ctDNA and Adjuvant Therapy for Colorectal Cancer: Time to Re-Invent Our Treatment Paradigm


Abstract
Simple Summary
Abstract
1. Introduction
2. Circulating Tumour DNA and Minimal Residual Disease
3. ctDNA and MRD Detection in Colorectal Cancer
4. ctDNA and Surveillance in CRC
5. ctDNA-Based Randomised Interventional Adjuvant Trials
6. Challenges and Future Directions
7. Conclusions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Douaiher, J.; Ravipati, A.; Grams, B.; Chowdhury, S.; Alatise, O.; Are, C. Colorectal cancer-global burden, trends, and geographical variations. J. Surg. Oncol. 2017, 115, 619–630. [Google Scholar] [CrossRef]
- Venook, A.P.; Niedzwiecki, N.; Lenz, H.-J.; Innocenti, F.; Fruth, B.; Meyerhardt, J.A.; Schrag, D.; Greene, C.; O’Neil, B.H.; Atkins, J.N.; et al. Effect of First-Line Chemotherapy Combined with Cetuximab or Bevacizumab on Overall Survival in Patients with KRAS Wild-Type Advanced or Metastatic Colorectal Cancer: A Randomized Clinical Trial. JAMA 2017, 317, 2392–2401. [Google Scholar] [CrossRef]
- Cremolini, C.; Loupakis, F.; Antoniotti, C.; Lupi, C.; Sensi, E.; Lonardi, S.; Mezi, S.; Tomasello, G.; Ronzoni, M.; Zaniboni, A.; et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: Updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015, 16, 1306–1315. [Google Scholar] [CrossRef]
- Heinemann, V.; von Weikersthal, L.F.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.-E.; Heintges, T.; Lerchenmüller, C.; Kahl, C.; Seipelt, G.; et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1065–1075. [Google Scholar] [CrossRef]
- Normanno, N.; Abate, R.E.; Lambiase, M.; Forgione, L.; Cardone, C.; Iannaccone, A.; Sacco, A.; Rachiglio, A.; Martinelli, E.; Rizzi, D.; et al. RAS testing of liquid biopsy correlates with the outcome of metastatic colorectal cancer patients treated with first line FOLFIRI plus cetuximab in the CAPRI-GOIM trial. Ann. Oncol. 2018, 29, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Vidal, J.; Muinelo, L.; Dalmases, A.; Jones, F.; Edelstein, D.; Iglesias, M.; Orrillo, M.; Abalo, A.; Rodríguez, C.; Brozos, E.; et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann. Oncol. 2017, 28, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, J.; Elez, E.; Caratù, G.; Matito, J.; Santos, C.; Macarulla, T.; Vidal, J.; Garcia, M.; Viéitez, J.; Paéz, D.; et al. Concordance of blood- and tumor-based detection of RAS mutations to guide anti-EGFR therapy in metastatic colorectal cancer. Ann. Oncol. 2017, 28, 1294–1301. [Google Scholar] [CrossRef]
- Sargent, D.; Sobrero, A.; Grothey, A.; O’Connell, M.J.; Buyse, M.; Andre, T.; Zheng, Y.; Green, E.; Labianca, R.; O’Callaghan, C.; et al. Evidence for Cure by Adjuvant Therapy in Colon Cancer: Observations Based on Individual Patient Data From 20,898 Patients on 18 Randomized Trials. J. Clin. Oncol. 2009, 27, 872–877. [Google Scholar] [CrossRef]
- Shah, M.A.; Renfro, L.A.; Allegra, C.J.; André, T.; de Gramont, A.; Schmoll, H.-J.; Haller, D.G.; Alberts, S.R.; Yothers, G.; Sargent, D.J. Impact of Patient Factors on Recurrence Risk and Time Dependency of Oxaliplatin Benefit in Patients with Colon Cancer: Analysis from Modern-Era Adjuvant Studies in the Adjuvant Colon Cancer End Points (ACCENT) Database. J. Clin. Oncol. 2016, 34, 843–853. [Google Scholar] [CrossRef]
- Labianca, R.; Nordlinger, B.; Beretta, G.D.; Mosconi, S.; Mandalà, M.; Cervantes, A.; Arnold, D. Early colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment, and follow-up. Ann. Oncol. 2013, 24, vi64–vi72. [Google Scholar] [CrossRef] [PubMed]
- Argilés, G.; Tabernero, J.; Labianca, R.; Hochhauser, D.; Salazar, R.; Iveson, T.; Laurent-Puig, P.; Quirke, P.; Yoshino, T.; Taieb, J.; et al. Localised colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment, and follow-up. Ann. Oncol. 2020, 31, 1291–1305. [Google Scholar] [CrossRef] [PubMed]
- Costas-Chavarri, A.; Temin, S.; Shah, M.A. Treatment of Patients with Early-Stage Colorectal Cancer: ASCO Resource-Stratified Guideline Summary. J. Oncol. Pr. 2019, 15, 290–292. [Google Scholar] [CrossRef]
- O’Connor, E.S.; Greenblatt, D.Y.; LoConte, N.K.; Gangnon, R.E.; Liou, J.-I.; Heise, C.P.; Smith, M.A. Adjuvant Chemotherapy for Stage II Colon Cancer with Poor Prognostic Features. J. Clin. Oncol. 2011, 29, 3381–3388. [Google Scholar] [CrossRef] [PubMed]
- Figueredo, A.; Charette, M.L.; Maroun, J.; Brouwers, M.C.; Zuraw, L. Adjuvant Therapy for Stage II Colon Cancer: A Systematic Review from the Cancer Care Ontario Program in Evidence-Based Care’s Gastrointestinal Cancer Disease Site Group. J. Clin. Oncol. 2004, 22, 3395–3407. [Google Scholar] [CrossRef]
- André, T.; Boni, C.; Navarro, M.; Tabernero, J.; Hickish, T.; Topham, C.; Bonetti, A.; Clingan, P.; Bridgewater, J.; Rivera, F.; et al. Improved Overall Survival with Oxaliplatin, Fluorouracil, and Leucovorin As Adjuvant Treatment in Stage II or III Colon Cancer in the MOSAIC Trial. J. Clin. Oncol. 2009, 27, 3109–3116. [Google Scholar] [CrossRef]
- André, T.; de Gramont, A.A.; Vernerey, D.; Chibaudel, B.B.; Bonnetain, F.; Tijeras-Raballand, A.A.; Scriva, A.A.; Hickish, T.T.; Tabernero, J.; van Laethem, J.L.; et al. Adjuvant Fluorouracil, Leucovorin, and Oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF Mutation and Mismatch Repair Status of the MOSAIC Study. J. Clin. Oncol. 2015, 33, 4176–4187. [Google Scholar] [CrossRef]
- Schmoll, H.-J.; Tabernero, J.; Maroun, J.; de Braud, F.; Price, T.; Van Cutsem, E.; Hill, M.; Hoersch, S.; Rittweger, K.; Haller, D.G. Capecitabine Plus Oxaliplatin Compared with Fluorouracil/Folinic Acid as Adjuvant Therapy for Stage III Colon Cancer: Final Results of the NO16968 Randomized Controlled Phase III Trial. J. Clin. Oncol. 2015, 33, 3733–3740. [Google Scholar] [CrossRef]
- Kidwell, K.M.; Yothers, G.; Ganz, P.A.; Land, S.R.; Ko, C.Y.; Cecchini, R.S.; Kopec, J.A.; Wolmark, N. Long-term neurotoxicity effects of oxaliplatin added to fluorouracil and leucovorin as adjuvant therapy for colon cancer: Results from National Surgical Adjuvant Breast and Bowel Project trials C-07 and LTS-01. Cancer 2012, 118, 5614–5622. [Google Scholar] [CrossRef]
- Mols, F.; Beijers, T.; Lemmens, V.; Hurk, C.J.V.D.; Vreugdenhil, G.; van de Poll-Franse, L.V. Chemotherapy-Induced Neuropathy and Its Association with Quality of Life Among 2- to 11-Year Colorectal Cancer Survivors: Results from the Population-Based PROFILES Registry. J. Clin. Oncol. 2013, 31, 2699–2707. [Google Scholar] [CrossRef]
- Pachman, D.R.; Qin, R.; Seisler, D.K.; Smith, E.M.; Beutler, A.S.; Ta, L.E.; Lafky, J.M.; Wagner-Johnston, N.D.; Ruddy, K.J.; Dakhil, S.R.; et al. Clinical Course of Oxaliplatin-Induced Neuropathy: Results from the Randomized Phase III Trial N08CB (Alliance). J. Clin. Oncol. 2015, 33, 3416–3422. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Sobrero, A.; Shields, A.F.; Yoshino, T.; Paul, J.; Taieb, J.; Souglakos, J.; Shi, Q.; Kerr, R.; Labianca, R.; et al. Duration of Adjuvant Chemotherapy for Stage III Colon Cancer. N. Engl. J. Med. 2018, 378, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Sobrero, A.F.; Andre, T.; Meyerhardt, J.A.; Grothey, A.; Iveson, T.; Yoshino, T.; Sougklakos, I.; Meyers, J.P.; Labianca, R.; Saunders, M.P.; et al. Overall survival (OS) and long-term disease-free survival (DFS) of three versus six months of adjuvant (adj) oxaliplatin and fluoropyrimidine-based therapy for patients (pts) with stage III colon cancer (CC): Results from the IDEA (International Duration Evaluation of Adj chemotherapy) collaboration. J. Clin. Oncol. 2020, 38, 4004. [Google Scholar] [CrossRef]
- Saltz, L.; Niedzwiecki, D.; Hollis, D.; Goldberg, R.M.; Hantel, A.; Thomas, J.P.; Fields, A.L.; Mayer, R.J. Irinotecan Fluorouracil Plus Leucovorin Is Not Superior to Fluorouracil Plus Leucovorin Alone as Adjuvant Treatment for Stage III Colon Cancer: Results of CALGB 89803. J. Clin. Oncol. 2007, 25, 3456–3461. [Google Scholar] [CrossRef] [PubMed]
- van Cutsem, E.; Labianca, R.; Bodoky, G.; Barone, C.; Aranda, E.; Nordlinger, B.; Topham, C.; Tabernero, J.; André, T.; Sobrero, A.F.; et al. Randomized Phase III Trial Comparing Biweekly Infusional Fluorouracil/Leucovorin Alone or With Irinotecan in the Adjuvant Treatment of Stage III Colon Cancer: PETACC-3. J. Clin. Oncol. 2009, 27, 3117–3125. [Google Scholar] [CrossRef]
- Ychou, M.; Raoul, J.-L.; Douillard, J.-Y.; Gourgou-Bourgade, S.; Bugat, R.; Mineur, L.; Viret, F.; Becouarn, Y.; Bouché, O.; Gamelin, E.; et al. A phase III randomised trial of LV5FU2 + irinotecan versus LV5FU2 alone in adjuvant high-risk colon cancer (FNCLCC Accord02/FFCD9802). Ann. Oncol. 2009, 20, 674–680. [Google Scholar] [CrossRef]
- Sargent, D.J.; Alberts, S.R.; Nair, S.; Mahoney, M.R.; Mooney, M.; Thibodeau, S.N.; Smyrk, T.C.; Sinicrope, F.A.; Chan, E.; Gill, S.; et al. Effect of Oxaliplatin, Fluorouracil, and Leucovorin With or Without Cetuximab on Survival Among Patients with Resected Stage III Colon Cancer. JAMA 2012, 307, 1383–1393. [Google Scholar] [CrossRef]
- Taieb, J.; Balogoun, R.; le Malicot, K.; Tabernero, J.; Mini, E.; Folprecht, G.; van Laethem, J.-L.; Emile, J.-F.; Mulot, C.; Fratté, S.; et al. Adjuvant FOLFOX +/− cetuximab in fullRAS andBRAF wildtype stage III colon cancer patients. Ann. Oncol. 2017, 28, 824–830. [Google Scholar] [CrossRef]
- de Gramont, A.; van Cutsem, E.; Schmoll, H.-J.; Tabernero, J.; Clarke, S.; Moore, M.J.; Cunningham, D.; Cartwright, T.H.; Hecht, J.R.; Rivera, F.; et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): A phase 3 randomised controlled trial. Lancet Oncol. 2012, 13, 1225–1233. [Google Scholar] [CrossRef]
- Kerr, R.; Love, S.; Segelov, E.; Johnstone, E.; Falcon, B.; Hewett, P.; Weaver, A.; Church, D.; Scudder, C.; Pearson, S.; et al. Adjuvant capecitabine plus bevacizumab versus capecitabine alone in patients with colorectal cancer (QUASAR 2): An open label, randomised phase 3 trial. Lancet Oncol. 2016, 17, 1543–1557. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar] [PubMed]
- Volik, S.; Alcaide, M.; Morin, R.D.; Collins, C.C. Cell-free DNA (cfDNA): Clinical Significance and Utility in Cancer Shaped by Emerging Technologies. Mol. Cancer Res. 2016, 14, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Mandel, P.; Metais, P. Nuclear Acids in Human Blood Plasma. C. R. Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar] [PubMed]
- Vasioukhin, V.; Anker, P.; Maurice, P.; Lyautey, J.; Lederrey, C.; Stroun, M. Point mutations of the N-ras gene in the blood plasma DNA of patients with myelodysplastic syndrome or acute myelogenous leukaemia. Br. J. Haematol. 1994, 86, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef]
- Bachet, J.; Bouché, O.; Taı̈eb, J.; Dubreuil, O.; Garcia, M.; Meurisse, A.; Normand, C.; Gornet, J.; Artru, P.; Louafi, S.; et al. RAS mutation analysis in circulating tumor DNA from patients with metastatic colorectal cancer: The AGEO RASANC prospective multicenter study. Ann. Oncol. 2018, 29, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Schmidt, K.; Choti, M.A.; E Romans, K.; Goodman, S.N.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.J.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Osumi, H.; Shinozaki, E.; Yamaguchi, K.; Zembutsu, H. Early change in circulating tumor DNA as a potential predictor of response to chemotherapy in patients with metastatic colorectal cancer. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Tie, J.; Kinde, I.; Wang, Y.; Wong, H.L.; Roebert, J.; Christie, M.; Tacey, M.; Wong, R.; Singh, M.; Karapetis, C.S.; et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann. Oncol. 2015, 26, 1715–1722. [Google Scholar] [CrossRef]
- Buccisano, F.; Maurillo, L.; del Principe, M.I.; del Poeta, G.; Sconocchia, G.; Lo-Coco, F.; Arcese, W.; Amadori, S.; Venditti, A. Prognostic and therapeutic implications of minimal residual disease detection in acute myeloid leukemia. Blood 2012, 119, 332–341. [Google Scholar] [CrossRef]
- Borowitz, M.J.; Devidas, M.; Hunger, S.P.; Bowman, W.P.; Carroll, A.J.; Carroll, W.L.; Linda, S.; Martin, P.L.; Pullen, D.J.; Viswanatha, D.; et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: A Children’s Oncology Group study. Blood 2008, 111, 5477–5485. [Google Scholar] [CrossRef] [PubMed]
- Brüggemann, M.; Raff, T.; Flohr, T.; Gökbuget, N.; Nakao, M.; Droese, J.; Lüschen, S.; Pott, C.; Ritgen, M.; Scheuring, U.; et al. Clinical significance of minimal residual disease quantification in adult patients with standard-risk acute lymphoblastic leukemia. Blood 2006, 107, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Landgren, O.; Devlin, S.; Boulad, M.; Mailankody, S. Role of MRD status in relation to clinical outcomes in newly diagnosed multiple myeloma patients: A meta-analysis. Bone Marrow Transplant. 2016, 51, 1565–1568. [Google Scholar] [CrossRef] [PubMed]
- Landgren, O.; Lu, S.X.; Hultcrantz, M. MRD Testing in Multiple Myeloma: The Main Future Driver for Modern Tailored Treatment. Semin. Hematol. 2018, 55, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Kwok, M.; Rawstron, A.C.; Varghese, A.; Evans, P.A.S.; O’Connor, S.J.M.; Doughty, C.; Newton, D.J.; Moreton, P.; Hillmen, P. Minimal residual disease is an independent predictor for 10-year survival in CLL. Blood 2016, 128, 2770–2773. [Google Scholar] [CrossRef] [PubMed]
- Hoelzer, D.; Bassan, R.; Dombret, H.; Fielding, A.; Ribera, J.M.; Buske, C. Acute lymphoblastic leukaemia in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, v69–v82. [Google Scholar] [CrossRef] [PubMed]
- Gorgannezhad, L.; Umer, M.; Islam, N.; Nguyen, N.-T.; Shiddiky, M.J. Circulating tumor DNA and liquid biopsy: Opportunities, challenges, and recent advances in detection technologies. Lab Chip 2018, 18, 1174–1196. [Google Scholar] [CrossRef]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, C.C.S.P.R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.J.; Lindeman, N.; Lockwood, C.M.; Rai, A.J.; Schilsky, R.L.; et al. Circulating Tumor DNA Analysis in Patients with Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. Arch. Pathol. Lab. Med. 2018, 142, 1242–1253. [Google Scholar] [CrossRef]
- Perakis, S.; Auer, M.; Belic, J.; Heitzer, E. Advances in Circulating Tumor DNA Analysis. In Advances in Clinical Chemistry; Elsevier BV: Amstedam, The Netherlands, 2017; Volume 80, pp. 73–153. [Google Scholar]
- Dasari, A.; Morris, V.K.; Allegra, C.J.; Atreya, C.; Benson, A.B.; Boland, P.; Chung, K.; Copur, M.S.; Corcoran, R.B.; Deming, D.A.; et al. ctDNA applications and integration in colorectal cancer: An NCI Colon and Rectal–Anal Task Forces whitepaper. Nat. Rev. Clin. Oncol. 2020, 17, 757–770. [Google Scholar] [CrossRef]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection, and quantification of rare mutations with massively parallel sequencing. Proc. Natil. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.Y.; Kaper, F.; Dawson, S.-J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive Identification and Monitoring of Cancer Mutations by Targeted Deep Sequencing of Plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.R.; Contente-Cuomo, T.; Sammut, S.-J.; Odenheimer-Bergman, A.; Ernst, B.; Perdigones, N.; Chin, S.-F.; Farooq, M.; Mejia, R.; Cronin, P.A.; et al. Personalized circulating tumor DNA analysis to detect residual disease after neoadjuvant therapy in breast cancer. Sci. Transl. Med. 2019, 11, eaax7392. [Google Scholar] [CrossRef]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.-T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients with Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef]
- Abbosh, C.; Frankell, A.; Garnett, A.; Harrison, T.; Weichert, M.; Licon, A.; Veeriah, S.; Daber, B.; Moreau, M.; Chesh, A.; et al. Abstract CT023: Phylogenetic tracking and minimal residual disease detection using ctDNA in early-stage NSCLC: A lung TRACERx study. In Proceedings of the Tumor Biology; American Association for Cancer Research (AACR): Philadelphia, PA, USA, 2020; Volume 80, p. CT023. [Google Scholar]
- Heider, K.; Gale, D.; Ruiz-Valdepenas, A.; Marsico, G.; Sharma, G.; Perry, M.; Osborne, R.; Howarth, K.; Lazarus, T.; Rundell, V.; et al. Abstract 735: Sensitive detection of ctDNA in early-stage non-small cell lung cancer patients with a personalized sequencing assay. In Proceedings of the Clinical Trials; American Association for Cancer Research (AACR): Philadelphia, PA, USA, 2020; Volume 80, p. 735. [Google Scholar]
- Postel, M.; Roosen, A.; Laurent-Puig, P.; Taly, V.; Wang-Renault, S.-F. Droplet-based digital PCR and next generation sequencing for monitoring circulating tumor DNA: A cancer diagnostic perspective. Expert Rev. Mol. Diagn. 2018, 18, 7–17. [Google Scholar] [CrossRef]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-Throughput Droplet Digital PCR System for Absolute Quantitation of DNA Copy Number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Commissioner of the, U.S. Food and Drug Administration. Available online: https://www.fda.gov/home (accessed on 11 January 2021).
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.-L.; Christie, M.; et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 2016, 8, 346ra92. [Google Scholar] [CrossRef]
- Schøler, L.V.; Reinert, T.; Ørntoft, M.-B.W.; Kassentoft, C.G.; Árnadóttir, S.S.; Vang, S.; Nordentoft, I.; Knudsen, M.; Lamy, P.; Andreasen, D.; et al. Clinical Implications of Monitoring Circulating Tumor DNA in Patients with Colorectal Cancer. Clin. Cancer Res. 2017, 23, 5437–5445. [Google Scholar] [CrossRef]
- Taieb, J.; Taly, V.; Vernerey, D.; Bourreau, C.; Bennouna, J.; Faroux, R.; Desrame, J.; Bouche, O.; Borg, C.; Egreteau, J.; et al. Analysis of circulating tumour DNA (ctDNA) from patients enrolled in the IDEA-FRANCE phase III trial: Prognostic and predictive value for adjuvant treatment duration. Ann. Oncol. 2019, 30, v867. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Wang, Y.; Li, L.; Christie, M.; Simons, K.; Elsaleh, H.; Kosmider, S.; Wong, R.; Yip, D.; et al. Serial circulating tumour DNA analysis during multimodality treatment of locally advanced rectal cancer: A prospective biomarker study. Gut 2019, 68, 663–671. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Wang, Y.; Christie, M.; Simons, K.; Lee, M.; Wong, R.; Kosmider, S.; Ananda, S.; McKendrick, J.; et al. Circulating Tumor DNA Analyses as Markers of Recurrence Risk and Benefit of Adjuvant Therapy for Stage III Colon Cancer. JAMA Oncol. 2019, 5, 1710–1717. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Wang, Y.; Springer, S.; Kinde, I.; Wong, H.-L.; Kosmider, S.; Tran, B.; Christie, M.; Thomson, B.N.; Wong, R.; et al. Serial circulating tumor DNA (ctDNA) and recurrence risk in patients (pts) with resectable colorectal liver metastasis (CLM). J. Clin. Oncol. 2016, 34, e15131. [Google Scholar] [CrossRef]
- Khakoo, S.; Carter, P.D.; Brown, G.; Valeri, N.; Picchia, S.; Bali, M.A.; Shaikh, R.; Jones, T.; Begum, R.; Rana, I.; et al. MRI Tumor Regression Grade and Circulating Tumor DNA as Complementary Tools to Assess Response and Guide Therapy Adaptation in Rectal Cancer. Clin. Cancer Res. 2020, 26, 183–192. [Google Scholar] [CrossRef]
- Parikh, A.R.; van Seventer, E.E.; Boland, G.M.; Hartwig, A.; Jaimovich, A.; Raymond, V.M.; Talasaz, A.; Corcoran, R.B. A plasma-only integrated genomic and epigenomic circulating tumor DNA (ctDNA) assay to inform recurrence risk in colorectal cancer (CRC). J. Clin. Oncol. 2019, 37, 3602. [Google Scholar] [CrossRef]
- Overman, M.J.; Vauthey, J.-N.; Aloia, T.A.; Conrad, C.; Chun, Y.S.; Pereira, A.; Jiang, Z.; Crosby, S.; Wei, S.; Raghav, K.P.S.; et al. Circulating tumor DNA (ctDNA) utilizing a high-sensitivity panel to detect minimal residual disease post liver hepatectomy and predict disease recurrence. J. Clin. Oncol. 2017, 35, 3522. [Google Scholar] [CrossRef]
- Cervantes, A.; Gimeno-Valiente, F.; Gambardella, V.; Zuñiga, S.; Rentero-Garrido, P.; Huerta, M.; Roselló, S.; Martinez-Ciarpaglini, C.; Carbonell-Asins, J.; Carrasco, F.; et al. Targeted next-generation sequencing of circulating-tumor DNA for tracking minimal residual disease in localized colon cancer. Ann. Oncol. 2019, 30, 1804–1812. [Google Scholar] [CrossRef]
- Meyerhardt, J.A.; Mangu, P.B.; Flynn, P.J.; Korde, L.; Loprinzi, C.L.; Minsky, B.D.; Petrelli, N.J.; Ryan, K.; Schrag, D.H.; Wong, S.L.; et al. Follow-Up Care, Surveillance Protocol, and Secondary Prevention Measures for Survivors of Colorectal Cancer: American Society of Clinical Oncology Clinical Practice Guideline Endorsement. J. Clin. Oncol. 2013, 31, 4465–4470. [Google Scholar] [CrossRef]
- Primrose, J.N.; Perera, R.; Gray, A.; Rose, P.; Fuller, A.; Corkhill, A.; George, S.; Mant, D. Effect of 3 to 5 Years of Scheduled CEA and CT Follow-up to Detect Recurrence of Colorectal Cancer. JAMA 2014, 311, 263–270. [Google Scholar] [CrossRef]
- Rosati, G.; Ambrosini, G.; Barni, S.; Andreoni, B.; Corradini, G.; Luchena, G.; Daniele, B.; Gaion, F.; Oliverio, G.; Duro, M.; et al. A randomized trial of intensive versus minimal surveillance of patients with resected Dukes B2-C colorectal carcinoma. Ann. Oncol. 2016, 27, 274–280. [Google Scholar] [CrossRef]
- Lepage, C.; Phelip, J.; Cany, L.; Barbier, E.; Manfredi, S.; Deguiral, P.; Faroux, R.; Baconnier, M.; Pezet, D.; Duchmann, J.; et al. Effect of 5 years of imaging and CEA follow-up to detect recurrence of colorectal cancer (CRC)—PRODIGE 13 a FFCD phase III trial. Ann. Oncol. 2020, 31, S410. [Google Scholar] [CrossRef]
- Litvak, A.; Cercek, A.; Segal, N.; Reidy-Lagunes, D.; Stadler, Z.K.; Yaeger, R.D.; Kemeny, N.E.; Weiser, M.R.; Pessin, M.S.; Saltz, L. False-Positive Elevations of Carcinoembryonic Antigen in Patients with a History of Resected Colorectal Cancer. J. Natl. Compr. Cancer Netw. 2014, 12, 907–913. [Google Scholar] [CrossRef]
- Newton, K.F.; Newman, W.; Hill, J. Review of biomarkers in colorectal cancer. Color. Dis. 2011, 14, 3–17. [Google Scholar] [CrossRef]
- Goldstein, M.J.; Mitchell, E.P. Carcinoembryonic Antigen in the Staging and Follow-up of Patients with Colorectal Cancer. Cancer Investig. 2005, 23, 338–351. [Google Scholar] [CrossRef]
- Anandappa, G.; Starling, N.; Peckitt, C.; Bryant, A.; Begum, R.; Carter, P.; Hatt, S.; Khakoo, S.S.; Turner, A.; Kidd, S.; et al. TRACC: Tracking mutations in cell-free DNA to predict relapse in early colorectal cancer—A randomized study of circulating tumour DNA (ctDNA) guided adjuvant chemotherapy versus standard of care chemotherapy after curative surgery in patients with high-risk stage II or stage III colorectal cancer (CRC). J. Clin. Oncol. 2020, 38, TPS4120. [Google Scholar] [CrossRef]
- Schraa, S.J.; on behalf of the PLCRC-MEDOCC group; Van Rooijen, K.L.; Van Der Kruijssen, D.E.W.; Alarcón, C.R.; Phallen, J.; Sausen, M.; Simmons, J.; Coupé, V.M.H.; Van Grevenstein, W.M.U.; et al. Circulating tumor DNA guided adjuvant chemotherapy in stage II colon cancer (MEDOCC-CrEATE): Study protocol for a trial within a cohort study. BMC Cancer 2020, 20, 1–10. [Google Scholar] [CrossRef]
- Morris, V.K.; Yothers, G.; Kopetz, S.; Jacobs, S.A.; Lucas, P.C.; Iqbal, A.; Boland, P.M.; Deming, D.A.; Scott, A.J.; Lim, H.J.; et al. Phase II/III study of circulating tumor DNA as a predictive biomarker in adjuvant chemotherapy in patients with stage II colon cancer: NRG-GI005 (COBRA). J. Clin. Oncol. 2020, 38, TPS4121. [Google Scholar] [CrossRef]
- Folprecht, G.; Reinacher-Schick, A.; Tannapfel, A.; Weitz, J.; Kossler, T.; Weiss, L.; Aust, D.E.; Von Bubnoff, N.; Kramer, M.; Thiede, C. Circulating tumor DNA-based decision for adjuvant treatment in colon cancer stage II evaluation: (CIRCULATE-trial) AIO-KRK-0217. J. Clin. Oncol. 2020, 38, TPS273. [Google Scholar] [CrossRef]
- Taieb, J.; Benhaim, L.; Puig, P.L.; le Malicot, K.; Emile, J.F.; Geillon, F.; Tougeron, D.; Manfredi, S.; Chauvenet, M.; Taly, V.; et al. Decision for adjuvant treatment in stage II colon cancer based on circulating tumor DNA:The CIRCULATE-PRODIGE 70 trial. Dig. Liver Dis. 2020, 52, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Yukami, H.; Mishima, S.; Kotani, D.; Oki, E.; Taniguchi, H.; Nakamura, Y.; Kato, T.; Takemasa, I.; Yamanaka, T.; Shirasu, H.; et al. P-120 Prospective observational study monitoring circulating tumor DNA in resectable colorectal cancer patients undergoing radical surgery: GALAXY study in CIRCULATE-Japan (trial in progress). Ann. Oncol. 2020, 31, S128–S129. [Google Scholar] [CrossRef]
- Yukami, H.; Saori, M.; Kotani, D.; Oki, E.; Taniguchi, H.; Nakamura, Y.; Kato, T.; Takemasa, I.; Yamanaka, T.; Shirasu, H.; et al. 113TiP Prospective observational study monitoring circulating tumour DNA in resectable colorectal cancer patients undergoing radical surgery: GALAXY study in CIRCULATE-Japan. Ann. Oncol. 2020, 31, S1285–S1286. [Google Scholar] [CrossRef]
- Nors, J.; Henriksen, T.V.; Gotschalck, K.A.; Juul, T.; Søgaard, J.; Iversen, L.H.; Andersen, C.L. IMPROVE-IT2: Implementing noninvasive circulating tumor DNA analysis to optimize the operative and postoperative treatment for patients with colorectal cancer—Intervention trial 2. Study protocol. Acta Oncol. 2020, 59, 336–341. [Google Scholar] [CrossRef]
- Sargent, D.J.; Conley, B.A.; Allegra, C.; Collette, L. Clinical Trial Designs for Predictive Marker Validation in Cancer Treatment Trials. J. Clin. Oncol. 2005, 23, 2020–2027. [Google Scholar] [CrossRef]
- Lonardi, S.; Montagut, C.; Pietrantonio, F.; Elez, E.; Sartore-Bianchi, A.; Tarazona, N.; Sciallero, S.; Zampino, M.G.; Mosconi, S.; Muñoz, S.; et al. The PEGASUS trial: Post-surgical liquid biopsy-guided treatment of stage III and high-risk stage II colon cancer patients. J. Clin. Oncol. 2020, 38, TPS4124. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Lo, S.N.; Wang, Y.; Li, L.; Christie, M.; Lee, M.; Wong, R.; Kosmider, S.; Skinner, I.; et al. Prognostic significance of postsurgery circulating tumor DNA in nonmetastatic colorectal cancer: Individual patient pooled analysis of three cohort studies. Int. J. Cancer 2021, 148, 1014–1026. [Google Scholar] [CrossRef]
- Gai, W.; Sun, K. Epigenetic Biomarkers in Cell-Free DNA and Applications in Liquid Biopsy. Genes 2019, 10, 32. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Nishida, N.; Calin, G.A.; Pantel, K. Clinical relevance of circulating cell-free microRNAs in cancer. Nat. Rev. Clin. Oncol. 2014, 11, 145–156. [Google Scholar] [CrossRef]
- Henriksen, T.V.; Reinert, T.; Christensen, E.; Sethi, H.; Birkenkamp-Demtröder, K.; Gögenur, M.; Gögenur, I.; Zimmermann, B.G.; Dyrskjøt, L.; Andersen, C.L.; et al. The effect of surgical trauma on circulating free DNA levels in cancer patients—Implications for studies of circulating tumor DNA. Mol. Oncol. 2020, 14, 1670–1679. [Google Scholar] [CrossRef]
- Kasi, P.M.; Dayyani, F.; Morris, V.K.; Kopetz, S.; Parikh, A.R.; Starr, J.S.; Cohen, S.; Grothey, A.; Lieu, C.H.; O’Hara, M.H.; et al. Tumor-informed assessment of molecular residual disease and its incorporation into practice for patients with early and advanced-stage colorectal cancer (CRC-MRD Consortia). J. Clin. Oncol. 2020, 38, 4108. [Google Scholar] [CrossRef]
- Garcia-Murillas, I.; Chopra, N.; Comino-Méndez, I.; Beaney, M.; Tovey, H.; Cutts, R.J.; Swift, C.; Kriplani, D.; Afentakis, M.; Hrebien, S.; et al. Assessment of Molecular Relapse Detection in Early-Stage Breast Cancer. JAMA Oncol. 2019, 5, 1473–1478. [Google Scholar] [CrossRef]
- Gray, R.; Quirke, P.; Handley, K.; Lopatin, M.; Magill, L.; Baehner, F.L.; Beaumont, C.; Clark-Langone, K.M.; Yoshizawa, C.N.; Lee, M.; et al. Validation Study of a Quantitative Multigene Reverse Transcriptase–Polymerase Chain Reaction Assay for Assessment of Recurrence Risk in Patients with Stage II Colon Cancer. J. Clin. Oncol. 2011, 29, 4611–4619. [Google Scholar] [CrossRef] [PubMed]
- Niedzwiecki, D.; Frankel, W.L.; Venook, A.P.; Ye, X.; Friedman, P.N.; Goldberg, R.M.; Mayer, R.J.; Colacchio, T.A.; Mulligan, J.M.; Davison, T.S.; et al. Association Between Results of a Gene Expression Signature Assay and Recurrence-Free Interval in Patients with Stage II Colon Cancer in Cancer and Leukemia Group B 9581 (Alliance). J. Clin. Oncol. 2016, 34, 3047–3053. [Google Scholar] [CrossRef]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.-S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Mlecnik, B.; Bifulco, C.; Bindea, G.; Marliot, F.; Lugli, A.; Lee, J.J.; Zlobec, I.; Rau, T.T.; Berger, M.D.; Nagtegaal, I.D.; et al. Multicenter International Society for Immunotherapy of Cancer Study of the Consensus Immunoscore for the Prediction of Survival and Response to Chemotherapy in Stage III Colon Cancer. J. Clin. Oncol. 2020, 38, 3638–3651. [Google Scholar] [CrossRef]
- Galon, J.; Hermitte, F.; Mlecnik, B.; Lugli, A.; Bifulco, C.B.; Nagtegaal, I.D.; Hartmann, A.; Marliot, F.; Eynde, M.V.D.; Roehrl, M.H.A.; et al. Immunoscore as a parameter predicting time to recurrence and disease-free survival in T4N0 stage II colon cancer patients. J. Clin. Oncol. 2020, 38, 4105. [Google Scholar] [CrossRef]


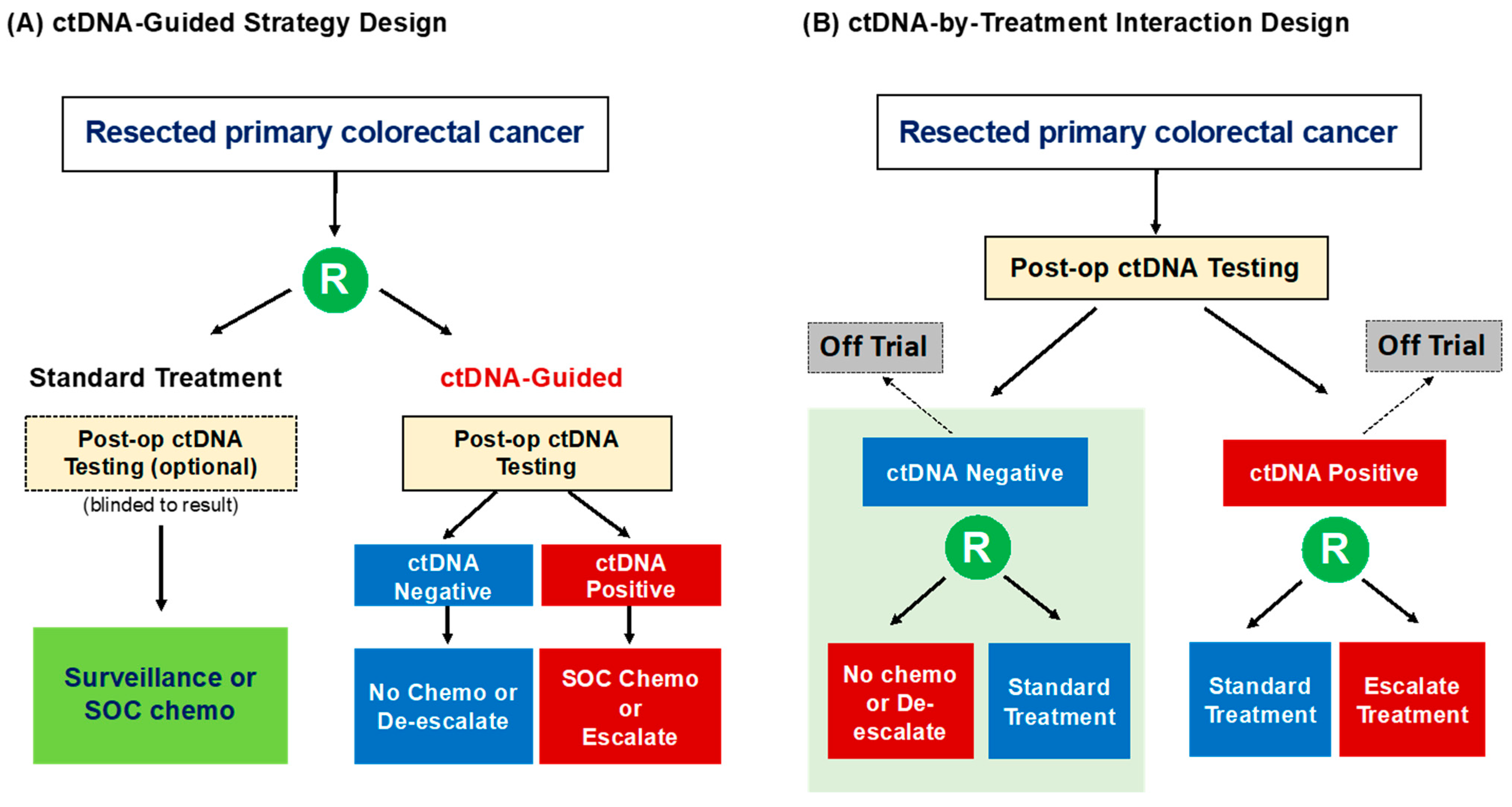{kind=link}
| Reference | No. of Patients | Stages Evaluated | Method for ctDNA Analysis | Adjuvant Chemo Given | Key Results | % of Patients ctDNA Positive |
|---|---|---|---|---|---|---|
| Tie et al., 2016 [62] | 230 | II | Safe-SeqS | 23% | In patients not treated with adjuvant treatment, presence of ctDNA after surgery was associated with an inferior recurrence-free survival (HR, 18; p = 0.001) 85% of patients were ctDNA-positive up to or at the time of radiologic recurrence, CEA was only elevated in 41% of patients. The median lead time from ctDNA detection to radiological recurrence was 167 days; range 81–279 days | Post-op: 7.9% Post-Treatment: 11% Surveillance: 11.7% |
| Reinert et al., 2019 [56] | 130 | I–III | Signatera | 62% | Post-op ctDNA-positive patients were more than 7 times more likely to experience disease recurrence than ctDNA-negative patients (HR, 7.2; p < 0.001) Lead time to detect disease recurrence compared with standard surveillance: Mean 8.7 months; range 0.8–16.5 months | Post-op: 10.6% Post-Treatment: 12% Surveillance: 20% |
| Schøler et al., 2017 [63] | 45 | I–IV | ddPCR | 36.8% | Longitudinal samples from 27 patients revealed ctDNA detection postoperatively in all relapsing patients (n = 14), but not in any of the non-relapsing patients. Of 21 patients treated for localised disease, all 6 ctDNA-positive patients (within 3 months of surgery) relapsed compared with 4 of the remaining patients (HR, 37.7; 95% CI; 4.2–335.5; p < 0.001). Time to detect disease recurrence of standard surveillance: Median lead time of 9.4 months, ranging from 0.4 to 14.9 months | Post-op: 28.6% (stages I–III) Post-Treatment: not reported Surveillance: not reported |
| Taieb et al., 2019 [64] | 805 | II–III | ddPCR (2 methylation markers) | All patients | 2-year DFS was 64% vs. 82% in ctDNA-positive and -negative patients, respectively (HR, 1.75; 95% CI, 1.25–2.45; p < 0.001). Post-surgical plasma ctDNA predicted metastatic relapse a median of 10 months before recurrence was visible on radiological scans (HR, 11.33; p = 0.0001 | Post-op: 13.5% Post-Treatment: not reported Surveillance: not reported |
| Tie et al., 2019 [65] | 159 | Locally advanced | Safe-SeqS | 35.8% patients | Significantly worse recurrence-free survival was seen if ctDNA was detectable after chemoradiotherapy (HR, 6.6; p < 0.001) or after surgery (HR, 13.0; p < 0.001). Estimated 3-year recurrence-free survival was 33% for post-operative ctDNA-positive patients and 87% for the postoperative ctDNA-negative patients. | Post-op: 11.9% Post-Treatment: not reported Surveillance: not reported |
| Tie et al., 2019 [66] | 96 | III | Safe-SeqS | All patients | Estimated 3-year RFS was 30% when ctDNA was detectable after chemotherapy and 77% when ctDNA was undetectable (HR, 6.8; 95% CI, 11.0–157.0; p < 0.001) | Post-op: 21% Post-Treatment: 17% Surveillance: Not tested |
| Tie et al., 2016 [67] | 37 | IV (resectable colorectal liver metastases) | Safe-SeqS | 70% | ctDNA detectable at a median of 3 months prior to clinical recurrence. Ten of 10 pts (100%) with positive post-treatment (surgery and chemotherapy) ctDNA experienced recurrence vs. 4 of 27 (15%) with negative post-treatment ctDNA (HR, 13.16, p < 0.0001) | Post-op: 24.3% Post-Treatment: 27% Surveillance: 32.4% |
| Khakoo et al., 2020 [68] | 47 | Locally advanced rectal cancer | ddPCR | 91.3% | All 3 patients with detectable ctDNA post-surgery relapsed compared with none of the 20 patients with undetectable ctDNA (p = 0.001) | Post-op: 13% Post-Treatment: not reported Surveillance: not reported |
| Parikh et al., 2019 [69] | 72 | Stage II–III | Guardant health NGS | 41.2% | Patients who were ctDNA-positive after standard therapy completion had a recurrence positive predictive value 93%, negative predictive value 80%, (HR, 11.29; p < 0.0001) | Post-op: 19% (surgery arm only) Post-Treatment: 22.2% (chemotherapy arm only) Surveillance: not reported |
| Overman et al., 2017 [70] | 54 | IV (resectable liver metastases) | 30 kb ctDNA digital sequencing panel (Guardant Health) covering SNVs in 21 genes | Not reported | In 43 patients who underwent successful resection of all visible disease, post-op detection of ctDNA significantly correlated with RFS (HR, 3.1; 95% CI, 1.7–9.1; p = 0.002) with 2-year RFS of 0% vs. 47%. ctDNA detected at median of 5.1 months prior to radiographic recurrence. | Post-op: 44% Post-Treatment: Not reported Surveillance: Not reported |
| Tarazona et al., 2019 [71] | 150 | Stage II–III | ddPCR | 37.2% | Detection of ctDNA after surgery and in serial plasma samples during follow-up were associated with poorer DFS (HR, 17.56; log-rank p = 0.0014 and HR, 11.33; log-rank p = 0.0001, respectively) In patients treated with adjuvant chemotherapy, presence of ctDNA after therapy was associated with early relapse (HR, 10.02; log-rank p < 0.0001) | Post-op: 20.3% Post-Treatment: 28% (patients receiving adjuvant chemotherapy) Surveillance: Not reported |
| Trial Name/Country | Patient Population | Sample Size | ctDNA Assay | Timing of ctDNA Testing | Trial Intervention | Primary Objective |
|---|---|---|---|---|---|---|
| ctDNA-Guided Strategy Design | ||||||
| DYNAMIC (ACTRN-12615000381583) Australia | Stage II colon cancer | 450 | Safe-SeqS | Week 4 and 7 post-op | Standard of care: clinician determined management (surveillance or adjuvant chemotherapy) based on standard clinicopathological features ctDNA-guided: ctDNA-positive → adjuvant chemotherapy; ctDNA-negative → surveillance | To demonstrate that an adjuvant therapy strategy based on post-op ctDNA results will reduce the number of patients receiving adjuvant chemotherapy without compromising recurrence-free survival |
| DYNAMIC-III (ACTRN-12617001566325) Australia/New Zealand | Stage III colon cancer | 1000 | Safe-SeqS | Week 5–6 post-op | Standard of care: clinician determined standard of care adjuvant chemotherapy based on clinical risk ctDNA-guided: ctDNA-positive → escalated chemotherapy regimen from pre-planned treatment (increase duration or number of agents); ctDNA-negative → de-escalated chemotherapy regimen from pre-planned treatment (reduction in duration or number of agents) | To evaluate the impact of de-escalation/escalation treatment strategies as informed by post-op ctDNA-informed management
|
| DYNAMIC-RECTAL (ACTRN-12617001560381) Australia/New Zealand | Locally advanced rectal cancer | 408 | Safe-SeqS | Week 4 and 7 post-op | Standard of care: clinician determined management (surveillance or adjuvant chemotherapy) based on standard clinicopathological features ctDNA-guided: ctDNA-positive → adjuvant chemotherapy; ctDNA-negative and ypN0 → surveillance; ctDNA-negative and ypN+ → surveillance or adjuvant chemotherapy at clinician’s choice | To demonstrate that an adjuvant therapy strategy incorporating ctDNA results in addition to standard pathologic risk assessment will reduce the number of patients receiving adjuvant chemotherapy without compromising recurrence-free survival |
| TRACC (NCT04050345) [79] United Kingdom | High risk stage II, stage III colorectal cancer | 1621 | NGS-based 22-gene colorectal panel | <8 weeks post-op, 3 months post-op | Standard of care: 6 months of capecitabine or 3 months of CAPOX ctDNA-guided: ctDNA-positive → standard adjuvant chemotherapy; ctDNA-negative → de-escalate treatment but re-escalate if ctDNA becomes positive at 3 months |
|
| MEDOCC-CrEATE (NL6281/NTR6455) [80] Netherlands | Stage II colon cancer | 1320 | PGDx elio™ | 4–21 days post-op | Standard of care: surveillance ctDNA-guided: ctDNA-positive → 6 months of CAPOX; ctDNA-negative → surveillance | To investigate the willingness of patients to receive adjuvant chemotherapy after detection of ctDNA post-surgery |
| Marker-by-Treatment Interaction Design | ||||||
| NRG GI-005 (COBRA) NCT04068103 [81] United States/Canada | Stage IIA colon cancer | 1408 | Guardant LUNAR-1™ | Post-op | Standard of care: Surveillance ctDNA-guided: ctDNA-positive → adjuvant FOLFOX/CAPOX; ctDNA-negative → surveillance |
|
| CIRCULATE AIO-KRK-0217 (NCT04089631) [82] Germany | Stage II colon cancer (MSS tumours) | 4812 | Not reported | Post-op | ctDNA-positive patients randomised to: Standard of care: surveillance Experimental: adjuvant chemotherapy (capecitabine or CAPOX) | To compare the disease-free survival in patients who are positive for postoperative ctDNA treated with or without adjuvant chemotherapy |
| CIRCULATE PRODIGE 70 (NCT04120701) [83] France | Stage II colon cancer | 1980 | ddPCR (2 methylated markers WIF1 and NPY) | Week 2–8 post-op | 198 ctDNA-positive patients randomised to: Standard of care: surveillance Experimental: adjuvant FOLFOX | To demonstrate a 17.5% gain in 3-year DFS in post-op ctDNA-positive patients treated with adjuvant FOLFOX compared to observation alone |
| VEGA (UMIN000039205) [84] Japan | High-risk stage II, low-risk stage III colon cancer—ctDNA-negative | 1240 | Signatera™ | 1-month post-op | Post-op ctDNA-negative patients randomised to: Standard of care: 3 months of CAPOX Experimental: Surveillance
| To demonstrate the non-inferiority of observation vs. adjuvant CAPOX with absence of ctDNA at 1 month post-surgery |
| ctDNA-Enriched 2nd Line Adjuvant Therapy Trial | ||||||
| ALTAIR (UMIN000039205) [85] Japan | Stage II/III colorectal cancer or stage IV with resectable metastases | 240 | Signatera™ | 1-month post-op and after 3 months of standard adjuvant CAPOX | Patients who are ctDNA-positive after completion of 3 months adjuvant CAPOX are randomised to: Standard of care: placebo/surveillance Experimental: trifluridine/tipiracil | To demonstrate the superiority of trifluridine/tipiracil over placebo in patients with ctDNA that remains positive after standard adjuvant therapy |
| ACT-3 (NCT04259944) United States | Stage III colorectal cancer | 500 | Guardant LUNAR-1™ | 3–6 weeks post adjuvant chemo | Patients who are ctDNA-positive after completion of 3–6 months of adjuvant FOLFOX/CAPOX are randomised to: Standard of care: surveillance Experimental:
| To demonstrate the superiority of FOLFIRI over surveillance in patients with positive ctDNA after standard adjuvant therapy |
| ctDNA-Guided Surveillance Strategy Design | ||||||
| IMPROVE-IT2 (NCT04084249) [86] Denmark | High risk stage II, stage III colorectal cancer | 254 | Droplet digital PCR (colorectal panel) | Every 4 months post-op for 24 months | Standard of care: standard Danish follow-up program (CT scans at 12 and 36 months) ctDNA-guided surveillance: 3-monthly FDG-PET/CT for patient with positive ctDNA during surveillance | To demonstrate that ctDNA guided post-operative surveillance combining ctDNA and radiological assessments could result in earlier detection of recurrent disease and identify more patients eligible for curative treatment |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naidoo, M.; Gibbs, P.; Tie, J. ctDNA and Adjuvant Therapy for Colorectal Cancer: Time to Re-Invent Our Treatment Paradigm. Cancers 2021, 13, 346. https://doi.org/10.3390/cancers13020346
Naidoo M, Gibbs P, Tie J. ctDNA and Adjuvant Therapy for Colorectal Cancer: Time to Re-Invent Our Treatment Paradigm. Cancers. 2021; 13(2):346. https://doi.org/10.3390/cancers13020346
Chicago/Turabian StyleNaidoo, Mahendra, Peter Gibbs, and Jeanne Tie. 2021. "ctDNA and Adjuvant Therapy for Colorectal Cancer: Time to Re-Invent Our Treatment Paradigm" Cancers 13, no. 2: 346. https://doi.org/10.3390/cancers13020346
APA StyleNaidoo, M., Gibbs, P., & Tie, J. (2021). ctDNA and Adjuvant Therapy for Colorectal Cancer: Time to Re-Invent Our Treatment Paradigm. Cancers, 13(2), 346. https://doi.org/10.3390/cancers13020346