
Reduction of Cell Proliferation by Acute C2H6O Exposure

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animal Procedures
2.2. In Vivo and Ex Vivo Optical Bioluminescence Imaging
2.3. Complete Blood Counts
2.4. Proteome Analysis
2.5. Immunoblot Analysis
2.6. Statistical Analysis
3. Results
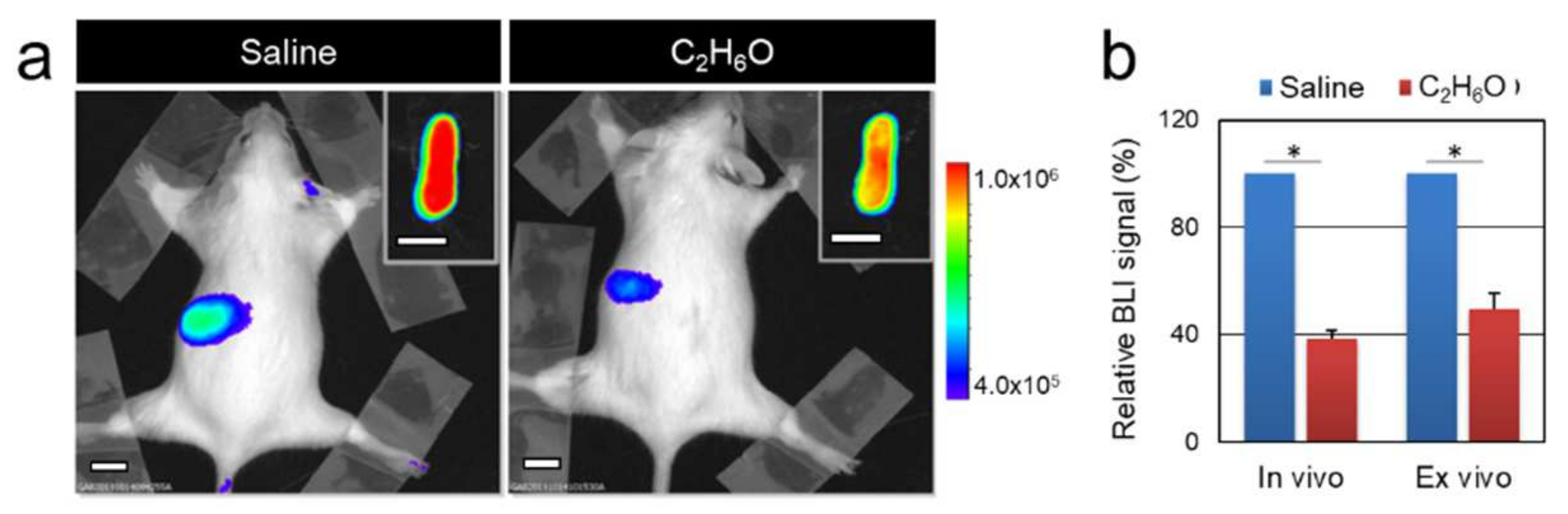
3.1. Alcohol Intake Transiently Modulates Bioluminescence Signals Associated with Cell Proliferation in the MITO-Luc Mouse
3.1.1. Modulation of Aldehyde Dehydrogenases Activity Affects Ethanol-Induced Alteration of Bioluminescence
3.1.2. Polyphenol Administration Partially Restores Bioluminescence upon Ethanol Exposure
3.2. Acute Methanol Administration Transiently Modulates Cell Proliferation in the MITO-Luc Mice
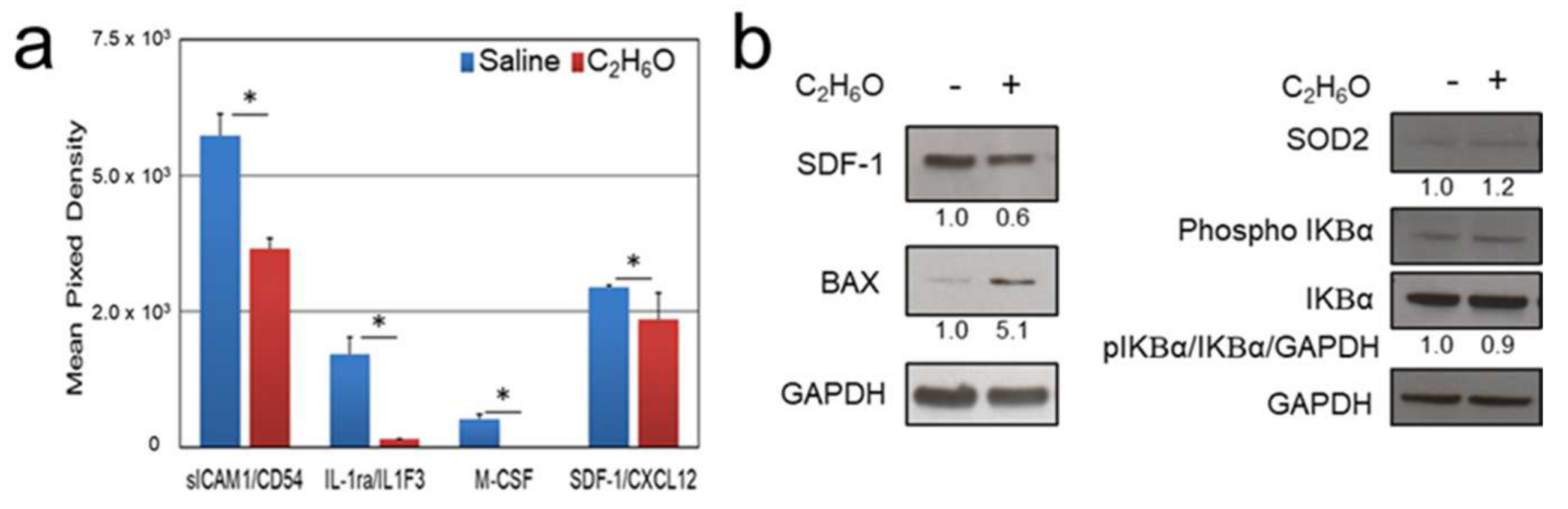
3.3. Proteome Analysis after Ethanol Administration Indicate Serum and Splenic Reduction of CXCL12 Levels
3.4. Immunoblot Analysis after Ethanol Administration Indicate Increased Splenic Expression of Oxidative Stress and Apoptosis Markers
3.5. Short-Term Activation of NF-Y Driven Luciferase after Acute Ethanol Administration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pflaum, T.; Hausler, T.; Baumung, C.; Ackermann, S.; Kuballa, T.; Rehm, J.; Lachenmeier, D.W. Carcinogenic compounds in alcoholic beverages: An update. Arch. Toxicol. 2016, 90, 2349–2367. [Google Scholar] [CrossRef]
- Szabo, G.; Mandrekar, P. A recent perspective on alcohol, immunity, and host defense. Alcohol Clin. Exp. Res. 2009, 33, 220–232. [Google Scholar] [CrossRef]
- Blot, W.J. Alcohol and cancer. Cancer Res. 1992, 52, 2119s–2123s. [Google Scholar]
- Jung, M.K.; Callaci, J.J.; Lauing, K.L.; Otis, J.S.; Radek, K.A.; Jones, M.K.; Kovacs, E.J. Alcohol exposure and mechanisms of tissue injury and repair. Alcohol Clin. Exp. Res. 2011, 35, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Molina, P.E.; Happel, K.I.; Zhang, P.; Kolls, J.K.; Nelson, S. Focus on: Alcohol and the immune system. Alcohol Res. Health 2010, 33, 97–108. [Google Scholar] [PubMed]
- Messingham, K.A.; Faunce, D.E.; Kovacs, E.J. Alcohol, injury, and cellular immunity. Alcohol 2002, 28, 137–149. [Google Scholar] [CrossRef]
- Brown, L.A.; Cook, R.T.; Jerrells, T.R.; Kolls, J.K.; Nagy, L.E.; Szabo, G.; Wands, J.R.; Kovacs, E.J. Acute and chronic alcohol abuse modulate immunity. Alcohol Clin. Exp. Res. 2006, 30, 1624–1631. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.H.; Ho, P.S.; Yeh, Y.W.; Liang, C.S.; Kuo, S.C.; Huang, C.C.; Chen, C.Y.; Shih, M.C.; Ma, K.H.; Sung, Y.F.; et al. Differential cytokine levels between early withdrawal and remission states in patients with alcohol dependence. Psychoneuroendocrinology 2017, 76, 183–191. [Google Scholar] [CrossRef]
- Barr, T.; Helms, C.; Grant, K.; Messaoudi, I. Opposing effects of alcohol on the immune system. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 65, 242–251. [Google Scholar] [CrossRef] [Green Version]
- Thinnahanumaih, M.; Puranik, N.; Bidare, C.; Kammar, K.F.; Venkatakrishniah, O.K.; Maitri, V. Moderate alcohol intake even a short duration has deleterious effects on hematologic profile in Indian men. Int. J. Med. Sci. Public Health 2012, 1, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, T.W. Prenatal alcohol exposure and the developing immune system. Alcohol Res. 2015, 37, 279–285. [Google Scholar]
- Lindenbaum, J. Hematologic complications of alcohol abuse. Semin Liver Dis 1987, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhu, Z.; Zhang, F.; Meadows, G.G. Alcohol consumption and antitumor immunity: Dynamic changes from activation to accelerated deterioration of the immune system. Adv. Exp. Med. Biol. 2015, 815, 313–331. [Google Scholar] [CrossRef]
- Scherübl, H. Alcohol use and gastrointestinal cancer risk. Visc. Med. 2020, 36, 175–181. [Google Scholar] [CrossRef]
- Ballard, H.S. The hematological complications of alcoholism. Alcohol Health Res. World 1997, 21, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz, L.E.; Montero, A.; González-Gross, M.; Vallejo, A.I.; Romeo, J.; Marcos, A. Influence of alcohol consumption on immunological status: A review. Eur. J. Clin. Nutr. 2002, 56, S50–S53. [Google Scholar] [CrossRef]
- Pasala, S.; Barr, T.; Messaoudi, I. Impact of alcohol abuse on the adaptive immune system. Alcohol Res. 2015, 37, 185–197. [Google Scholar]
- Fröbel, J.; Landspersky, T.; Percin, G.; Schreck, C.; Rahmig, S.; Ori, A.; Nowak, D.; Essers, M.; Waskow, C.; Oostendorp, R.A.J. The hematopoietic bone marrow niche ecosystem. Front. Cell Dev. Biol. 2021, 9, 705410. [Google Scholar] [CrossRef]
- Huff, N.K.; Spencer, N.D.; Gimble, J.M.; Bagby, G.J.; Nelson, S.; Lopez, M.J. Impaired expansion and multipotentiality of adult stromal cells in a rat chronic alcohol abuse model. Alcohol 2011, 45, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Taffe, M.A.; Kotzebue, R.W.; Crean, R.D.; Crawford, E.F.; Edwards, S.; Mandyam, C.D. Long-lasting reduction in hippocampal neurogenesis by alcohol consumption in adolescent nonhuman primates. Proc. Natl. Acad. Sci. USA 2010, 107, 11104–11109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meagher, R.C.; Sieber, F.; Spivak, J.L. Suppression of hematopoietic-progenitor-cell proliferation by ethanol and acetaldehyde. N. Engl. J. Med. 1982, 307, 845–849. [Google Scholar] [CrossRef]
- Di Rocco, G.; Baldari, S.; Pani, G.; Toietta, G. Stem cells under the influence of alcohol: Effects of ethanol consumption on stem/progenitor cells. Cell. Mol. Life Sci. 2019, 76, 231–244. [Google Scholar] [CrossRef] [Green Version]
- Boffetta, P.; Hashibe, M. Alcohol and cancer. Lancet Oncol. 2006, 7, 149–156. [Google Scholar] [CrossRef]
- Hao, H.N.; Parker, G.C.; Zhao, J.; Barami, K.; Lyman, W.D. Differential responses of human neural and hematopoietic stem cells to ethanol exposure. J. Hematother. Stem Cell Res. 2003, 12, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Varlamov, O.; Bucher, M.; Myatt, L.; Newman, N.; Grant, K.A. Daily ethanol drinking followed by an abstinence period impairs bone marrow niche and mitochondrial function of hematopoietic stem/progenitor cells in rhesus macaques. Alcohol Clin. Exp. Res. 2020, 44, 1088–1098. [Google Scholar] [CrossRef]
- Crabbe, J.C.; Phillips, T.J.; Belknap, J.K. The complexity of alcohol drinking: Studies in rodent genetic models. Behav. Genet. 2010, 40, 737–750. [Google Scholar] [CrossRef] [Green Version]
- Goeman, F.; Manni, I.; Artuso, S.; Ramachandran, B.; Toietta, G.; Bossi, G.; Rando, G.; Cencioni, C.; Germoni, S.; Straino, S.; et al. Molecular imaging of nuclear factor-Y transcriptional activity maps proliferation sites in live animals. Mol. Biol. Cell 2012, 23, 1467–1474. [Google Scholar] [CrossRef]
- Manni, I.; Di Rocco, G.; Fusco, S.; Leone, L.; Barbati, S.A.; Carapella, C.M.; Grassi, C.; Piaggio, G.; Toietta, G. Monitoring the response of hyperbilirubinemia in the mouse brain by in vivo bioluminescence imaging. Int. J. Mol. Sci. 2017, 18, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzi, N.; Manni, I.; Vantaggiato, C.; Delledonne, G.A.; Gentileschi, M.P.; Maggi, A.; Piaggio, G.; Ciana, P. In vivo imaging of cell proliferation for a dynamic, whole body, analysis of undesired drug effects. Toxicol. Sci. 2015, 145, 296–306. [Google Scholar] [CrossRef] [Green Version]
- Courties, G.; Herisson, F.; Sager, H.B.; Heidt, T.; Ye, Y.; Wei, Y.; Sun, Y.; Severe, N.; Dutta, P.; Scharff, J.; et al. Ischemic stroke activates hematopoietic bone marrow stem cells. Circ. Res. 2015, 116, 407–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, R.S.; Marbourg, J.M.; Brennan, F.H.; Mifflin, K.A.; Hall, J.C.E.; Jiang, R.R.; Mo, X.M.; Karunasiri, M.; Burke, M.H.; Dorrance, A.M.; et al. Spinal cord injury causes chronic bone marrow failure. Nat. Commun. 2020, 11, 3702. [Google Scholar] [CrossRef] [PubMed]
- Goral, J.; Karavitis, J.; Kovacs, E.J. Exposure-dependent effects of ethanol on the innate immune system. Alcohol 2008, 42, 237–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, W.; Zhang, W.; Li, Q.; Xie, G.; Sun, Q.; Sun, X.; Tan, X.; Jia, W.; Zhou, Z. Pharmacological activation of aldehyde dehydrogenase 2 by Alda-1 reverses alcohol-induced hepatic steatosis and cell death in mice. J. Hepatol. 2015, 62, 1375–1381. [Google Scholar] [CrossRef]
- Hillbom, M.E.; Sarviharju, M.S.; Lindros, K.O. Potentiation of ethanol toxicity by cyanamide in relation to acetaldehyde accumulation. Toxicol. Appl. Pharmacol 1983, 70, 133–139. [Google Scholar] [CrossRef]
- Carito, V.; Ceccanti, M.; Cestari, V.; Natella, F.; Bello, C.; Coccurello, R.; Mancinelli, R.; Fiore, M. Olive polyphenol effects in a mouse model of chronic ethanol addiction. Nutrition 2017, 33, 65–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, K.W.; Perkins, R.A.; Kawagoe, J.L.; Pollack, G.M. Comparative toxicokinetics of methanol in the female mouse and rat. Fundam. Appl. Toxicol. 1995, 26, 258–264. [Google Scholar] [CrossRef]
- Baldari, S.; Di Rocco, G.; Trivisonno, A.; Samengo, D.; Pani, G.; Toietta, G. Promotion of survival and engraftment of transplanted adipose tissue-derived stromal and vascular cells by overexpression of manganese superoxide dismutase. Int. J. Mol. Sci. 2016, 17, 1082. [Google Scholar] [CrossRef] [Green Version]
- Di Rocco, G.; Gentile, A.; Antonini, A.; Truffa, S.; Piaggio, G.; Capogrossi, M.; Toietta, G. Analysis of biodistribution and engraftment into the liver of genetically modified mesenchymal stromal cells derived from adipose tissue. Cell Transpl. 2012, 21, 1997–2008. [Google Scholar] [CrossRef] [Green Version]
- Wands, J.R.; Carter, E.A.; Bucher, N.L.; Isselbacher, K.J. Inhibition of hepatic regeneration in rats by acute and chronic ethanol intoxication. Gastroenterology 1979, 77, 528–531. [Google Scholar] [CrossRef]
- Smith, C.; Gasparetto, M.; Jordan, C.; Pollyea, D.A.; Vasiliou, V. The effects of alcohol and aldehyde dehydrogenases on disorders of hematopoiesis. Adv. Exp. Med. Biol. 2015, 815, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Casagrande, G.; Michot, F. Alcohol-induced bone marrow damage: Status before and after a 4-week period of abstinence from alcohol with or without disulfiram. A randomized bone marrow study in alcohol-dependent individuals. Blut 1989, 59, 231–236. [Google Scholar] [CrossRef]
- Guo, R.; Ren, J. Alcohol and acetaldehyde in public health: From marvel to menace. Int. J. Environ. Res. Public Health 2010, 7, 1285–1301. [Google Scholar] [CrossRef] [Green Version]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Daly, M.; Arends, M.J.; Patel, K.J. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 2012, 489, 571–575. [Google Scholar] [CrossRef]
- Joenje, H. Metabolism: Alcohol, DNA and disease. Nature 2011, 475, 45–46. [Google Scholar] [CrossRef]
- Langevin, F.; Crossan, G.P.; Rosado, I.V.; Arends, M.J.; Patel, K.J. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 2011, 475, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Jamal, M.; Ameno, K.; Tanaka, N.; Ito, A.; Takakura, A.; Kumihashi, M.; Kinoshita, H. Ethanol and acetaldehyde after intraperitoneal administration to Aldh2-knockout mice-reflection in blood and brain levels. Neurochem. Res. 2016, 41, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Vasudevan, D.M. Alcohol-induced oxidative stress. Life Sci. 2007, 81, 177–187. [Google Scholar] [CrossRef]
- Koch, O.R.; Pani, G.; Borrello, S.; Colavitti, R.; Cravero, A.; Farrè, S.; Galeotti, T. Oxidative stress and antioxidant defenses in ethanol-induced cell injury. Mol. Aspects Med. 2004, 25, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Koop, D.R. Alcohol metabolism’s damaging effects on the cell: A focus on reactive oxygen generation by the enzyme cytochrome P450 2E1. Alcohol Res. Health 2006, 29, 274–280. [Google Scholar] [PubMed]
- Brocardo, P.S.; Gil-Mohapel, J.; Christie, B.R. The role of oxidative stress in fetal alcohol spectrum disorders. Brain Res. Rev. 2011, 67, 209–225. [Google Scholar] [CrossRef]
- Arteel, G.E. Oxidants and antioxidants in alcohol-induced liver disease. Gastroenterology 2003, 124, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Joya, X.; Garcia-Algar, O.; Salat-Batlle, J.; Pujades, C.; Vall, O. Advances in the development of novel antioxidant therapies as an approach for fetal alcohol syndrome prevention. Birth Defects Res. A Clin. Mol. Teratol. 2015, 103, 163–177. [Google Scholar] [CrossRef]
- De Nicoló, S.; Tarani, L.; Ceccanti, M.; Maldini, M.; Natella, F.; Vania, A.; Chaldakov, G.N.; Fiore, M. Effects of olive polyphenols administration on nerve growth factor and brain-derived neurotrophic factor in the mouse brain. Nutrition 2013, 29, 681–687. [Google Scholar] [CrossRef]
- Alirezaei, M.; Dezfoulian, O.; Neamati, S.; Rashidipour, M.; Tanideh, N.; Kheradmand, A. Oleuropein prevents ethanol-induced gastric ulcers via elevation of antioxidant enzyme activities in rats. J. Physiol. Biochem. 2012, 68, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Pontel, L.B.; Rosado, I.V.; Burgos-Barragan, G.; Garaycoechea, J.I.; Yu, R.; Arends, M.J.; Chandrasekaran, G.; Broecker, V.; Wei, W.; Liu, L.; et al. Endogenous formaldehyde is a hematopoietic stem cell genotoxin and metabolic carcinogen. Mol. Cell 2015, 60, 177–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.; Wen, H.; Yuan, L.; McHale, C.M.; Li, H.; Wang, K.; Yuan, J.; Yang, X.; Zhang, L. Formaldehyde induces toxicity in mouse bone marrow and hematopoietic stem/progenitor cells and enhances benzene-induced adverse effects. Arch. Toxicol. 2017, 91, 921–933. [Google Scholar] [CrossRef]
- Szende, B.; Tyihák, E. Effect of formaldehyde on cell proliferation and death. Cell Biol. Int. 2010, 34, 1273–1282. [Google Scholar] [CrossRef]
- Achur, R.N.; Freeman, W.M.; Vrana, K.E. Circulating cytokines as biomarkers of alcohol abuse and alcoholism. J. Neuroimmune Pharmacol. 2010, 5, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crews, F.T.; Bechara, R.; Brown, L.A.; Guidot, D.M.; Mandrekar, P.; Oak, S.; Qin, L.; Szabo, G.; Wheeler, M.; Zou, J. Cytokines and alcohol. Alcohol Clin. Exp. Res. 2006, 30, 720–730. [Google Scholar] [CrossRef] [PubMed]
- García-Marchena, N.; Araos, P.F.; Barrios, V.; Sánchez-Marín, L.; Chowen, J.A.; Pedraz, M.; Castilla-Ortega, E.; Romero-Sanchiz, P.; Ponce, G.; Gavito, A.L.; et al. Plasma chemokines in patients with alcohol use disorders: Association of CCL11 (Eotaxin-1) with psychiatric comorbidity. Front. Psychiatry 2016, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [Green Version]
- Koch, O.R.; De Leo, M.E.; Borrello, S.; Palombini, G.; Galeotti, T. Ethanol treatment up-regulates the expression of mitochondrial manganese superoxide dismutase in rat liver. Biochem. Biophys. Res. Commun. 1994, 201, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Slukvin, I.I.; Jerrells, T.R. Different pathways of in vitro ethanol-induced apoptosis in thymocytes and splenic T and B lymphocytes. Immunopharmacology 1995, 31, 43–57. [Google Scholar] [CrossRef]
- Nowak, A.J.; Relja, B. The impact of acute or chronic alcohol intake on the NF-κB signaling pathway in alcohol-related liver disease. Int. J. Mol. Sci. 2020, 21, 9407. [Google Scholar] [CrossRef]
- von Haefen, C.; Sifringer, M.; Menk, M.; Spies, C.D. Ethanol enhances susceptibility to apoptotic cell death via down-regulation of autophagy-related proteins. Alcohol Clin. Exp. Res. 2011, 35, 1381–1391. [Google Scholar] [CrossRef]
- Eid, N.A.; Ito, Y.; Li, Z.; Abe, H.; Kusakabe, K.; Shibata, M.A.; Otsuki, Y. The relationship between apoptosis and splenocyte depletion in rats following ethanol treatment. Med. Electron. Microsc. 2000, 33, 89–95. [Google Scholar] [CrossRef]
- Saad, A.J.; Jerrells, T.R. Flow cytometric and immunohistochemical evaluation of ethanol-induced changes in splenic and thymic lymphoid cell populations. Alcohol Clin. Exp. Res. 1991, 15, 796–803. [Google Scholar] [CrossRef]
- Singhal, P.C.; Reddy, K.; Ding, G.; Kapasi, A.; Franki, N.; Ranjan, R.; Nwakoby, I.E.; Gibbons, N. Ethanol-induced macrophage apoptosis: The role of TGF-beta. J. Immunol. 1999, 162, 3031–3036. [Google Scholar]
- Zhang, Y.; Sun, Y.; Miao, Q.; Wang, Q.; Yang, B.; Li, Y.; Li, L.; Zhang, R. Nuclear factor Y participates in alcoholic liver disease by activating SREBP1 expression in mice. Biochem. Biophys. Res. Commun. 2021, 541, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Uddin, R.K.; Singh, S.M. cis-Regulatory sequences of the genes involved in apoptosis, cell growth, and proliferation may provide a target for some of the effects of acute ethanol exposure. Brain Res. 2006, 1088, 31–44. [Google Scholar] [CrossRef]
- Poznyak, V.; Rekve, D. Global Status Report on Alcohol and Health 2018. Available online: https://apps.who.int/iris/handle/10665/274603 (accessed on 25 July 2021).
- Manthey, J.; Shield, K.D.; Rylett, M.; Hasan, O.S.M.; Probst, C.; Rehm, J. Global alcohol exposure between 1990 and 2017 and forecasts until 2030: A modelling study. Lancet 2019, 393, 2493–2502. [Google Scholar] [CrossRef]
- Azagba, S.; Shan, L.; Latham, K.; Manzione, L. Trends in binge and heavy drinking among adults in the United States, 2011–2017. Subst. Use Misuse 2020, 55, 990–997. [Google Scholar] [CrossRef]
- Cservenka, A.; Brumback, T. The burden of binge and heavy drinking on the brain: Effects on adolescent and young adult neural structure and function. Front. Psychol. 2017, 8, 1111. [Google Scholar] [CrossRef]
- Jones, S.A.; Lueras, J.M.; Nagel, B.J. Effects of binge drinking on the developing brain. Alcohol Res. 2018, 39, 87–96. [Google Scholar] [PubMed]
- Pascual, M.; Montesinos, J.; Guerri, C. Role of the innate immune system in the neuropathological consequences induced by adolescent binge drinking. J. Neurosci. Res. 2018, 96, 765–780. [Google Scholar] [CrossRef] [PubMed]
- Hermens, D.F.; Lagopoulos, J. Binge drinking and the young brain: A mini review of the neurobiological underpinnings of alcohol-induced blackout. Front. Psychol. 2018, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.; Lee, J.-E. Impact of drinking alcohol on gut microbiota: Recent perspectives on ethanol and alcoholic beverage. Curr. Opin. Food Sci. 2021, 37, 91–97. [Google Scholar] [CrossRef]
- Praud, D.; Rota, M.; Rehm, J.; Shield, K.; Zatoński, W.; Hashibe, M.; La Vecchia, C.; Boffetta, P. Cancer incidence and mortality attributable to alcohol consumption. Int. J. Cancer 2016, 138, 1380–1387. [Google Scholar] [CrossRef]
- Rehm, J.; Gmel, G.E.; Gmel, G.; Hasan, O.S.M.; Imtiaz, S.; Popova, S.; Probst, C.; Roerecke, M.; Room, R.; Samokhvalov, A.V.; et al. The relationship between different dimensions of alcohol use and the burden of disease—An update. Addiction 2017, 112, 968–1001. [Google Scholar] [CrossRef] [Green Version]
- Parkin, D.M. Cancers attributable to consumption of alcohol in the UK in 2010. Br. J. Cancer 2011, 105, S14–S18. [Google Scholar] [CrossRef]
- Nelson, D.E.; Jarman, D.W.; Rehm, J.; Greenfield, T.K.; Rey, G.; Kerr, W.C.; Miller, P.; Shield, K.D.; Ye, Y.; Naimi, T.S. Alcohol-attributable cancer deaths and years of potential life lost in the United States. Am. J. Public Health 2013, 103, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Rumgay, H.; Shield, K.; Charvat, H.; Ferrari, P.; Sornpaisarn, B.; Obot, I.; Islami, F.; Lemmens, V.E.P.P.; Rehm, J.; Soerjomataram, I. Global burden of cancer in 2020 attributable to alcohol consumption: A population-based study. Lancet Oncol. 2021, 22, 1071–1080. [Google Scholar] [CrossRef]
- Ratna, A.; Mandrekar, P. Alcohol and cancer: Mechanisms and therapies. Biomolecules 2017, 7, 61. [Google Scholar] [CrossRef] [Green Version]
- Nieminen, M.T.; Salaspuro, M. Local acetaldehyde—An essential role in alcohol-related upper gastrointestinal tract carcinogenesis. Cancers 2018, 10, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stornetta, A.; Guidolin, V.; Balbo, S. Alcohol-derived acetaldehyde exposure in the oral cavity. Cancers 2018, 10, 20. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; Jahanzaib Anwar, M.; Usman, A.; Keshavarzian, A.; Bishehsari, F. Colorectal cancer and alcohol consumption-populations to molecules. Cancers 2018, 10, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milsom, M.D. Stem cells spirited away by alcohol-induced DNA damage. Hemasphere 2018, 2, e36. [Google Scholar] [CrossRef]
- Bungartz, G.; Land, H.; Scadden, D.T.; Emerson, S.G. NF-Y is necessary for hematopoietic stem cell proliferation and survival. Blood 2012, 119, 1380–1389. [Google Scholar] [CrossRef]
- Radomska, H.S.; Satterthwaite, A.B.; Taranenko, N.; Narravula, S.; Krause, D.S.; Tenen, D.G. A nuclear factor Y (NFY) site positively regulates the human CD34 stem cell gene. Blood 1999, 94, 3772–3780. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, Y.; Joe, G.J.; Pompetti, R.; Emerson, S.G. NF-Ya activates multiple hematopoietic stem cell (HSC) regulatory genes and promotes HSC self-renewal. Proc. Natl. Acad. Sci. USA 2005, 102, 11728–11733. [Google Scholar] [CrossRef] [Green Version]
- Hicks, S.D.; Lewis, L.; Ritchie, J.; Burke, P.; Abdul-Malak, Y.; Adackapara, N.; Canfield, K.; Shwarts, E.; Gentile, K.; Meszaros, Z.S.; et al. Evaluation of cell proliferation, apoptosis, and DNA-repair genes as potential biomarkers for ethanol-induced CNS alterations. BMC Neurosci. 2012, 13, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachini, N.; Papamatheakis, J. NF-Y and the immune response: Dissecting the complex regulation of MHC genes. Biochim Biophys. Acta Gene Regul. Mech. 2017, 1860, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Valledor, A.F.; Borràs, F.E.; Cullell-Young, M.; Celada, A. Transcription factors that regulate monocyte/macrophage differentiation. J. Leukoc. Biol. 1998, 63, 405–417. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Crews, F.T. Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp. Neurol. 2008, 210, 349–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, S.A.; Geil, C.R.; Nixon, K. Prior binge ethanol exposure potentiates the microglial response in a model of alcohol-induced neurodegeneration. Brain Sci. 2016, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- McClain, J.A.; Morris, S.A.; Deeny, M.A.; Marshall, S.A.; Hayes, D.M.; Kiser, Z.M.; Nixon, K. Adolescent binge alcohol exposure induces long-lasting partial activation of microglia. Brain Behav. Immun. 2011, 25, S120–S128. [Google Scholar] [CrossRef] [Green Version]
- Nixon, K.; Crews, F.T. Binge ethanol exposure decreases neurogenesis in adult rat hippocampus. J. Neurochem. 2002, 83, 1087–1093. [Google Scholar] [CrossRef]
- Sutherland, G.T.; Sheahan, P.J.; Matthews, J.; Dennis, C.V.; Sheedy, D.S.; McCrossin, T.; Curtis, M.A.; Kril, J.J. The effects of chronic alcoholism on cell proliferation in the human brain. Exp. Neurol. 2013, 247, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Chang, C.C.; Basson, M.D.; Upham, B.L.; Wei, L.; Zhang, P. Alcohol disrupts human liver stem/progenitor cell proliferation and differentiation. J. Stem Cell Res. Ther. 2014, 4, 205. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhou, H.; Chervenak, R.; Moscatello, K.M.; Brunson, L.E.; Chervenak, D.C.; Wolcott, R.M. Ethanol exhibits specificity in its effects on differentiation of hematopoietic progenitors. Cell Immunol. 2009, 255, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Latvala, J.; Parkkila, S.; Melkko, J.; Niemelä, O. Acetaldehyde adducts in blood and bone marrow of patients with ethanol-induced erythrocyte abnormalities. Mol. Med. 2001, 7, 401–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hira, A.; Yabe, H.; Yoshida, K.; Okuno, Y.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; Miyano, S.; Nakamura, J.; Kojima, S.; et al. Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients. Blood 2013, 122, 3206–3209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Wassenhove, L.D.; Mochly-Rosen, D.; Weinberg, K.I. Aldehyde dehydrogenase 2 in aplastic anemia, Fanconi anemia and hematopoietic stem cells. Mol. Genet. Metab. 2016, 119, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhou, H.; Moscatello, K.M.; Dixon, C.; Brunson, L.E.; Chervenak, R.; Chervenak, D.C.; Zhao, X.; Wolcott, R.M. In utero exposure to alcohol alters cell fate decisions by hematopoietic progenitors in the bone marrow of offspring mice during neonatal development. Cell Immunol. 2006, 239, 75–85. [Google Scholar] [CrossRef]
- Moscatello, K.M.; Biber, K.L.; Jennings, S.R.; Chervenak, R.; Wolcott, R.M. Effects of in utero alcohol exposure on B cell development in neonatal spleen and bone marrow. Cell Immunol. 1999, 191, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Latino-Martel, P.; Chan, D.S.; Druesne-Pecollo, N.; Barrandon, E.; Hercberg, S.; Norat, T. Maternal alcohol consumption during pregnancy and risk of childhood leukemia: Systematic review and meta-analysis. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1238–1260. [Google Scholar] [CrossRef] [Green Version]
- Scheer, M.A.; Schneider, K.J.; Finnigan, R.L.; Maloney, E.P.; Wells, M.A.; Clemens, D.L. The involvement of acetaldehyde in ethanol-induced cell cycle impairment. Biomolecules 2016, 6, 17. [Google Scholar] [CrossRef] [Green Version]
- Gentry, T.; Foster, S.; Winstead, L.; Deibert, E.; Fiordalisi, M.; Balber, A. Simultaneous isolation of human BM hematopoietic, endothelial and mesenchymal progenitor cells by flow sorting based on aldehyde dehydrogenase activity: Implications for cell therapy. Cytotherapy 2007, 9, 259–274. [Google Scholar] [CrossRef]
- Vassalli, G. Aldehyde dehydrogenases: Not just markers, but functional regulators of stem cells. Stem Cells Int. 2019, 2019, 3904645. [Google Scholar] [CrossRef] [Green Version]
- Chute, J.P.; Muramoto, G.G.; Whitesides, J.; Colvin, M.; Safi, R.; Chao, N.J.; McDonnell, D.P. Inhibition of aldehyde dehydrogenase and retinoid signaling induces the expansion of human hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 2006, 103, 11707–11712. [Google Scholar] [CrossRef] [Green Version]
- Dollé, L.; Gao, B. Pharmacological chaperone therapies: Can aldehyde dehydrogenase activator make us healthier? J. Hepatol. 2015, 62, 1228–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Wang, C.; Xu, H.; Gao, Y. Aldehyde dehydrogenase, liver disease and cancer. Int. J. Biol. Sci. 2020, 16, 921–934. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Ferreira, J.C.B.; Joshi, A.U.; Stevens, M.C.; Li, S.J.; Hsu, J.H.; Maclean, R.; Ferreira, N.D.; Cervantes, P.R.; Martinez, D.D.; et al. Novel and prevalent non-East Asian ALDH2 variants; Implications for global susceptibility to aldehydes’ toxicity. EBioMedicine 2020, 55, 102753. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldari, S.; Manni, I.; Di Rocco, G.; Paolini, F.; Palermo, B.; Piaggio, G.; Toietta, G. Reduction of Cell Proliferation by Acute C2H6O Exposure. Cancers 2021, 13, 4999. https://doi.org/10.3390/cancers13194999
Baldari S, Manni I, Di Rocco G, Paolini F, Palermo B, Piaggio G, Toietta G. Reduction of Cell Proliferation by Acute C2H6O Exposure. Cancers. 2021; 13(19):4999. https://doi.org/10.3390/cancers13194999
Chicago/Turabian StyleBaldari, Silvia, Isabella Manni, Giuliana Di Rocco, Francesca Paolini, Belinda Palermo, Giulia Piaggio, and Gabriele Toietta. 2021. "Reduction of Cell Proliferation by Acute C2H6O Exposure" Cancers 13, no. 19: 4999. https://doi.org/10.3390/cancers13194999
APA StyleBaldari, S., Manni, I., Di Rocco, G., Paolini, F., Palermo, B., Piaggio, G., & Toietta, G. (2021). Reduction of Cell Proliferation by Acute C2H6O Exposure. Cancers, 13(19), 4999. https://doi.org/10.3390/cancers13194999