisfeld, R.A. Serological and biochemical analyses of monoclonal antibodies to human melanoma-associated antigens. Hybridoma 1982, 1, 423–432. [Google Scholar] [CrossRef]
- Nishiyama, A.; Stallcup, W.B. Expression of NG2 Proteoglycan Causes Retention of Type VI Collagen on the Cell Surface. Mol. Biol. Cell 1993, 4, 1097–1108. [Google Scholar] [CrossRef] [Green Version]
- Svendsen, A.; Verhoeff, J.J.C.; Immervoll, H.; Brøgger, J.C.; Kmiecik, J.; Poli, A.; Netland, I.A.; Prestegarden, L.; Planagumà, J.; Torsvik, A.; et al. Expression of the progenitor marker NG2/CSPG4 predicts poor survival and resistance to ionising radiation in glioblastoma. Acta Neuropathol. 2011, 122, 495–510. [Google Scholar] [CrossRef] [Green Version]
- Chekenya, M.; Krakstad, C.; Svendsen, A.; Netland, I.A.; Staalesen, V.; Tysnes, B.B.; Selheim, F.; Wang, J.; Sakariassen, P.Ø.; Sandal, T.; et al. The progenitor cell marker NG2/MPG promotes chemoresistance by activation of integrin-dependent PI3K/Akt signaling. Oncogene 2008, 27, 5182–5194. [Google Scholar] [CrossRef] [Green Version]
- McCoy, M.K.; Tansey, M.G. TNF signaling inhibition in the CNS: Implications for normal brain function and neurodegenerative disease. J Neuroinflamm. 2008, 5, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Usheva, A.; Erat, A.; Chen, J.-F.; Enjyoji, K.; Linden, J.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczmarek, E.; Koziak, K.; Sévigny, J.; Siegel, J.B.; Anrather, J.; Beaudoin, A.R.; Bach, F.H.; Robson, S.C. Identification and characterization of CD39/vascular ATP diphosphohydrolase. J. Biol. Chem. 1996, 271, 33116–33122. [Google Scholar] [CrossRef] [Green Version]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.C.; Gutierrez-Vazquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A.; et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 2019, 22, 729–740. [Google Scholar] [CrossRef]
- Sun, X.; Wu, Y.; Gao, W.; Enjyoji, K.; Csizmadia, E.; Muller, C.E.; Murakami, T.; Robson, S.C. CD39/ENTPD1 expression by CD4+Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology 2010, 139, 1030–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Vijayan, D.; Li, X.Y.; Robson, S.C.; Geetha, N.; Teng, M.W.L.; Smyth, M.J. The role of NK cells and CD39 in the immunological control of tumor metastases. Oncoimmunology 2019, 8, e1593809. [Google Scholar] [CrossRef] [Green Version]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.-L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Wilhelm, D.; Rajky, O.; Kurscheid, S.; Kresl, P.; Wöhrer, A.; Marosi, C.; Hegi, M.E.; et al. Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro. Oncol. 2017, 19, 1460–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, L.G.; Choi, B.D.; Curry, W.T. (R)-2-hydroxyglutarate drives immune quiescence in the tumor microenvironment of IDH-mutant gliomas. Transl. Cancer Res. 2019, 8, S167–s170. [Google Scholar] [CrossRef]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef]
- Wiewrodt, D.; Nagel, G.; Dreimüller, N.; Hundsberger, T.; Perneczky, A.; Kaina, B. MGMT in primary and recurrent human glioblastomas after radiation and chemotherapy and comparison with p53 status and clinical outcome. Int. J. Cancer 2008, 122, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Rabe, M.; Dumont, S.; Alvarez-Arenas, A.; Janati, H.; Belmonte-Beitia, J.; Calvo, G.F.; Thibault-Carpentier, C.; Sery, Q.; Chauvin, C.; Joalland, N.; et al. Identification of a transient state during the acquisition of temozolomide resistance in glioblastoma. Cell Death Dis. 2020, 11, 19. [Google Scholar] [CrossRef]
- Perrot, I.; Michaud, H.A.; Giraudon-Paoli, M.; Augier, S.; Docquier, A.; Gros, L.; Courtois, R.; Déjou, C.; Jecko, D.; Becquart, O.; et al. Blocking Antibodies Targeting the CD39/CD73 Immunosuppressive Pathway Unleash Immune Responses in Combination Cancer Therapies. Cell Rep. 2019, 27, 2411–2425.e2419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Stasi, A.; Tey, S.-K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [Green Version]
- Tsai, R.K.; Discher, D.E. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J. Cell Biol. 2008, 180, 989–1003. [Google Scholar] [CrossRef] [Green Version]
- Vanmeerbeek, I.; Sprooten, J.; De Ruysscher, D.; Tejpar, S.; Vandenberghe, P.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; et al. Trial watch: Chemotherapy-induced immunogenic cell death in immuno-oncology. Oncoimmunology 2020, 9, 1703449. [Google Scholar] [CrossRef] [Green Version]
- Grossman, S.A.; Ye, X.; Lesser, G.; Sloan, A.; Carraway, H.; Desideri, S.; Piantadosi, S. Immunosuppression in patients with high-grade gliomas treated with radiation and temozolomide. Clin. Cancer Res. 2011, 17, 5473–5480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.B.; Sohn, S.; Krown, S.E.; Livingston, P.O.; Wolchok, J.D.; Quinn, C.; Williams, L.; Foster, T.; Sepkowitz, K.A.; Chapman, P.B. Selective CD4+ lymphopenia in melanoma patients treated with temozolomide: A toxicity with therapeutic implications. J. Clin. Oncol. 2004, 22, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.I.; Rhodin, K.E.; Dechant, C.; Cui, X.; Chongsathidkiet, P.; Wilkinson, D.; Waibl-Polania, J.; Sanchez-Perez, L.; Fecci, P.E. 4-1BB Agonism Averts TIL Exhaustion and Licenses PD-1 Blockade in Glioblastoma and Other Intracranial Cancers. Clin. Cancer Res. 2020, 26, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Wolburg, H.; Lippoldt, A. Tight junctions of the blood-brain barrier: Development, composition and regulation. Vasc. Pharmacol. 2002, 38, 323–337. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Kivisakk, P.; Kidd, G. Three or more routes for leukocyte migration into the central nervous system. Nat. Rev. Immunol. 2003, 3, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Updat. 2015, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Pardridge, W.M. Blood-brain barrier delivery. Drug Discov. Today 2007, 12, 54–61. [Google Scholar] [CrossRef]
- Seitz, R.J.; Wechsler, W. Immunohistochemical demonstration of serum proteins in human cerebral gliomas. Acta Neuropathol. 1987, 73, 145–152. [Google Scholar] [CrossRef]
- Roberts, H.C.; Roberts, T.P.; Brasch, R.C.; Dillon, W.P. Quantitative measurement of microvascular permeability in human brain tumors achieved using dynamic contrast-enhanced MR imaging: Correlation with histologic grade. Am. J. Neuroradiol. 2000, 21, 891–899. [Google Scholar] [PubMed]
- Pronin, I.N.; Holodny, A.I.; Petraikin, A.V. MRI of high-grade glial tumors: Correlation between the degree of contrast enhancement and the volume of surrounding edema. Neuroradiology 1997, 39, 348–350. [Google Scholar] [CrossRef] [PubMed]
- van Hooren, L.; Vaccaro, A.; Ramachandran, M.; Vazaios, K.; Libard, S.; van de Walle, T.; Georganaki, M.; Huang, H.; Pietilä, I.; Lau, J.; et al. Agonistic CD40 therapy induces tertiary lymphoid structures but impairs responses to checkpoint blockade in glioma. Nat. Commun. 2021, 12, 4127. [Google Scholar] [CrossRef] [PubMed]
- Waziri, A.; Killory, B.; Ogden, A.T., 3rd; Canoll, P.; Anderson, R.C.; Kent, S.C.; Anderson, D.E.; Bruce, J.N. Preferential in situ CD4+CD56+ T cell activation and expansion within human glioblastoma. J. Immunol. 2008, 180, 7673–7680. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.L.; Hughes, J.T.; Esiri, M.M.; Coakham, H.B.; Brownell, D.B. Immunohistological study of mononuclear cell infiltrate in malignant gliomas. Acta Neuropathol. 1987, 74, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Jung, T.Y.; Jung, S.; Jang, W.Y.; Moon, K.S.; Kim, I.Y.; Lee, M.C.; Lee, J.J. Tumour-infiltrating T-cell subpopulations in glioblastomas. Br. J. Neurosurg. 2012, 26, 21–27. [Google Scholar] [CrossRef]
- Dunn, G.P.; Dunn, I.F.; Curry, W.T. Focus on TILs: Prognostic significance of tumor infiltrating lymphocytes in human glioma. Cancer Immun. 2007, 7, 12. [Google Scholar]
- Yang, I.; Tihan, T.; Han, S.J.; Wrensch, M.R.; Wiencke, J.; Sughrue, M.E.; Parsa, A.T. CD8+ T-cell infiltrate in newly diagnosed glioblastoma is associated with long-term survival. J. Clin. Neurosci. 2010, 17, 1381–1385. [Google Scholar] [CrossRef] [Green Version]
- Lohr, J.; Ratliff, T.; Huppertz, A.; Ge, Y.; Dictus, C.; Ahmadi, R.; Grau, S.; Hiraoka, N.; Eckstein, V.; Ecker, R.C.; et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin. Cancer Res. 2011, 17, 4296–4308. [Google Scholar] [CrossRef] [Green Version]
- Hayek, S.M.; Deer, T.R.; Pope, J.E.; Panchal, S.J.; Patel, V.B. Intrathecal therapy for cancer and non-cancer pain. Pain Physician 2011, 14, 219–248. [Google Scholar] [CrossRef]
- Serwer, L.P.; James, C.D. Challenges in drug delivery to tumors of the central nervous system: An overview of pharmacological and surgical considerations. Adv. Drug Deliv. Rev. 2012, 64, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Dakhil, S.; Ensminger, W.; Kindt, G.; Niederhuber, J.; Chandler, W.; Greenberg, H.; Wheeler, R. Implanted system for intraventricular drug infusion in central nervous system tumors. Cancer Treat. Rep. 1981, 65, 401–411. [Google Scholar] [PubMed]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef]
- Press, M.F.; Cordon-Cardo, C.; Slamon, D.J. Expression of the HER-2/neu proto-oncogene in normal human adult and fetal tissues. Oncogene 1990, 7, 953–962. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
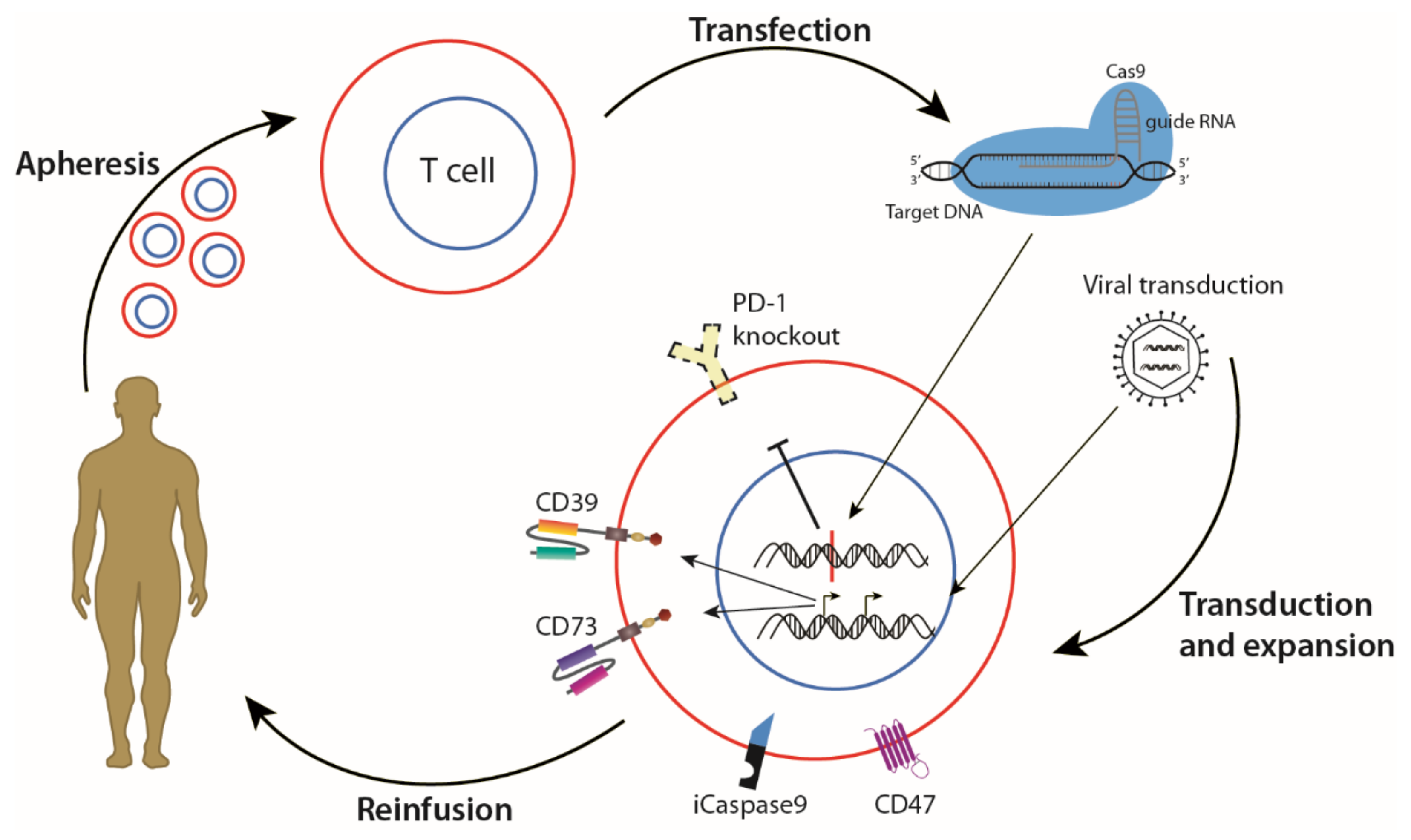{kind=link}
| Comparisons to Consider | CAR T | CAR NK |
|---|---|---|
| Cell source | Autologous setting | Allogeneic setting or cell lines [108] |
| Off-the-shelf | Not eligible | Eligible [100] |
| GvHD | Induces GvHD | Not known to induce GvHD |
| Gene delivery | Effective by transfection or transduction | Effective by Cas9 RNP |
| Patients as candidates | Restricted to patient’s condition | All patients are potential candidates |
| Off-target effects | ICANS/CRS [70,71] | No/minimal ICANS or CRS |
| Response to antigen loss | No alternative mechanism | Response by existing NK cell receptors |
| Infusion dose | Single/few doses | Multiple or high |
| In vivo persistency | Longer (several months) [123] | Shorter (days to weeks) [106] |
| FDA/EMA approval | Approved [63,64] | Not yet approved |
| Cancer indication | Haematological malignancies [61] | Alternative for solid tumours |
| Clinical Trial ID | Target | Cell Type/Expression | Trial Method | Trial Status | Reference |
|---|---|---|---|---|---|
| NCT03726515, NCT01454596 | EGFRvIII | Glioma cells, 64% of GBM cases | CAR T | Completed, completed | [138] |
| NCT03383978, NCT01109095 | HER2/ErbB2 | Tumour cells, normal endothelial and neuronal brain cells (low expression) | CAR NK | Recruiting, completed | [139] |
| NCT04003649, NCT02208362 | IL13Rα2 | Tumour cells, TAMs, MDSCs, overexpressed in 50% of GBM cases | CAR T | Recruiting, recruiting | [138] |
| NCT04077866, NCT04385173 | B7H3 | DC, T cells, overexpression on GBM tumour cells | CAR T | Recruiting, recruiting | [140] |
| NCT04270461 | NKG2DL | Receptor expressed on NK, NKT and T cells, ligands expressed on tumour cells | CAR T | Withdrawn | [90] |
| Potential New Targets | Significance of Target | Reference |
|---|---|---|
| CSPG4 | Pre-clinical data suggests combination CAR T therapy with TNFα treatment for GBM. | [170] |
| MR1 | A pan-population MHC class 1-related protein. T cells with TCRs targeting MR1 potently kill cancer cells, and not healthy cells. | [171] |
| NKG2D | Multiple ligands, less likely to lead to immune escape. | [156] |
| CD39 | Promotes T cell proliferation and Th1 cytokine release. | [172] |
| 2-HG | In IDH-mutant gliomas, IDH inhibition combined with immunotherapy might enhance T cell activity. | [173] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnesen, V.S.; Gras Navarro, A.; Chekenya, M. Challenges and Prospects for Designer T and NK Cells in Glioblastoma Immunotherapy. Cancers 2021, 13, 4986. https://doi.org/10.3390/cancers13194986
Arnesen VS, Gras Navarro A, Chekenya M. Challenges and Prospects for Designer T and NK Cells in Glioblastoma Immunotherapy. Cancers. 2021; 13(19):4986. https://doi.org/10.3390/cancers13194986
Chicago/Turabian StyleArnesen, Victoria Smith, Andrea Gras Navarro, and Martha Chekenya. 2021. "Challenges and Prospects for Designer T and NK Cells in Glioblastoma Immunotherapy" Cancers 13, no. 19: 4986. https://doi.org/10.3390/cancers13194986
APA StyleArnesen, V. S., Gras Navarro, A., & Chekenya, M. (2021). Challenges and Prospects for Designer T and NK Cells in Glioblastoma Immunotherapy. Cancers, 13(19), 4986. https://doi.org/10.3390/cancers13194986