The Transcriptomic Landscape of Prostate Cancer Development and Progression: An Integrative Analysis

 ,
,  , , , ,
, , , ,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Source Data
2.2. Data QC
2.3. Non-Malignant Tissue Assessment
2.4. Tumor Content Estimation
2.5. Stromal Genes Filtering
2.6. Biological and Laboratory Effects
2.7. Microarray and RNAseq Data Pre-Processing
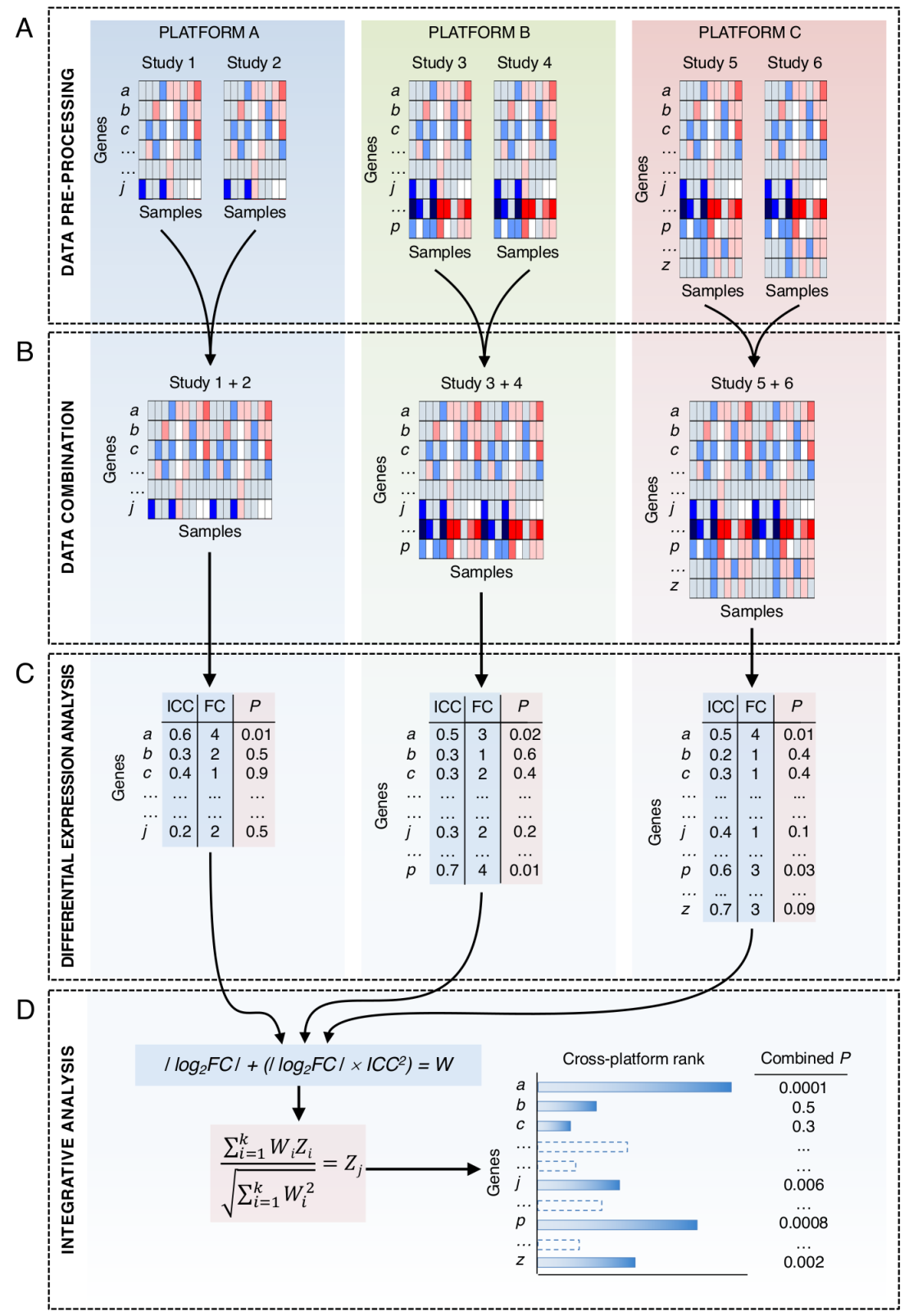
2.8. Data Integration and Molecular Alterations Map Assembly
2.9. Canonical Pathways Analysis
2.10. Survival Analysis
2.11. Experimental Validation—Patient Samples
2.12. Experimental Validation—quantitative PCR(qPCR) Analysis
2.13. Quantification and Statistical Analysis
3. Results and Discussion
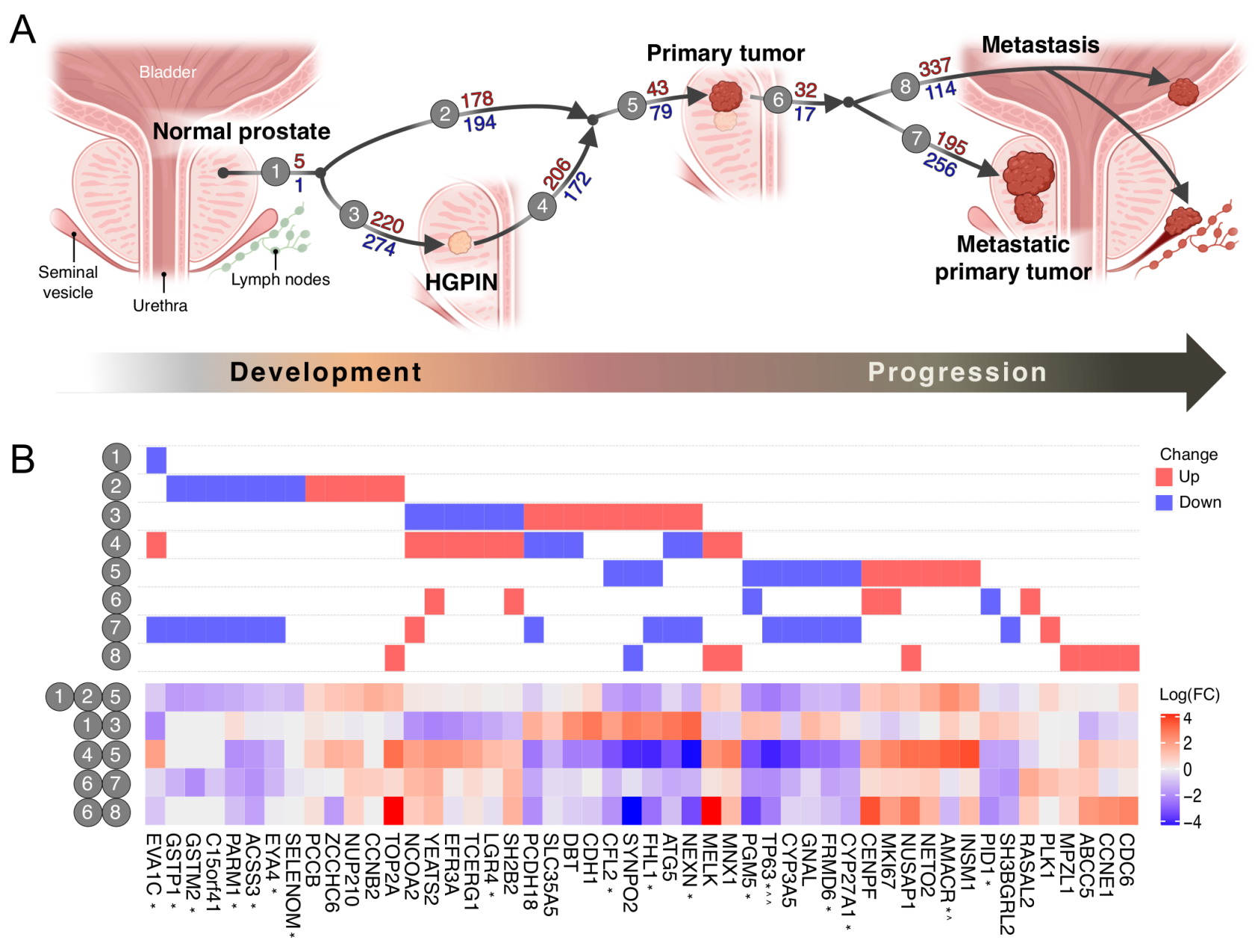
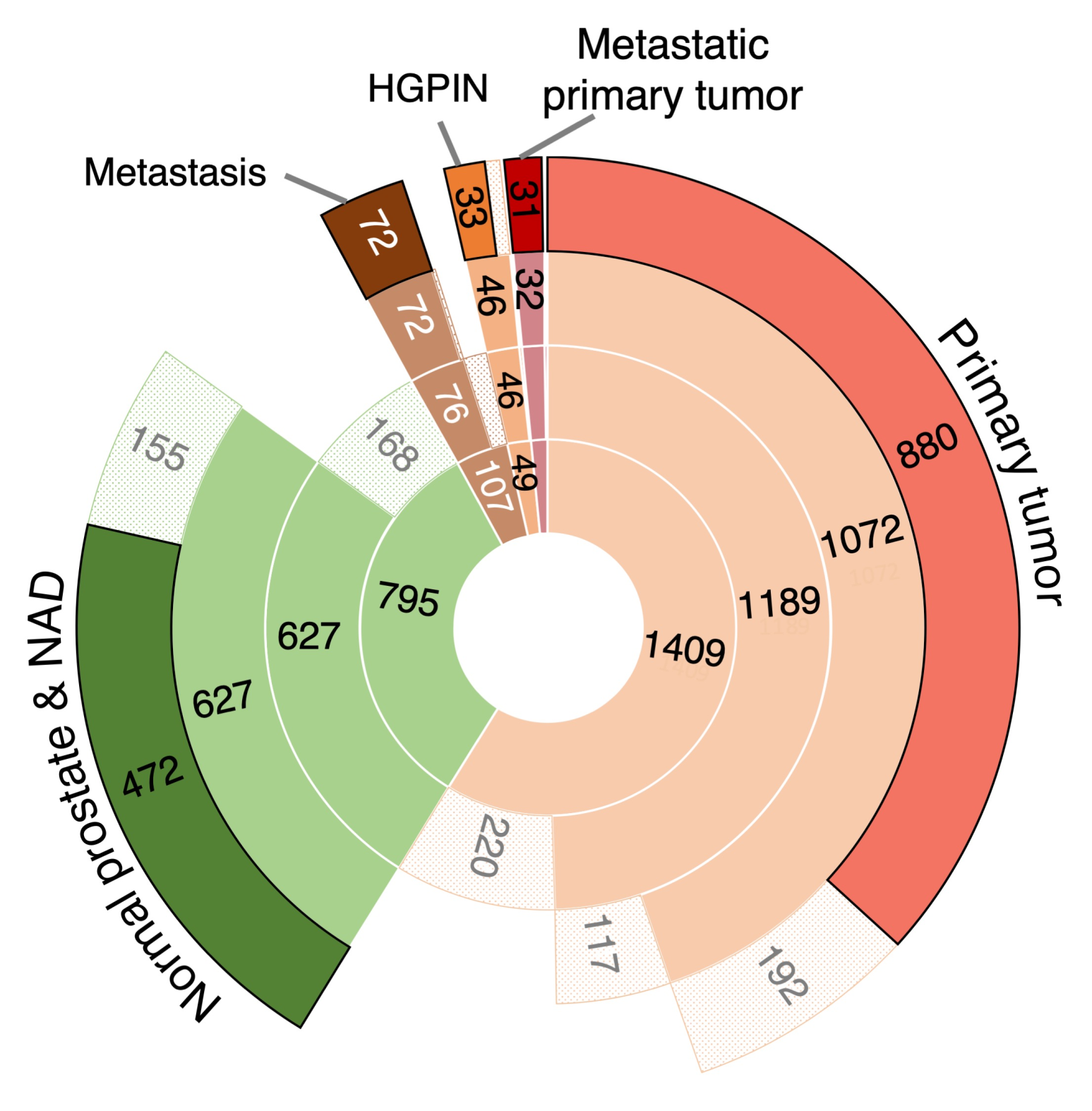
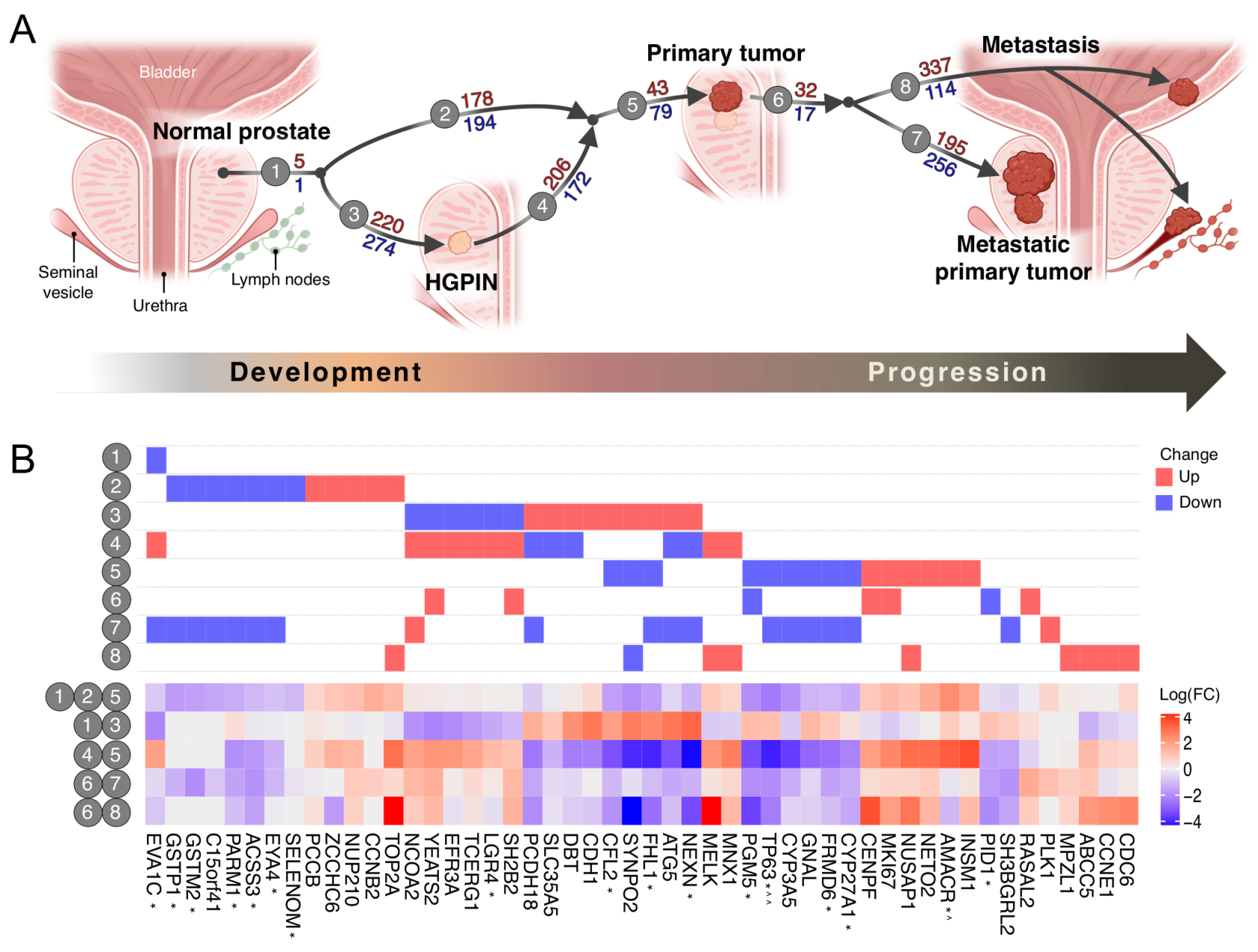
3.1. The Transcriptomic Landscape of Prostate Cancer
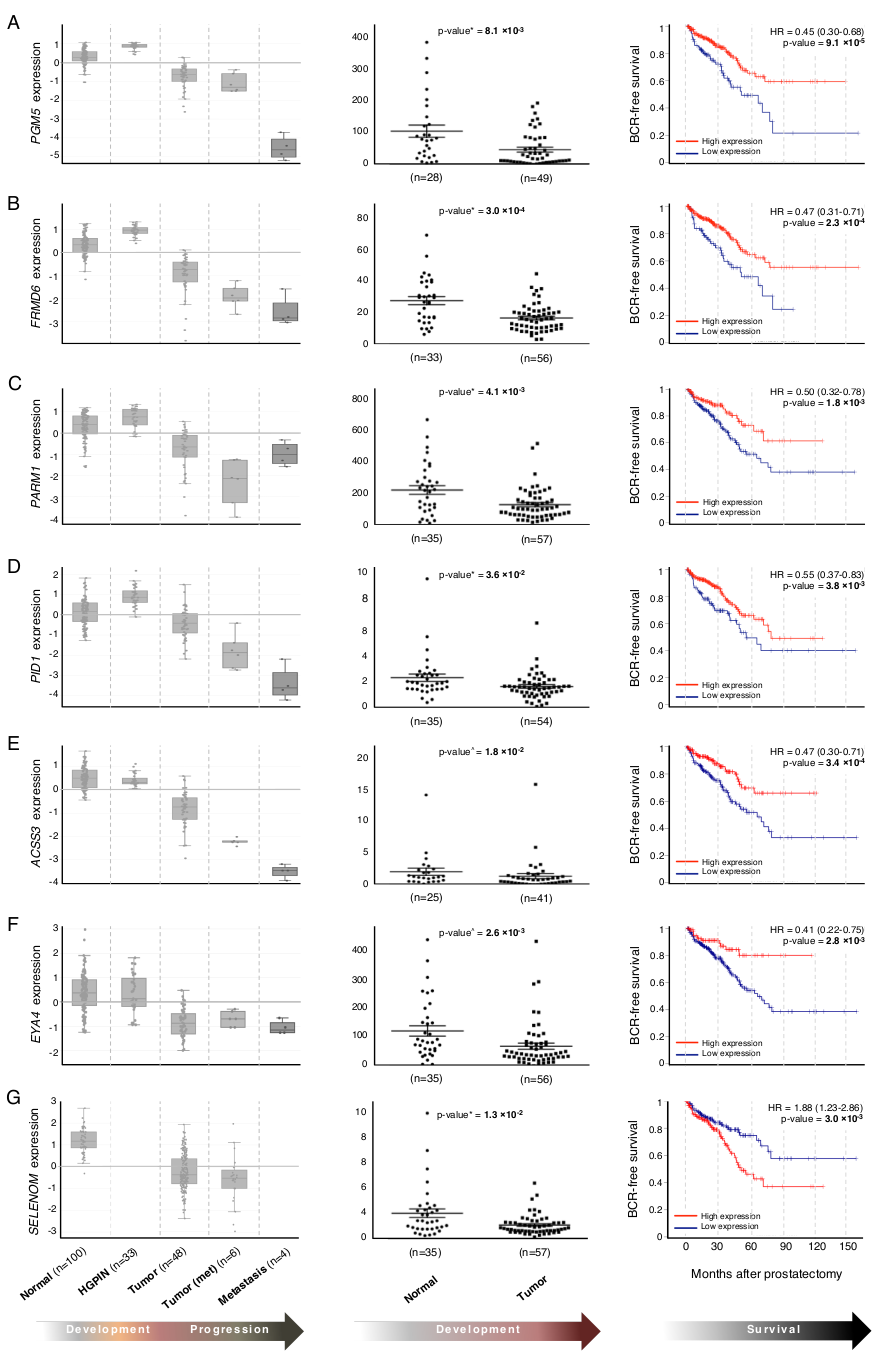
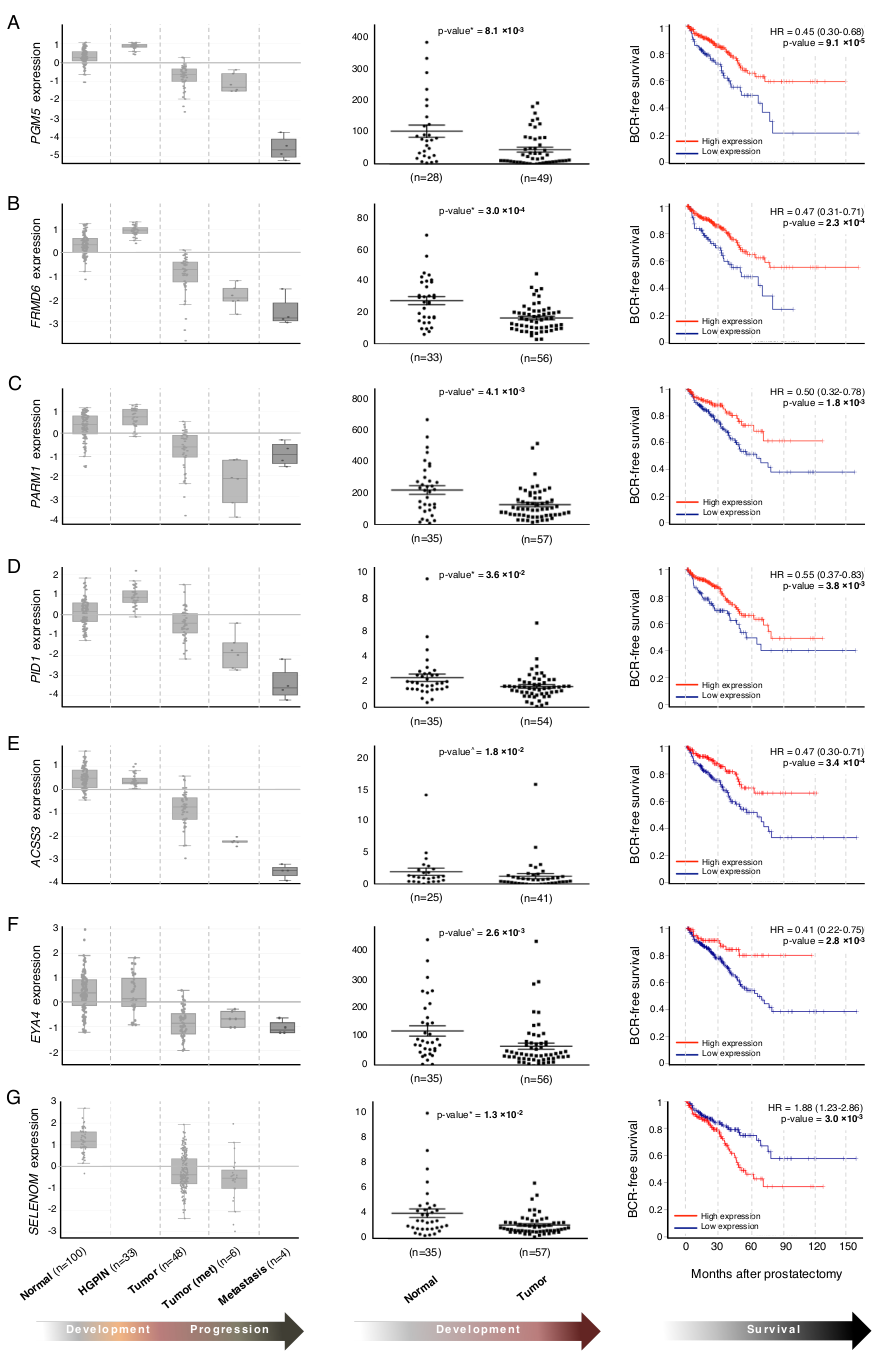
3.2. In Silico Predictions Validate in Clinical Samples
3.3. HGPIN Molecular Changes
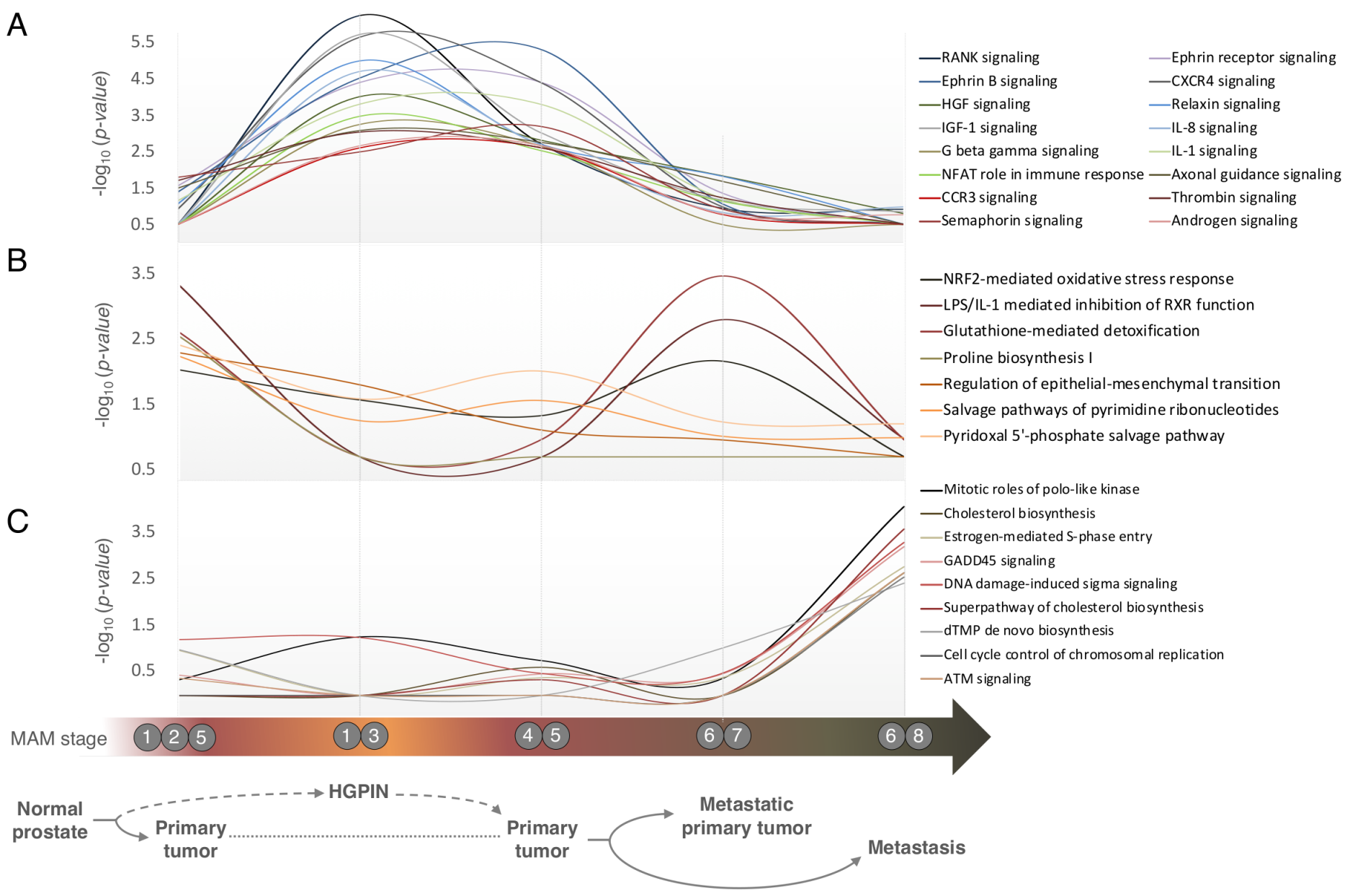
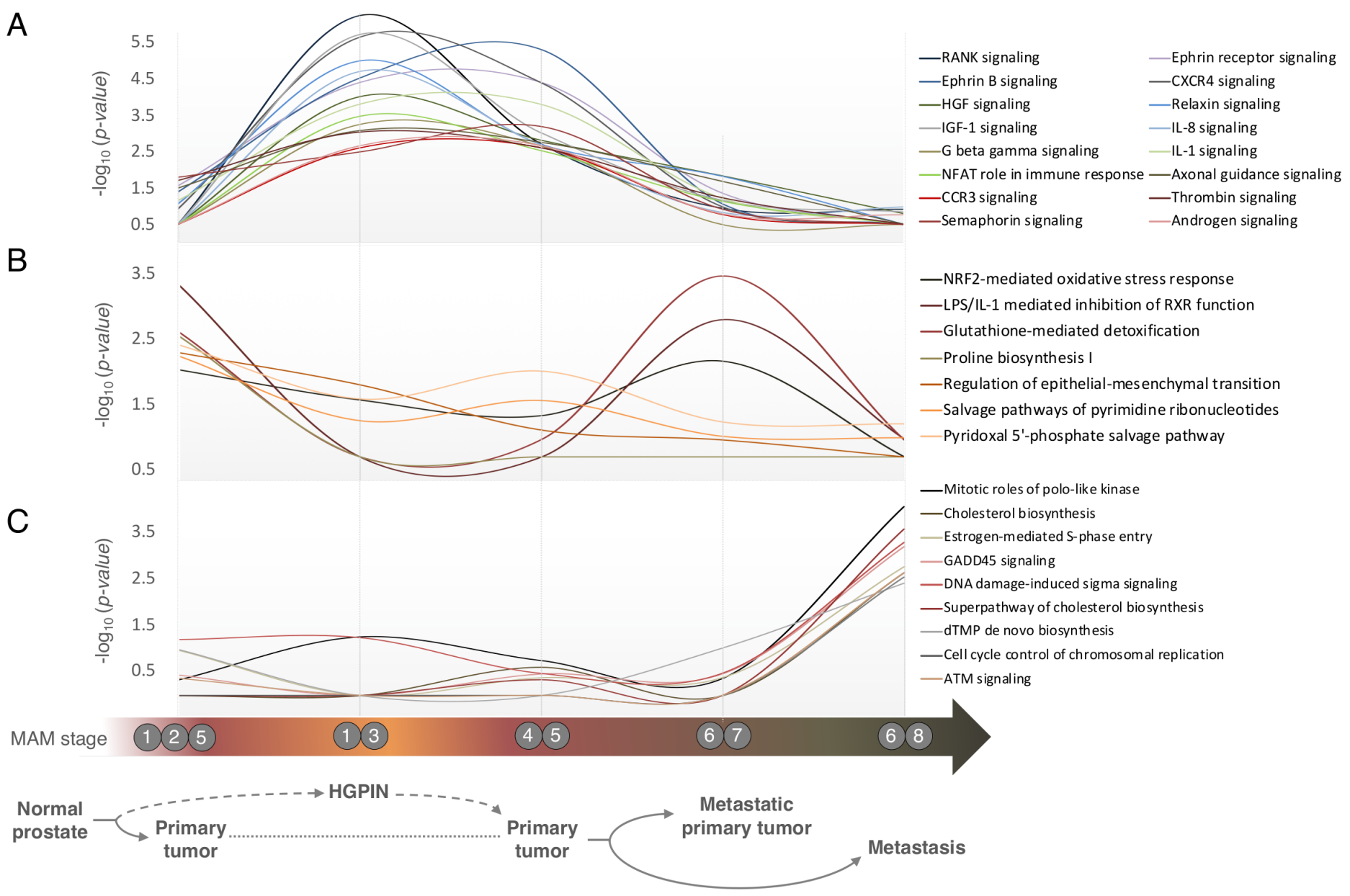
3.4. Pathways Involved in Prostate Cancer Development and Progression are Distinct
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spratt, D.E.; Zumsteg, Z.S.; Feng, F.Y.; Tomlins, S.A. Translational and clinical implications of the genetic landscape of prostate cancer. Nat. Rev. Clin. Oncol. 2016, 13, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Lange, L.A.; Hu, Y.; Zhang, H.; Xue, C.; Schmidt, E.M.; Tang, Z.Z.; Bizon, C.; Lange, E.M.; Smith, J.D.; Turner, E.H.; et al. Whole-exome sequencing identifies rare and low-frequency coding variants associated with LDL cholesterol. Am. J. Hum. Genet. 2014, 94, 233–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Marzo, A.M.; Haffner, M.C.; Lotan, T.L.; Yegnasubramanian, S.; Nelson, W.G. Premalignancy in prostate cancer: Rethinking what we know. Cancer Prev. Res. 2016, 9, 648–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, X.; Wang, M.; McElyea, S.D.; Sherman, S.; House, M.; Korc, M. A microRNA signature in circulating exosomes is superior to exosomal glypican-1 levels for diagnosing pancreatic cancer. Cancer Lett. 2017, 393, 86–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [Green Version]
- Fraser, M.; Sabelnykova, V.Y.; Yamaguchi, T.N.; Heisler, L.E.; Livingstone, J.; Huang, V.; Shiah, Y.J.; Yousif, F.; Lin, X.; Masella, A.P.; et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017, 541, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Horning, A.M.; Wang, Y.; Lin, C.K.; Louie, A.D.; Jadhav, R.R.; Hung, C.N.; Wang, C.M.; Lin, C.L.; Kirma, N.B.; Liss, M.A.; et al. Single-Cell RNA-seq reveals a subpopulation of prostate cancer cells with enhanced cell-Cycle–Related transcription and attenuated androgen response. Cancer Res. 2018, 78, 853–864. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.; Manu, V.; Malik, A.; Dutta, V.; Mani, N.S.; Patrikar, S. Diagnostic utility of p63 and α-methyl acyl Co A racemase in resolving suspicious foci in prostatic needle biopsy and transurethral resection of prostate specimens. J. Cancer Res. Ther. 2014, 10, 686–692. [Google Scholar]
- Sequeiros, T.; Bastarós, J.M.; Sánchez, M.; Rigau, M.; Montes, M.; Placer, J.; Planas, J.; De Torres, I.; Reventõs, J.; Pegtel, D.M.; et al. Urinary biomarkers for the detection of prostate cancer in patients with high-grade prostatic intraepithelial neoplasia. Prostate 2015, 75, 1102–1113. [Google Scholar] [CrossRef]
- Asada, K.; Ando, T.; Niwa, T.; Nanjo, S.; Watanabe, N.; Okochi-Takada, E.; Yoshida, T.; Miyamoto, K.; Enomoto, S.; Ichinose, M.; et al. FHL1 on chromosome X is a single-hit gastrointestinal tumor-suppressor gene and contributes to the formation of an epigenetic field defect. Oncogene 2013, 32, 2140–2149. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Tang, W.; Su, G.; Cai, M.; An, H.X.; Zhang, Y. PCDH18 is frequently inactivated by promoter methylation in colorectal cancer. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Yan, H.; Sun, J.; Yang, F.; Song, C.; Jiang, F.; Li, Y.; Dong, J.; Zheng, G.Y.; Tian, X.L.; et al. NEXN is a novel susceptibility gene for coronary artery disease in Han Chinese. PLoS ONE 2013, 8, e82135. [Google Scholar] [CrossRef] [PubMed]
- Kai, F.; Fawcett, J.P.; Duncan, R. Synaptopodin-2 induces assembly of peripheral actin bundles and immature focal adhesions to promote lamellipodia formation and prostate cancer cell migration. Oncotarget 2015, 6, 11162–11174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uzozie, A.C.; Selevsek, N.; Wahlander, A.; Nanni, P.; Grossmann, J.; Weber, A.; Buffoli, F.; Marra, G. Targeted proteomics for multiplexed verification of markers of colorectal tumorigenesis. Mol. Cell. Proteom. 2017, 16, 407–427. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, S.K.; Olsen, S.N.; Dake, B.; De Raedt, T.; Lim, E.; Bronson, R.T.; Beroukhim, R.; Polyak, K.; Brown, M.; Kuperwasser, C.; et al. The RasGAP Gene, RASAL2, Is a Tumor and Metastasis Suppressor. Cancer Cell 2013, 24, 365–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Tan, P.; Rodriguez, M.; He, L.; Tan, K.; Zeng, L.; Siwko, S.; Liu, M. Leucine-rich repeat-containing G protein-coupled receptor 4 (Lgr4) is necessary for prostate cancer metastasis via epithelial-mesenchymal transition. J. Biol. Chem. 2017, 292, 15525–15537. [Google Scholar] [CrossRef] [Green Version]
- Visser-Grieve, S.; Hao, Y.; Yang, X. Human homolog of Drosophila expanded, hEx, functions as a putative tumor suppressor in human cancer cell lines independently of the Hippo pathway. Oncogene 2012, 31, 1189–1195. [Google Scholar] [CrossRef] [Green Version]
- Charfi, C.; Levros, L.C.; Edouard, E.; Rassart, E. Characterization and identification of PARM-1 as a new potential oncogene. Mol. Cancer 2013, 12, 84. [Google Scholar] [CrossRef] [Green Version]
- Scaltriti, M.; Eichhorn, P.J.; Cortés, J.; Prudkin, L.; Aurac, C.; Jiménez, J.; Chandarlapaty, S.; Serra, V.; Prat, A.; Ibrahim, Y.H.; et al. Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proc. Natl. Acad. Sci. USA 2011, 108, 3761–3766. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, L.K.; Wang, X.; Li, Y.; Ekholm-Reed, S.; Wu, X.; Wang, P.; Reed, S.I. Cyclin e deregulation promotes loss of specific genomic regions. Curr. Biol. 2015, 25, 1327–1333. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, J.N.; Guo, Z.; Baus, R.M.; Werner, H.; Rehrauer, W.M.; Lloyd, R. V INSM1: A Novel Immunohistochemical and Molecular Marker for Neuroendocrine and Neuroepithelial Neoplasms. Am. J. Clin. Pathol. 2015, 144, 579–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuner, R.; Fälth, M.; Pressinotti, N.C.; Brase, J.C.; Puig, S.B.; Metzger, J.; Gade, S.; Schäfer, G.; Bartsch, G.; Steiner, E.; et al. The maternal embryonic leucine zipper kinase (MELK) is upregulated in high-grade prostate cancer. J. Mol. Med. 2013, 91, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.A.; Gong, X.; Ganesh, D.; Brooks, J.D. NUSAP1 promotes invasion and metastasis of prostate cancer. Oncotarget 2017, 8, 29935–29950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labbé, D.P.; Sweeney, C.J.; Brown, M.; Galbo, P.; Rosario, S.; Wadosky, K.M.; Ku, S.-Y.; Sjöström, M.; Alshalalfa, M.; Erho, N.; et al. TOP2A and EZH2 Provide Early Detection of an Aggressive Prostate Cancer Subgroup. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 7072–7083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barros-Silva, J.D.; Linn, D.E.; Steiner, I.; Guo, G.; Ali, A.; Pakula, H.; Ashton, G.; Peset, I.; Brown, M.; Clarke, N.W.; et al. Single-Cell Analysis Identifies LY6D as a Marker Linking Castration-Resistant Prostate Luminal Cells to Prostate Progenitors and Cancer. Cell Rep. 2018, 25, 3504–3518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montes, M.; Coiras, M.; Becerra, S.; Moreno-Castro, C.; Mateos, E.; Majuelos, J.; Oliver, F.J.; Hernández-Munain, C.; Alcamí, J.; Suñé, C. Functional consequences for apoptosis by transcription elongation regulator 1 (TCERG1)-Mediated Bcl-x and Fas/CD95 Alternative Splicing. PLoS ONE 2015, 10, e0139812. [Google Scholar] [CrossRef] [Green Version]
- Morell, C.; Bort, A.; Vara-Ciruelos, D.; Ramos-Torres, Á.; Altamirano-Dimas, M.; Díaz-Laviada, I.; Rodríguez-Henche, N. Up-Regulated Expression of LAMP2 and Autophagy Activity during Neuroendocrine Differentiation of Prostate Cancer LNCaP Cells. PLoS ONE 2016, 11, e0162977. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Zhu, Q.; Li, Z.; Peng, Y.; Yu, X.; Yuan, B.; Liu, Y.; Liu, Y.; Yin, L.; Peng, Y.; et al. The FOXM1-ABCC5 axis contributes to paclitaxel resistance in nasopharyngeal carcinoma cells. Cell Death Dis. 2017, 8, e2659. [Google Scholar] [CrossRef] [Green Version]
- Ashour, N.; Angulo, J.C.; Andrés, G.; Alelú, R.; González-Corpas, A.; Toledo, M.V.; Rodríguez-Barbero, J.M.; López, J.I.; Sánchez-Chapado, M.; Ropero, S. A DNA hypermethylation profile reveals new potential biomarkers for prostate cancer diagnosis and prognosis. Prostate 2014, 74, 1171–1182. [Google Scholar] [CrossRef]
- Njiaju, U.O.; Gamazon, E.R.; Gorsic, L.K.; Delaney, S.M.; Wheeler, H.E.; Im, H.K.; Dolan, M.E. Whole-genome studies identify solute carrier transporters in cellular susceptibility to paclitaxel. Pharmacogenet. Genom. 2012, 22, 498–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Møller, M.; Strand, S.H.; Mundbjerg, K.; Liang, G.; Gill, I.; Haldrup, C.; Borre, M.; Høyer, S.; Ørntoft, T.F.; Sørensen, K.D. Heterogeneous patterns of DNA methylation-based field effects in histologically normal prostate tissue from cancer patients. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornton, J.E.; Du, P.; Jing, L.; Sjekloca, L.; Lin, S.; Grossi, E.; Sliz, P.; Zon, L.I.; Gregory, R.I. Selective microRNA uridylation by Zcchc6 (TUT7) and Zcchc11 (TUT4). Nucleic Acids Res. 2014, 42, 11777–11791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, D.; Jing, Y.; Zhang, Z.; Liu, L.; Ding, J.; Zhao, F.; Ge, C.; Wang, Q.; Chen, T.; Yao, M.; et al. Amplification of MPZL1/PZR promotes tumor cell migration through Src-mediated phosphorylation of cortactin in hepatocellular carcinoma. Cell Res. 2014, 24, 204–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Chen, H.Y.; Cai, J.; Yang, G.Z.; Feng, D.; Zhai, Y.X.; Gong, H.; Qi, C.Y.; Zhang, Y.; Fu, H.; et al. Upregulation of NETO2 expression correlates with tumor progression and poor prognosis in colorectal carcinoma. BMC Cancer 2015, 15, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Erdreich-Epstein, A.; Robison, N.; Ren, X.; Zhou, H.; Xu, J.; Davidson, T.B.; Schur, M.; Gilles, F.H.; Ji, L.; Malvar, J.; et al. PID1 (NYGGF4), a new growth-inhibitory gene in embryonal brain tumors and gliomas. Clin. Cancer Res. 2014, 20, 827–836. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Hara, N.; Nishiyama, T.; Tasaki, M.; Ishizaki, F.; Tomita, Y. Corepressive function of nuclear receptor coactivator 2 in androgen receptor of prostate cancer cells treated with antiandrogen. BMC Cancer 2016, 16, 332. [Google Scholar] [CrossRef] [Green Version]
- Mitra, R.; Goodman, O.B. CYP3A5 regulates prostate cancer cell growth by facilitating nuclear translocation of AR. Prostate 2015, 75, 527–538. [Google Scholar] [CrossRef]
- Decock, A.; Ongenaert, M.; Hoebeeck, J.; De Preter, K.; Van Peer, G.; Van Criekinge, W.; Ladenstein, R.; Schulte, J.H.; Noguera, R.; Stallings, R.L.; et al. Genome-wide promoter methylation analysis in neuroblastoma identifies prognostic methylation biomarkers. Genome Biol. 2012, 13, R95. [Google Scholar] [CrossRef] [Green Version]
- Chang, W.C.; Cheng, W.C.; Cheng, B.H.; Chen, L.; Ju, L.J.; Ou, Y.J.; Jeng, L.B.; Yang, M.D.; Hung, Y.C.; Ma, W.L. Mitochondrial Acetyl-CoA Synthetase 3 is Biosignature of Gastric Cancer Progression. Cancer Med. 2018, 7, 1240–1252. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, J.; Wang, Y.; Zhang, Y.; Castro, P.; Shao, L.; Sreekumar, A.; Putluri, N.; Guha, N.; Deepak, S.; et al. MNX1 Is Oncogenically Upregulated in African-American Prostate Cancer. Cancer Res. 2016, 76, 6290–6298. [Google Scholar] [CrossRef] [Green Version]
- Alfaqih, M.A.; Nelson, E.R.; Liu, W.; Safi, R.; Jasper, J.S.; Macias, E.; Geradts, J.; Thompson, J.W.; Dubois, L.G.; Freeman, M.R.; et al. CYP27A1 loss dysregulates cholesterol homeostasis in prostate cancer. Cancer Res. 2017, 77, 1662–1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, E.; Totoki, Y.; Nakamura, H.; Morizane, C.; Nara, S.; Hama, N.; Suzuki, M.; Furukawa, E.; Kato, M.; Hayashi, H.; et al. Clinical utility of circulating tumor DNA for molecular assessment in pancreatic cancer. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaafar, N.; Moleirinho, A.; Kerkeni, E.; Monastiri, K.; Seboui, H.; Amorim, A.; Prata, M.J.; Quental, S. Molecular characterization of maple syrup urine disease patients from Tunisia. Gene 2013, 517, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Cherbonnel-Lasserre, C.L.; Linares-Cruz, G.; Rigaut, J.P.; Sabatier, L.; Dutrillaux, B. Strong decrease in biotin content may correlate with metabolic alterations in colorectal adenocarcinoma. Int. J. Cancer 1997, 72, 768–775. [Google Scholar] [CrossRef]
- Rajkumar, T.; Sabitha, K.; Vijayalakshmi, N.; Shirley, S.; Bose, M.V.; Gopal, G.; Selvaluxmy, G. Identification and validation of genes involved in cervical tumourigenesis. BMC Cancer 2011, 11, 80. [Google Scholar] [CrossRef] [Green Version]
- James, G.; Foster, S.R.; Key, B.; Beverdam, A. The Expression Pattern of EVA1C, a Novel Slit Receptor, Is Consistent with an Axon Guidance Role in the Mouse Nervous System. PLoS ONE 2013, 8, e74115. [Google Scholar] [CrossRef] [Green Version]
- Bian, Y.; Guo, J.; Qiao, L.; Sun, X. miR-3189-3p mimics enhance the effects of S100A4 siRNA on the inhibition of proliferation and migration of gastric cancer cells by targeting CFL2. Int. J. Mol. Sci. 2018, 19, 236. [Google Scholar] [CrossRef] [Green Version]
- Karanika, S.; Karantanos, T.; Li, L.; Wang, J.; Park, S.; Yang, G.; Zuo, X.; Song, J.H.; Maity, S.N.; Manyam, G.C.; et al. Targeting DNA Damage Response in Prostate Cancer by Inhibiting Androgen Receptor-CDC6-ATR-Chk1 Signaling. Cell Rep. 2017, 18, 1970–1981. [Google Scholar] [CrossRef]
- Towle, R.; Truong, D.; Garnis, C. Epigenetic mediated silencing of EYA4 contributes to tumorigenesis in oral dysplastic cells. Genes Chromosom. Cancer 2016, 55, 568–576. [Google Scholar] [CrossRef]
- Zhang, Y.; Qiu, Z.; Wei, L.; Tang, R.; Lian, B.; Zhao, Y.; He, X.; Xie, L. Integrated analysis of mutation data from various sources identifies key genes and signaling pathways in hepatocellular carcinoma. PLoS ONE 2014, 9, e100854. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Yang, L.; Zheng, L.; Ge, W.; Li, D.; Zhang, Y.; Hu, X.; Gao, Z.; Xu, J.; Huang, Y.; et al. Exome Capture Sequencing of Adenoma Reveals Genetic Alterations in Multiple Cellular Pathways at the Early Stage of Colorectal Tumorigenesis. PLoS ONE 2013, 8, 1–8. [Google Scholar] [CrossRef]
- Babbs, C.; Roberts, N.A.; Sanchez-Pulido, L.; McGowan, S.J.; Ahmed, M.R.; Brown, J.M.; Sabry, M.A.; Bentley, D.R.; McVean, G.A.; Donnelly, P.; et al. Homozygous mutations in a predicted endonuclease are a novel cause of congenital dyserythropoietic anemia type I. Haematologica 2013, 98, 1383–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerriero, E.; Accardo, M.; Capone, F.; Colonna, G.; Castello, G.; Costantini, S. Assessment of the Selenoprotein M (SELM) over-expression on human hepatocellular carcinoma tissues by immunohistochemistry. Eur. J. Histochem. 2014, 58, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Cuzick, J.; Swanson, G.P.; Fisher, G.; Brothman, A.R.; Berney, D.M.; Reid, J.E.; Mesher, D.; Speights, V.O.; Stankiewicz, E.; Foster, C.S.; et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: A retrospective study. Lancet Oncol. 2011, 12, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Rajan, P.; Stockley, J.; Sudbery, I.M.; Fleming, J.T.; Hedley, A.; Kalna, G.; Sims, D.; Ponting, C.P.; Heger, A.; Robson, C.N.; et al. Identification of a candidate prognostic gene signature by transcriptome analysis of matched pre- and post-treatment prostatic biopsies from patients with advanced prostate cancer. BMC Cancer 2014, 14, 977. [Google Scholar] [CrossRef] [Green Version]
- Feng, M.; Bao, Y.; Li, Z.; Li, J.; Gong, M.; Lam, S.; Wang, J.; Marzese, D.M.; Donovan, N.; Yu Tan, E.; et al. RASAL2 activates RAC1 to promote triple-negative breast cancer progression. J. Clin. Investig. 2014, 124, 5291–5304. [Google Scholar] [CrossRef] [Green Version]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Fladeby, C.; Gupta, S.N.; Barois, N.; Lorenzo, P.I.; Simpson, J.C.; Saatcioglu, F.; Bakke, O. Human PARM-1 is a novel mucin-like, androgen-regulated gene exhibiting proliferative effects in prostate cancer cells. Int. J. Cancer 2008, 122, 1229–1235. [Google Scholar] [CrossRef]
- Li, Y.; Yan, M.; Yang, J.; Raman, I.; Du, Y.; Min, S.; Fang, X.; Mohan, C.; Li, Q.Z. Glutathione S-transferase Mu 2-transduced mesenchymal stem cells ameliorated anti-glomerular basement membrane antibody-induced glomerulonephritis by inhibiting oxidation and inflammation. Stem Cell Res. Ther. 2014, 5, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Gonzalgo, M.L.; Pavlovich, C.P.; Lee, S.M.; Nelson, W.G. Prostate cancer detection by GSTP1 methylation analysis of postbiopsy urine specimens. Clin. Cancer Res. 2003, 9, 2673–2677. [Google Scholar] [PubMed]
- Li, X.; Jia, Z.; Shen, Y.; Ichikawa, H.; Jarvik, J.; Nagele, R.G.; Goldberg, G.S. Coordinate suppression of Sdpr and Fhl1 expression in tumors of the breast, kidney, and prostate. Cancer Sci. 2008, 99, 1326–1333. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wei, X.; Yuan, Y.; Sun, Q.; Zhan, J.; Zhang, J.; Tang, Y.; Li, F.; Ding, L.; Ye, Q.; et al. Src-mediated phosphorylation converts FHL1 from tumor suppressor to tumor promoter. J. Cell Biol. 2018, 217, 1335–1351. [Google Scholar] [CrossRef] [PubMed]
- Haffner, M.C.; Barbieri, C.E. Shifting Paradigms for High-grade Prostatic Intraepithelial Neoplasia. Eur. Urol. 2016, 69, 831–833. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.S.; Eeles, R.; Wedge, D.C.; Van Loo, P.; Gundem, G.; Alexandrov, L.B.; Kremeyer, B.; Butler, A.; Lynch, A.G.; Camacho, N.; et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nat. Genet. 2015, 47, 367–372. [Google Scholar] [CrossRef]
- Gorlov, I.P.; Byun, J.; Logothetis, C.J. In silico functional profiling of individual prostate cancer tumors: Many genes, few functions. Cancer Genom. Proteom. 2012, 9, 109–114. [Google Scholar]
- Tomlins, S.A.; Mehra, R.; Rhodes, D.R.; Cao, X.; Wang, L.; Dhanasekaran, S.M.; Kalyana-Sundaram, S.; Wei, J.T.; Rubin, M.A.; Pienta, K.J.; et al. Integrative molecular concept modeling of prostate cancer progression. Nat. Genet. 2007, 39, 41–51. [Google Scholar] [CrossRef]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.; Robinson, D.R.; Cao, X.; Saravana, M.; Khan, A.P.; Quist, M.J.; Jing, X.; Robert, J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2013, 487, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Gorlov, I.P.; Byun, J.; Gorlova, O.Y.; Aparicio, A.M.; Efstathiou, E.; Logothetis, C.J. Candidate pathways and genes for prostate cancer: A meta-analysis of gene expression data. BMC Med. Genom. 2009, 2, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hankinson, S.J.; Fam, M.; Patel, N.N. A review for clinicians: Prostate cancer and the antineoplastic properties of metformin. Urol. Oncol. 2017, 35, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Crawley, D.; Chandra, A.; Loda, M.; Gillett, C.; Cathcart, P.; Challacombe, B.; Cook, G.; Cahill, D.; Santa Olalla, A.; Cahill, F.; et al. Metformin and longevity (METAL): A window of opportunity study investigating the biological effects of metformin in localised prostate cancer. BMC Cancer 2017, 17, 494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehlen, P.; Delloye-Bourgeois, C.; Chédotal, A. Novel roles for Slits and netrins: Axon guidance cues as anticancer targets? Nat. Rev. Cancer 2011, 11, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Denduluri, S.K.; Idowu, O.; Wang, Z.; Liao, Z.; Yan, Z.; Mohammed, M.K.; Ye, J.; Wei, Q.; Wang, J.; Zhao, L.; et al. Insulin-like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis. 2015, 2, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Hancock, C.N.; Fischer, J.W.; Harman, M.; Phang, J.M. Proline biosynthesis augments tumor cell growth and aerobic glycolysis: Involvement of pyridine nucleotides. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phang, J.M.; Liu, W.; Hancock, C.N.; Fischer, J.W. Proline metabolism and cancer: Emerging links to glutamine and collagen. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Lam, B.; Neogi, S.G.; Yeo, G.S.H.; Azizan, E.A.B.; Brown, M.J. Transcriptome Pathway Analysis of Pathological and Physiological Aldosterone-Producing Human Tissues. Hypertension 2016, 68, 1424–1431. [Google Scholar] [CrossRef]
- Davalieva, K.; Kiprijanovska, S.; Kostovska, I.M.; Stavridis, S.; Stankov, O.; Komina, S.; Petrusevska, G.; Polenakovic, M. Comparative proteomics analysis of urine reveals down-regulation of acute phase response signaling and LXR/RXR activation pathways in prostate cancer. Proteomes 2018, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.H.; Kwak, M.K. Shadows of NRF2 in cancer: Resistance to chemotherapy. Curr. Opin. Toxicol. 2016, 1, 20–28. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef] [Green Version]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondria, cholesterol and cancer cell metabolism. Clin. Transl. Med. 2016, 5, 1–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelton, K.; Freeman, M.R.; Solomon, K.R. Cholesterol and prostate cancer. Curr. Opin. Pharmacol. 2012, 12, 751–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leon, C.G.; Locke, J.A.; Adomat, H.H.; Etinger, S.L.; Twiddy, A.L.; Neumann, R.D.; Nelson, C.C.; Guns, E.S.; Wasan, K.M. Alterations in cholesterol regulation contribute to the production of intratumoral androgens during progression to castration-resistant prostate cancer in a mouse xenograft model. Prostate 2010, 70, 390–400. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sample Ns (T,HGPIN,N) | Validation p-Value | Confirms in Silico Results? | Log-Rank Test p-Value (TCGA) | Biological Role | Association with other Malignancies | Reference |
|---|---|---|---|---|---|---|---|
| AMACR a | 55, 0, 36 | 1.9 × 10−2 | Yes (T vs. N) | 6.3 × 10−2 | Racemase processing bile for degradation | Luminal cell marker in PC | [9] |
| TP63 b | 22, 23, 22 | 6.9 × 10−3 | Yes (T vs. N) | 1.3 × 10−3 | Tumor suppressor | Basal cell marker in PC | [9] |
| CDH1 c | 11, 12, 7 | 0.26 | - | 1.5 × 10−4 | Cell adhesion | Marker of PC in patients with HGPIN | [10] |
| FHL1 c,f | 40, 0, 25 | 1.1 × 10−3 | Yes (T vs. N) | 4.7 × 10−2 | Cell adhesion, migration, colony formation | Forms epigenetic field defect in GISTs | [11] |
| PCDH18 d | 51, 0, 26 | 0.3 | - | - | Cell adhesion, migration, colony formation | Hypermethylated in CRC | [12] |
| NEXN d,f | 55, 0, 35 | 7.3 × 10−3 | Yes (T vs. N) | 8.3 × 10−2 | Cell adhesion, migration | Cardiomyopathy | [13] |
| SYNPO2 f | 23, 22, 24 | 1.2 × 10−2 | Yes (T vs. N) | 5.5 × 10−4 | Cell migration | Invasive cancer biomarker | [14] |
| PGM5 d | 49, 0, 28 | 8.1 × 10−3 | Yes (T vs. N) | 9.1 × 10−5 | Adherens-type cellular junctions | Downregulated in CRC | [15] |
| RASAL2 d | 21, 20, 18 | 0.5 | - | - | RHO-GAP; cell invasion | Key roles in tumor progression and metastasis | [16] |
| LGR4 f | 55, 0, 35 | 3.4 × 10−3 | Yes (T vs. N) | 1.7 × 10−1 | Activates Wnt signaling | Key protein in PC metastasis | [17] |
| SH2B2 d,f | 21, 15, 14 | 0.63 | - | - | Cellular proliferation | ||
| FRMD6 d | 56, 0, 33 | 3 × 10−4 | Yes (T vs. N) | 2.3 × 10−4 | Cellular proliferation | Tumor suppressor in BrCa | [18] |
| PARM1 d | 57, 0, 35 | 4.1 × 10−3 | Yes (T vs. N) | 1.8 × 10−3 | Cellular proliferation | Oncogenic in leukemias | [19] |
| CCNE1 d | 10, 12, 14 | 0.65 | - | - | Cell cycle progression | Chromosomal instability and trastuzumab resistance; poor prognosis in multiple cancers | [20,21] |
| INSM1 d | 24, 24, 22 | 0.61 | - | - | Cell cycle progression | Regulates NE differentiation in several tumor types | [22] |
| CCNB2 | 8, 3, 9 | 0.79 | - | - | Cell cycle progression | Recurrent PC | [8] |
| MKI67 | 26, 0, 7 | 5.1 × 10−3 | - | - | Cell cycle progression | Recurrent PC | [8] |
| MELK | 49, 0, 29 | 0.14 | - | 3.5 × 10−6 | Cell cycle progression | High-grade PC | [23] |
| NUSAP1 e | 45, 0, 22 | 0.83 | - | - | Cell cycle progression | Promotes invasion and metastasis PC | [24] |
| PLK1 e | 11, 13, 10 | 0.8 | - | - | Cell cycle progression | Recurrent PC | [8] |
| CENPF e | 54, 0, 29 | 0.56 | - | 3.3 × 10−9 | Cell cycle progression | Recurrent PC | [8] |
| TOP2A e | 45, 0, 21 | 0.46 | - | - | Cell cycle progression | Marker of aggressive PC | [25] |
| MYC | 14, 15, 10 | 0.39 | - | - | Cell cycle progression; apoptosis | Upregulated in HGPIN and PC | [26] |
| TCERG1 d,f | 7, 8, 6 | 0.22 | - | - | Transcriptional elongation and splicing; lipid homeostasis (C.elegans) | Sensitizes cell to apoptosis | [27] |
| ATG5 f | 19, 23, 21 | 0.63 | - | - | Apoptosis; autophagy | Increased levels in NE PC | [28] |
| ABCC5 d | 25, 25, 26 | 0.75 | - | 1.9 × 10−8 | Cellular export of cyclic nucleotides | Paclitaxel resistance in nasopharyngeal cancer | [29] |
| GSTM2 | 57, 0, 35 | 1.8 × 10−3 | Yes (T vs. N) | 2.9 × 10−2 | Detoxification of electrophilic compounds | Prognostic marker in PC | [30] |
| SLC35A5 d,f | 19, 19, 18 | 0.77 | - | - | Transmembrane protein | SNPs associated with paclitaxel sensitivity | [31] |
| GSTP1 c | 56, 0, 33 | 6 × 10−4 | Yes (T vs. N) | 1.2 × 10−2 | Drug metabolism; cell cycle regulation | Hypermethylated in PC | [32] |
| ZCCHC6 d | 24, 23, 21 | 0.67 | - | - | Uridylation of mRNA | Loss (C.elegans homologue) leads to chromosomal instability | [33] |
| MPZL1 d | 23, 23, 23 | 0.69 | - | - | Cell signaling via c-Src | Amplification promotes cell migration in HCC | [34] |
| NETO2 d | 18, 17, 10 | 0.41 | - | - | Glutamate signaling in neurons | Prognostic in CRC | [35] |
| PID1 d | 54, 0, 34 | 3.6 × 10−2 | Yes (T vs. N) | 3.8 × 10−3 | Insulin signaling | Tumor suppressor in brain tumors and gliomas | [36] |
| NCOA2 f | 21, 23, 21 | 0.47 | - | - | Transcriptional coactivator of nuclear hormone receptors | AR co-repressor with antiandrogens | [37] |
| CYP3A5 | 14, 0, 13 | 0.4 | - | - | Nuclear translocation of AR | Regulates growth PC | [38] |
| EZH2 | 40, 0, 20 | 0.81 | - | - | Transcriptional regulator | Marker of aggressive PC | [25] |
| ACSS3 d | 41, 0, 25 | 1.8 × 10−2 | Yes (T vs. N) | 3.4 × 10−4 | Cholesterogenesis | Prognostic marker in neuroblastoma, gastric cancer | [39,40] |
| MNX1 | 33, 0, 13 | 0.33 | - | - | Lipid synthesis | Upregulated in African American PC | [41] |
| CYP27A1 | 54, 0, 30 | 1.1 × 10−2 | Yes (T vs. N) | 5.6 × 10−5 | Cellular cholesterol homeostasis | Associated with poor prognosis in PC | [42] |
| YEATS2 d,f | 44, 0, 24 | 0.29 | - | - | Cellular metabolism; epigenetic regulation | Mutated in multiple tumor types | [43] |
| DBT d,f | 43, 0, 23 | 0.77 | - | - | Branched-chain amino acid metabolism | Mutated in maple syrup urine disease | [44] |
| PCCB d | 56, 0, 32 | 0.77 | - | - | Catabolism of propionyl-CoA | Reduced expression in CRC | [45] |
| NUP210 d | 53, 0, 35 | 0.2 | - | 7.6 × 10−3 | Muscle and neuronal differentiation | Upregulated in cervical cancer | [46] |
| EVA1C d | 53, 0, 28 | 1.3 × 10−2 | Yes (T vs. N) | 7.6 × 10−2 | Axon guidance (mouse) | [47] | |
| CFL2 d, f | 21, 21, 18 | 5.1 × 10−2 | Yes (T vs. N) | 1.5 × 10−2 | Axon guidance | Associated with progression in gastric cancer | [48] |
| CDC6 | 9, 16, 10 | 0.26 | - | - | DNA damage repair | Elevated in progression to PC | [49] |
| EYA4 d | 56, 0, 35 | 2.6 × 10−3 | Yes (T vs. N) | 2.8 × 10−3 | DNA damage repair | Hypermethylated in several cancers | [50] |
| GNAL d | 24, 0, 23 | 0.65 | - | - | Odorant signaling | Mutated in HCC | [51] |
| EFR3A d,f | 54, 0, 36 | 0.92 | - | - | Responsiveness of GPCRs | SNPs associated with CRC | [52] |
| SH3BGRL2 d | 31, 0, 54 | 0.34 | - | - | Unknown | ||
| C15orf41 d | 28, 0, 52 | 0.17 | - | - | Unknown | Congenital dyserythropoietic anemia | [53] |
| SELENOM d | 35, 0, 57 | 1.3 × 10−2 | Yes (T vs. N) | 3.0 × 10−3 | Unknown | IHC marker of aggressive HCC | [54] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marzec, J.; Ross-Adams, H.; Pirrò, S.; Wang, J.; Zhu, Y.; Mao, X.; Gadaleta, E.; Ahmad, A.S.; North, B.V.; Kammerer-Jacquet, S.-F.; et al. The Transcriptomic Landscape of Prostate Cancer Development and Progression: An Integrative Analysis. Cancers 2021, 13, 345. https://doi.org/10.3390/cancers13020345
Marzec J, Ross-Adams H, Pirrò S, Wang J, Zhu Y, Mao X, Gadaleta E, Ahmad AS, North BV, Kammerer-Jacquet S-F, et al. The Transcriptomic Landscape of Prostate Cancer Development and Progression: An Integrative Analysis. Cancers. 2021; 13(2):345. https://doi.org/10.3390/cancers13020345
Chicago/Turabian StyleMarzec, Jacek, Helen Ross-Adams, Stefano Pirrò, Jun Wang, Yanan Zhu, Xueying Mao, Emanuela Gadaleta, Amar S. Ahmad, Bernard V. North, Solène-Florence Kammerer-Jacquet, and et al. 2021. "The Transcriptomic Landscape of Prostate Cancer Development and Progression: An Integrative Analysis" Cancers 13, no. 2: 345. https://doi.org/10.3390/cancers13020345
APA StyleMarzec, J., Ross-Adams, H., Pirrò, S., Wang, J., Zhu, Y., Mao, X., Gadaleta, E., Ahmad, A. S., North, B. V., Kammerer-Jacquet, S.-F., Stankiewicz, E., Kudahetti, S. C., Beltran, L., Ren, G., Berney, D. M., Lu, Y.-J., & Chelala, C. (2021). The Transcriptomic Landscape of Prostate Cancer Development and Progression: An Integrative Analysis. Cancers, 13(2), 345. https://doi.org/10.3390/cancers13020345