
EZH2 Inhibition as New Epigenetic Treatment Option for Pancreatic Neuroendocrine Neoplasms (PanNENs)

 , , , , , ,
, , , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Collective
2.2. Cell Culture
2.3. In Vitro Drug Treatment
2.3.1. MTT Assay
2.3.2. IncuCyte Real-Time Cell Confluence
2.4. Western Blotting
2.5. EZH2 Silencing
2.6. In Vivo Experiments
2.7. Primary Cells Treatment
2.7.1. Primary Cell Culture
2.7.2. Primary Cell Isolation and Culture
2.7.3. Viability Measurement in Islet-Like Tumoroids
2.7.4. Micro-Cell-Blocks (MCBs) from Islet-Like Tumoroids
2.7.5. Curve Fitting and Drug Sensitivity Data
2.8. Correlation and Survival Analysis
3. Results
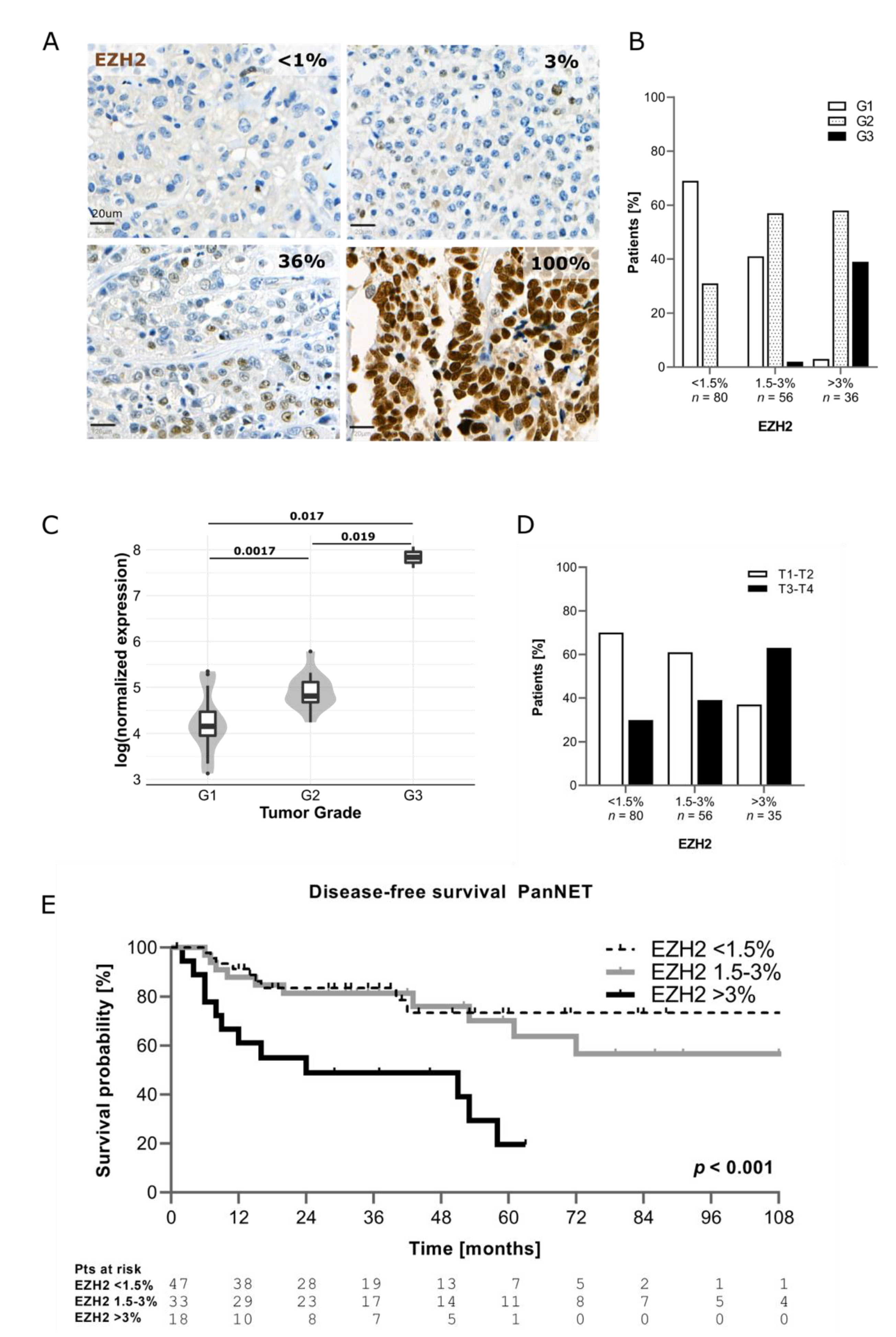
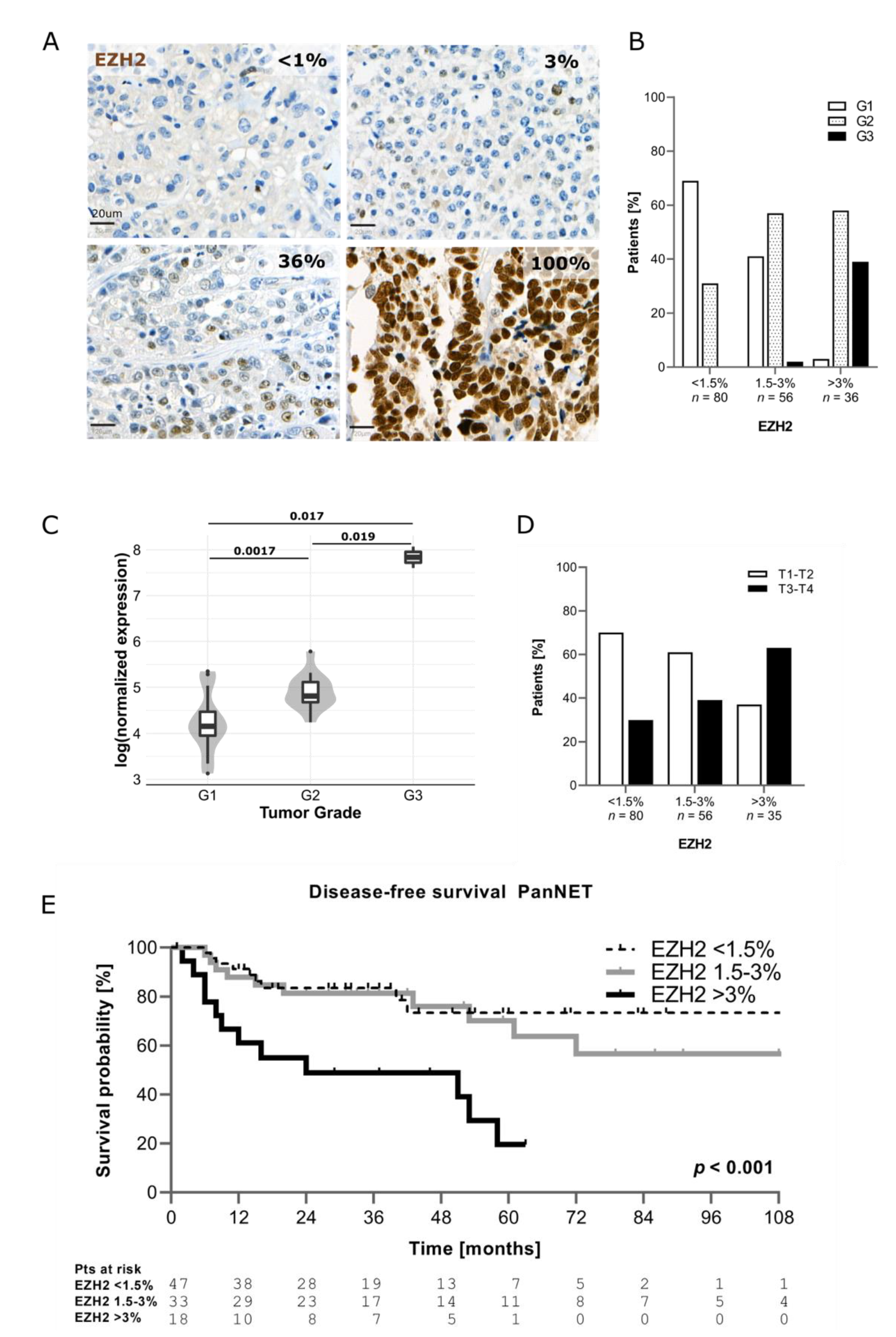
3.1. EZH2 Expression in PanNEN Correlates with Advanced Disease Status and Features of Aggressiveness
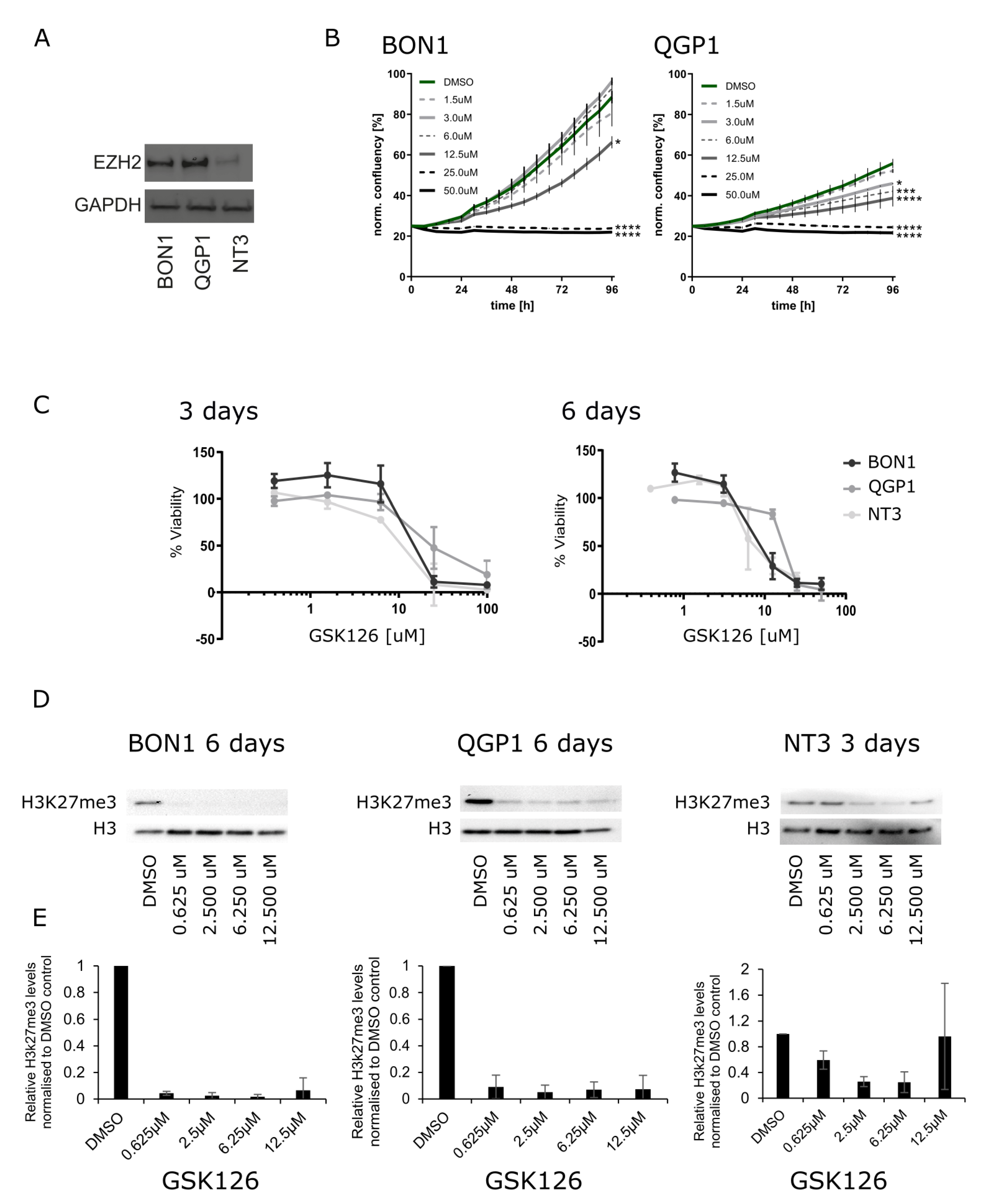
3.2. Inhibition of EZH2 in PanNEN Reduced Cell Viability and H3k27me3 Levels
3.3. Silencing of EZH2 in High-Grade PanNEN Cell Lines Impaired Cell Growth
3.4. Anti-EZH2 Treatment of Rip1TAG2 Mice Reduced H3K27me3 Levels and Tumor Burden
3.5. Treatment of Patient-Derived PanNET Tumoroids with EZH2 Inhibitors Reduced Cell Viability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Classification of Tumours Editorial Board. WHO Classification of Tumours. Digestive System Tumours, 5th ed.; IARC Press: Lyon, France, 2019. [Google Scholar]
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef]
- Elvebakken, H.; Perren, A.; Scoazec, J.-Y.; Tang, L.H.; Federspiel, B.; Klimstra, D.S.; Vestermark, L.W.; Ali, A.S.; Zlobec, I.; Myklebust, T.Å.; et al. A Consensus-Developed Morphological Re-Evaluation of 196 High-Grade Gastroenteropancreatic Neuroendocrine Neoplasms and Its Clinical Correlations. Neuroendocrinology 2021, 111, 883–894. [Google Scholar] [CrossRef]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.-M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Konukiewitz, B.; Jesinghaus, M.; Steiger, K.; Schlitter, A.M.; Kasajima, A.; Sipos, B.; Zamboni, G.; Weichert, W.; Pfarr, N.; Klöppel, G. Pancreatic neuroendocrine carcinomas reveal a closer relationship to ductal adenocarcinomas than to neuroendocrine tumors G3. Hum. Pathol. 2018, 77, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Rinke, A.; Auernhammer, C.J.; Bodei, L.; Kidd, M.; Krug, S.; Lawlor, R.; Marinoni, I.; Perren, A.; Scarpa, A.; Sorbye, H.; et al. Treatment of advanced gastroenteropancreatic neuroendocrine neoplasia, are we on the way to personalised medicine? Gut 2021, 70, 1768–1781. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, A.; Pipinikas, C.P.; Maire, R.S.; Bräutigam, K.; Simillion, C.; Dettmer, M.S.; Vassella, E.; Thirlwell, C.; Perren, A.; Marinoni, I. Epigenetic landscape of pancreatic neuroendocrine tumours reveals distinct cells of origin and means of tumour progression. Commun. Biol. 2020, 3, 740. [Google Scholar] [CrossRef]
- Di Domenico, A.; Wiedmer, T.; Marinoni, I.; Perren, A. Genetic and epigenetic drivers of neuroendocrine tumours (NET). Endocr. Relat. Cancer 2017, 24, R315–R334. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; LaFramboise, W.A.; Liu, T.-C.; Cao, D.; Luvison, A.; Miller, C.; Lyons, M.A.; O’Sullivan, R.J.; Zureikat, A.H.; Hogg, M.E.; et al. Loss of Chromatin-Remodeling Proteins and/or CDKN2A Associates With Metastasis of Pancreatic Neuroendocrine Tumors and Reduced Patient Survival Times. Gastroenterology 2018, 154, 2060–2063.e8. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, C.W.M. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.A.B.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Yap, T.A.; Winter, J.N.; Giulino-Roth, L.; Longley, J.; Lopez, J.; Michot, J.-M.; Leonard, J.P.; Ribrag, V.; McCabe, M.T.; Creasy, C.L.; et al. Phase I Study of the Novel Enhancer of Zeste Homolog 2 (EZH2) Inhibitor GSK2816126 in Patients with Advanced Hematologic and Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 7331–7339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gounder, M.; Schöffski, P.; Jones, R.L.; Agulnik, M.; Cote, G.M.; Villalobos, V.M.; Attia, S.; Chugh, R.; Chen, T.W.-W.; Jahan, T.; et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: An international, open-label, phase 2 basket study. Lancet. Oncol. 2020, 21, 1423–1432. [Google Scholar] [CrossRef]
- Morschhauser, F.; Tilly, H.; Chaidos, A.; McKay, P.; Phillips, T.; Assouline, S.; Batlevi, C.L.; Campbell, P.; Ribrag, V.; Damaj, G.L.; et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: An open-label, single-arm, multicentre, phase 2 trial. Lancet. Oncol. 2020, 21, 1433–1442. [Google Scholar] [CrossRef]
- Marinoni, I.; Kurrer, A.S.; Vassella, E.; Dettmer, M.; Rudolph, T.; Banz, V.; Hunger, F.; Pasquinelli, S.; Speel, E.J.; Perren, A. Loss of DAXX and ATRX are associated with chromosome instability and reduced survival of patients with pancreatic neuroendocrine tumors. Gastroenterology 2014, 146, 453–460.e5. [Google Scholar] [CrossRef]
- Lloyd, R.V.; Osamura, R.Y.; Klopper, G.; Rosai, J. (Eds.) WHO Classification of Tumours of Endocrine Organs, 4th ed.; IARC: Lyon, France, 2017. [Google Scholar]
- Brierly, J.D.; Gospodarowicz, M.K.; Witteking, C. (Eds.) TNM Classification of Malignant Tumours, 8th ed.; John Wiley & Sons: Oxford, UK; Hoboken, NJ, USA, 2017. [Google Scholar]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [Green Version]
- Benten, D.; Behrang, Y.; Unrau, L.; Weissmann, V.; Wolters-Eisfeld, G.; Burdak-Rothkamm, S.; Stahl, F.R.; Anlauf, M.; Grabowski, P.; Möbs, M.; et al. Establishment of the first well-differentiated human pancreatic neuroendocrine tumor model. Mol. Cancer Res. 2018, 16, 496–507. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.C.; Berkelman, T.; Yadav, G.; Hammond, M. A defined methodology for reliable quantification of Western blot data. Mol. Biotechnol. 2013, 55, 217–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschan, M.P.; Fischer, K.M.; Fung, V.S.; Pirnia, F.; Borner, M.M.; Fey, M.F.; Tobler, A.; Torbett, B.E. Alternative splicing of the human cyclin D-binding Myb-like protein (hDMP1) yields a truncated protein isoform that alters macrophage differentiation patterns. J. Biol. Chem. 2003, 278, 42750–42760. [Google Scholar] [CrossRef] [Green Version]
- April-Monn, S.L.; Wiedmer, T.; Magdalena, S.; Maire, R.S.; Schiavo Lena, M.; Trippel, M.; Di Domenico, A.; Muffatti, F.; Andreasi, V.; Capurso, G.; et al. 3D Primary Cell Culture: A Novel Preclinical Model For Pancreatic Neuroendocrine Tumors (PanNETs). Neuroendocrinology 2021, 111, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Sebaugh, J.L. Guidelines for accurate EC50/IC50 estimation. Pharm. Stat. 2011, 10, 128–134. [Google Scholar] [CrossRef]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature 1985, 315, 115–122. [Google Scholar] [CrossRef]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Li, W.; Hu, X.; Zhang, Q.; Sun, T.; Cui, S.; Wang, S.; Ouyang, Q.; Yin, Y.; Geng, C.; et al. Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 806–815. [Google Scholar] [CrossRef]
- Biran, N.; Siegel, D.S.; Vesole, D.H. The forgotten class of drugs for multiple myeloma: HDAC inhibitors. Lancet. Haematol. 2018, 5, e604–e605. [Google Scholar] [CrossRef]
- Stein, E.M.; Fathi, A.T.; DiNardo, C.D.; Pollyea, D.A.; Roboz, G.J.; Collins, R.; Sekeres, M.A.; Stone, R.M.; Attar, E.C.; Frattini, M.G.; et al. Enasidenib in patients with mutant IDH2 myelodysplastic syndromes: A phase 1 subgroup analysis of the multicentre, AG221-C-001 trial. Lancet Haematol. 2020, 7, e309–e319. [Google Scholar] [CrossRef]
- Duan, R.; Du, W.; Guo, W. EZH2: A novel target for cancer treatment. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Pasini, D.; Capra, M.; Prosperini, E.; Colli, E.; Helin, K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003, 22, 5323–5335. [Google Scholar] [CrossRef] [Green Version]
- Marconcini, R.; Faviana, P.; Campani, D.; Galli, L.; Antonuzzo, A.; Falcone, A.; Ricci, S. Enhancer of zeste homolog 2 (EZH2) expression in G1 -G2 Pancreatic Neuroendocrine Tumor (pNET). Ann. Oncol. 2016, 27, iv21. [Google Scholar] [CrossRef]
- Wang, H.; Bender, A.; Wang, P.; Karakose, E.; Inabnet, W.B.; Libutti, S.K.; Arnold, A.; Lambertini, L.; Stang, M.; Chen, H.; et al. Insights into beta cell regeneration for diabetes via integration of molecular landscapes in human insulinomas. Nat. Commun. 2017, 8, 767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Gu, X.; Su, I.; Bottino, R.; Contreras, J.L.; Tarakhovsky, A.; Kim, S.K. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 2009, 23, 975–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Qi, J.; Reyes, J.M.; Li, L.; Rao, P.K.; Li, F.; Lin, C.Y.; Perry, J.A.; Lawlor, M.A.; Federation, A.; et al. Oncogenic Deregulation of EZH2 as an Opportunity for Targeted Therapy in Lung Cancer. Cancer Discov. 2016, 6, 1006–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A., 3rd; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.-Y.; Wang, H.; Xiang, P.; Liu, Y.-W.; Li, H.-Z.; Lei, B.-X.; Yu, M.; Qi, S.-T. Inhibition of EZH2 reverses chemotherapeutic drug TMZ chemosensitivity in glioblastoma. Int. J. Clin. Exp. Pathol. 2014, 7, 6662–6670. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | n = 172 (%) |
|---|---|
| Gender | |
| Male | 92 (53) |
| Female | 80 (47) |
| Age, years | 59 (49; 69) * |
| Tumor function | |
| Non-functioning | 140 (81) |
| Insulinoma | 29 (17) |
| Gastrinoma | 2 (1) |
| Glucagonoma | 1 (1) |
| Tumor size, cm | 3 (2.4; 4.1) * |
| T stage ** | |
| T1–T2 | 103 (60) |
| T3–T4 | 68 (40) |
| N stage ** | |
| N0 | 77 (46) |
| N1 | 67 (40) |
| Nx | 24 (14) |
| M stage ** | |
| M0 | 128 (75) |
| M1 | 43 (25) |
| Tumor grade | |
| NET G1 | 79 (46) |
| NET G2 | 78 (45) |
| NET/NEC G3° | 15 (9) |
| Ki67, % | 3 (1.5; 8) * |
| DAXX/ATRX ** | |
| Negative | 59 (36) |
| Positive | 107 (64) |
| Patient | Gender | Age | Grade | Ki67 | Stage | DAXX/ATRX | EZH2 | Tumor Site | In Vitro Sensitivity |
|---|---|---|---|---|---|---|---|---|---|
| mP029 | Female | 55 | NET G2 | 4% | II | Lost | 6.3% | Primary | + |
| mP040 | Female | 55 | NET G2 | 10% | II | Preserved | 3% | Primary | +++ |
| mP044 | Female | 18 | NET G2 | 18% | III | Lost | 1.3% | Primary | + |
| mP055 | Female | 69 | NET G2 | 8% | III | Lost | 0.3% | Primary | + |
| aP321 | Male | 66 | NEC G3 | 50% | IV | Lost | 23% | Liver metastasis | ++ |
| aP476 | Male | 65 | NET G2 | 15% | IV | n.a. | 0% | Liver metastasis | +++ |
| Variables | EZH2 <1.5% n = 80 | EZH2 1.5–3% n = 56 | EZH2 >3% n = 36 | p |
|---|---|---|---|---|
| T Stage | 0.004 | |||
| T1–T2 | 56 (70) | 34 (61) | 13 (37) | |
| T3–T4 | 24 (30) | 22 (39) | 22 (63) | |
| N stage | 0.008 | |||
| N0 | 39 (50) | 28 (51) | 10 (29) | |
| N1 | 24 (30) | 21 (38) | 22 (65) | |
| Nx | 16 (20) | 6 (11) | 2 (6) | |
| M stage | <0.001 | |||
| M0 | 70 (88) | 41 (73) | 17 (49) | |
| M1 | 10 (12) | 15 (27) | 18 (51) | |
| DAXX/ATRX | 0.014 | |||
| Preserved | 57 (74) | 34 (63) | 16 (46) | |
| Lost | 20 (26) | 20 (37) | 19 (54) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
April-Monn, S.L.; Andreasi, V.; Schiavo Lena, M.; Sadowski, M.C.; Kim-Fuchs, C.; Buri, M.C.; Ketkar, A.; Maire, R.; Di Domenico, A.; Schrader, J.; et al. EZH2 Inhibition as New Epigenetic Treatment Option for Pancreatic Neuroendocrine Neoplasms (PanNENs). Cancers 2021, 13, 5014. https://doi.org/10.3390/cancers13195014
April-Monn SL, Andreasi V, Schiavo Lena M, Sadowski MC, Kim-Fuchs C, Buri MC, Ketkar A, Maire R, Di Domenico A, Schrader J, et al. EZH2 Inhibition as New Epigenetic Treatment Option for Pancreatic Neuroendocrine Neoplasms (PanNENs). Cancers. 2021; 13(19):5014. https://doi.org/10.3390/cancers13195014
Chicago/Turabian StyleApril-Monn, Simon Leonhard, Valentina Andreasi, Marco Schiavo Lena, Martin Carl Sadowski, Corina Kim-Fuchs, Michelle Claudine Buri, Avanee Ketkar, Renaud Maire, Annunziata Di Domenico, Jörg Schrader, and et al. 2021. "EZH2 Inhibition as New Epigenetic Treatment Option for Pancreatic Neuroendocrine Neoplasms (PanNENs)" Cancers 13, no. 19: 5014. https://doi.org/10.3390/cancers13195014
APA StyleApril-Monn, S. L., Andreasi, V., Schiavo Lena, M., Sadowski, M. C., Kim-Fuchs, C., Buri, M. C., Ketkar, A., Maire, R., Di Domenico, A., Schrader, J., Muffatti, F., Doglioni, C., Partelli, S., Falconi, M., Perren, A., & Marinoni, I. (2021). EZH2 Inhibition as New Epigenetic Treatment Option for Pancreatic Neuroendocrine Neoplasms (PanNENs). Cancers, 13(19), 5014. https://doi.org/10.3390/cancers13195014