
Transcriptomics and Metabolomics Integration Reveals Redox-Dependent Metabolic Rewiring in Breast Cancer Cells

 , , , , ,
, , , , ,  , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Proliferation Analysis
2.3. Metabolites Quantification in the Media Samples
2.4. Metabolite Extraction from Cell Culture
2.5. LC-MS Metabolic Profiling
2.6. Seahorse Oxygen Consumption Rate
2.7. NADH/NAD+ Quantification
2.8. ROS Levels Measurement
2.9. Stable Transfection
2.10. Real-Time PCR
2.11. Western Blot Analysis
2.12. Whole-Genome cDNA Microarray Expression Analysis
2.13. Transcriptomic Analysis
2.14. Integration Analysis between Transcriptomics and Metabolomics Data
2.15. Circos Plots Analysis
2.16. Chemical Synthesis
2.16.1. Synthesis of Sodium 2-Hydrazone-3-phenylpropanoate (P4)
2.16.2. Synthesis of Sodium 2-Hydroxyimino-3-phenylpropanoate (HIPP)
2.16.3. Studies of Stability of P4 and HIPP
2.17. Statistical Analysis
3. Results
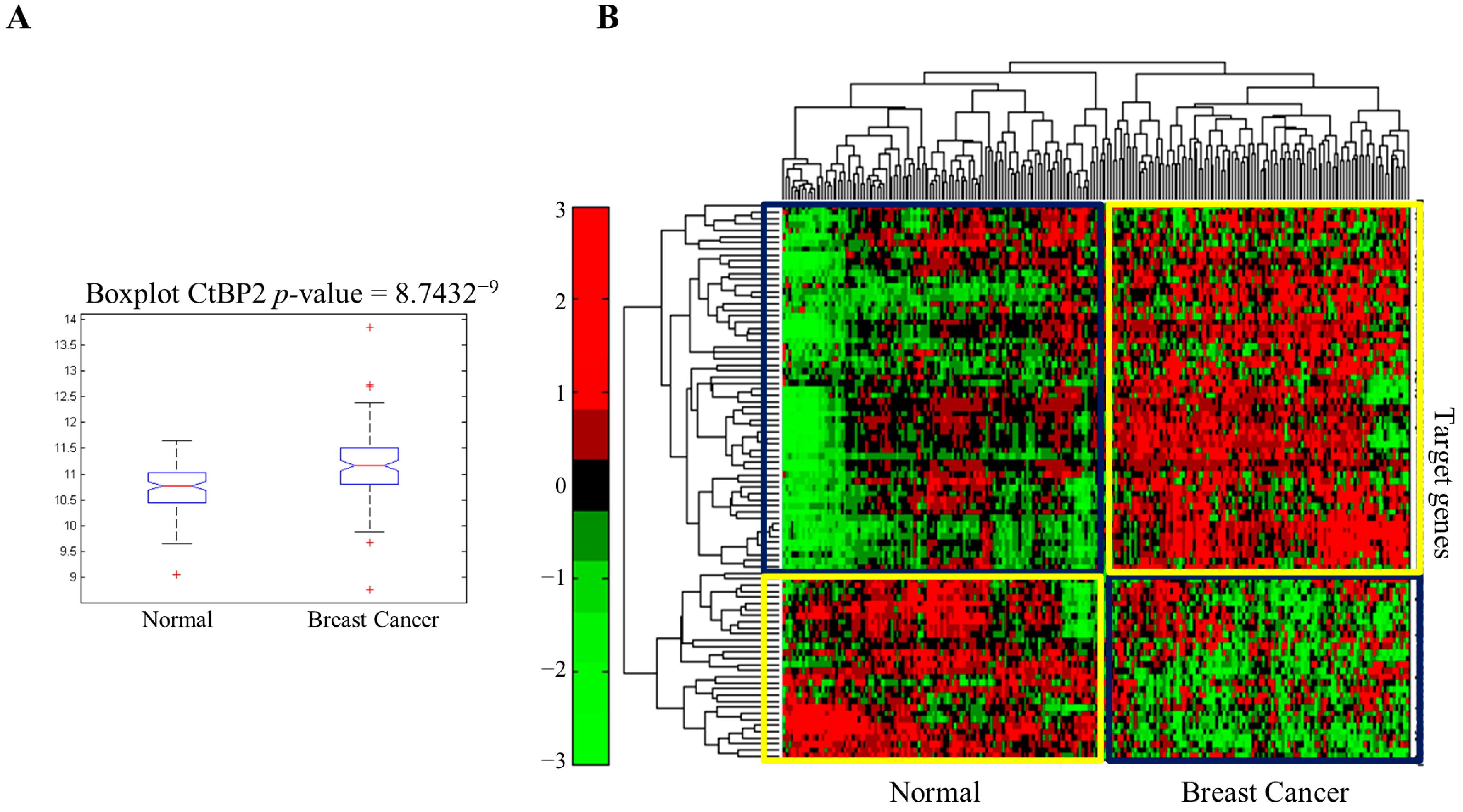
3.1. CtBP and Metabolic Rewiring of MDA-MB231 Triple-Negative Breast Tumor Cells
3.2. Metabolomics and Transcriptomics Integration Show Metabolism/Epigenetics Networking
3.3. Low Molecular Weight CtBP2 Inhibitors Validate the Power of Metabolic Rewiring
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Frezza, C. Metabolism and cancer: The future is now. Br. J. Cancer 2020, 122, 133–135. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, J. Systems Biology of Metabolism: A Driver for Developing Personalized and Precision Medicine. Cell Metab. 2017, 25, 572–579. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Alberghina, L.; Gaglio, D. Redox control of glutamine utilization in cancer. Cell Death Dis. 2014, 5, e1561. [Google Scholar] [CrossRef] [Green Version]
- Gaglio, D.; Bonanomi, M.; Valtorta, S.; Bharat, R.; Ripamonti, M.; Conte, F.; Fiscon, G.; Righi, N.; Napodano, E.; Papa, F.; et al. Disruption of redox homeostasis for combinatorial drug efficacy in K-Ras tumors as revealed by metabolic connectivity profiling. Cancer Metab. 2020, 8, 1–15. [Google Scholar] [CrossRef]
- Gaglio, D.; Valtorta, S.; Ripamonti, M.; Bonanomi, M.; Damiani, C.; Todde, S.; Negri, A.S.; Sanvito, F.; Mastroianni, F.; Di Campli, A.; et al. Divergent in vitro/in vivo responses to drug treatments of highly aggressive NIH-Ras cancer cells: A PET imaging and metabolomics-mass-spectrometry study. Oncotarget 2016, 7, 52017–52031. [Google Scholar] [CrossRef] [Green Version]
- Bott, A.; Peng, I.-C.; Fan, Y.; Faubert, B.; Zhao, L.; Li, J.; Neidler, S.; Sun, Y.; Jaber, N.; Krokowski, D.; et al. Oncogenic Myc Induces Expression of Glutamine Synthetase through Promoter Demethylation. Cell Metab. 2015, 22, 1068–1077. [Google Scholar] [CrossRef] [Green Version]
- Dejure, F.R.; Eilers, M. MYC and tumor metabolism: Chicken and egg. EMBO J. 2017, 36, 3409–3420. [Google Scholar] [CrossRef]
- Yuneva, M.; Zamboni, N.; Oefner, P.; Sachidanandam, R.; Lazebnik, Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 2007, 178, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Damiani, C.; Colombo, R.; Gaglio, D.; Mastroianni, F.; Pescini, D.; Westerhoff, H.V.; Mauri, G.; Vanoni, M.; Alberghina, L. A metabolic core model elucidates how enhanced utilization of glucose and glutamine, with enhanced glutamine-dependent lactate production, promotes cancer cell growth: The WarburQ effect. PLoS Comput. Biol. 2017, 13, e1005758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaglio, D.; Metallo, C.M.; A Gameiro, P.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 2011, 7, 523. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.S.; Manda, G. Metabolic Pathways of the Warburg Effect in Health and Disease: Perspectives of Choice, Chain or Chance. Int. J. Mol. Sci. 2017, 18, 2755. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, V.; He, J. Reactive Oxygen Species, Metabolic Plasticity, and Drug Resistance in Cancer. Int. J. Mol. Sci. 2020, 21, 3412. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnadurai, G. Transcriptional regulation by C-terminal binding proteins. Int. J. Biochem. Cell Biol. 2007, 39, 1593–1607. [Google Scholar] [CrossRef]
- Shi, Y.; Sawada, J.-I.; Sui, G.; Affar, E.B.; Whetstine, J.R.; Lan, F.; Ogawa, H.; Luke, M.P.-S.; Nakatani, Y.; Shi, Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nat. Cell Biol. 2003, 422, 735–738. [Google Scholar] [CrossRef]
- Subramanian, T.; Chinnadurai, G. Association of class I histone deacetylases with transcriptional corepressor CtBP. FEBS Lett. 2003, 540, 255–258. [Google Scholar] [CrossRef] [Green Version]
- Byun, J.S.; Gardner, K. C-Terminal Binding Protein: A Molecular Link between Metabolic Imbalance and Epigenetic Regulation in Breast Cancer. Int. J. Cell Biol. 2013, 2013, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Fjeld, C.C.; Birdsong, W.; Goodman, R.H. Differential binding of NAD+ and NADH allows the transcriptional corepressor carboxyl-terminal binding protein to serve as a metabolic sensor. Proc. Natl. Acad. Sci. 2003, 100, 9202–9207. [Google Scholar] [CrossRef] [Green Version]
- Dcona, M.; Morris, B.L.; Ellis, K.C.; Grossman, S.R. CtBP- an emerging oncogene and novel small molecule drug target: Advances in the understanding of its oncogenic action and identification of therapeutic inhibitors. Cancer Biol. Ther. 2017, 18, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Chinnadurai, G. The Transcriptional Corepressor CtBP: A Foe of Multiple Tumor Suppressors: Figure 1. Cancer Res. 2009, 69, 731–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, L.-J.; Byun, J.S.; Wong, M.M.; Wakano, C.; Taylor, T.; Bilke, S.; Baek, S.; Hunter, K.; Yang, H.; Lee, M.; et al. Genome-wide profiles of CtBP link metabolism with genome stability and epithelial reprogramming in breast cancer. Nat. Commun. 2013, 4, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Li, J.-J.; Guo, L.-Y.; Li, P.; Zhao, Z.; Zhou, H.; Di, L.-J. Molecular link between glucose and glutamine consumption in cancer cells mediated by CtBP and SIRT4. Oncog. 2018, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Bravatà, V.; Cammarata, F.P.; Minafra, L.; Musso, R.; Pucci, G.; Spada, M.; Fazio, I.; Russo, G.; Forte, G.I. Gene Expression Profiles Induced by High-dose Ionizing Radiation in MDA-MB-231 Triple-negative Breast Cancer Cell Line. Cancer Genom.-Proteom. 2019, 16, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Bravatà, V.; Cammarata, F.P.; Minafra, L.; Pisciotta, P.; Scazzone, C.; Manti, L.; Savoca, G.; Petringa, G.; Cirrone, G.A.P.; Cuttone, G.; et al. Proton-irradiated breast cells: Molecular points of view. J. Radiat. Res. 2019, 60, 451–465. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Klipper-Aurbach, Y.; Wasserman, M.; Braunspiegel-Weintrob, N.; Borstein, D.; Peleg, S.; Assa, S.; Karp, M.; Benjamini, Y.; Hochberg, Y.; Laron, Z. Mathematical formulae for the prediction of the residual beta cell function during the first two years of disease in children and adolescents with insulin-dependent diabetes mellitus. Med. Hypotheses 1995, 45, 486–490. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Santos, A.; von Mering, C.; Jensen, L.J.; Bork, P.; Kuhn, M. STITCH 5: Augmenting protein–chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016, 44, D380–D384. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Korwar, S.; Morris, B.L.; Parikh, H.; Coover, R.A.; Doughty, T.W.; Love, I.M.; Hilbert, B.J.; Royer, W.E.; Kellogg, G.; Grossman, S.R.; et al. Design, synthesis, and biological evaluation of substrate-competitive inhibitors of C-terminal Binding Protein (CtBP). Bioorg. Med. Chem. 2016, 24, 2707–2715. [Google Scholar] [CrossRef] [Green Version]
- Knapp, S.; Amorelli, B.; Darout, E.; Ventocilla, C.C.; Goldman, L.M.; Huhn, R.A.; Minnihan, E.C. A Family of Mycothiol Analogues. J. Carbohydr. Chem. 2005, 24, 103–130. [Google Scholar] [CrossRef]
- Hilbert, B.J.; Morris, B.L.; Ellis, K.C.; Paulsen, J.; Schiffer, C.A.; Grossman, S.R.; Royer, W.E. Structure-Guided Design of a High Affinity Inhibitor to Human CtBP. ACS Chem. Biol. 2015, 10, 1118–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilbert, B.J.; Grossman, S.R.; Schiffer, C.A.; Royer, W.E. Crystal structures of human CtBP in complex with substrate MTOB reveal active site features useful for inhibitor design. FEBS Lett. 2014, 588, 1743–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straza, M.W.; Paliwal, S.; Kovi, R.C.; Rajeshkumar, B.; Trenh, P.; Parker, D.; Whalen, G.F.; Lyle, S.; Schiffer, C.A.; Grossman, S.R. Therapeutic targeting of C-terminal binding protein in human cancer. Cell Cycle 2010, 9, 3764–3774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.-J.; Lin, M.-W.; Yen, C.-H.; Lin, Y.-T.; Wang, C.-K.; Huang, S.-F.; Chen, K.-H.; Yang, C.-P.; Chen, T.-L.; Hou, M.-F.; et al. Characterization of Niemann-Pick Type C2 Protein Expression in Multiple Cancers Using a Novel NPC2 Monoclonal Antibody. PLOS ONE 2013, 8, e77586. [Google Scholar] [CrossRef]
- Hemler, M.E. Tetraspanin proteins promote multiple cancer stages. Nat. Rev. Cancer 2014, 14, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Mikhailov, A.T.; Torrado, M. The enigmatic role of the ankyrin repeat domain 1 gene in heart development and disease. Int. J. Dev. Biol. 2008, 52, 811–821. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, M.; Yamashita, K.; Waraya, M.; Minatani, N.; Ushiku, H.; Kojo, K.; Ema, A.; Kosaka, Y.; Katoh, H.; Sengoku, N.; et al. Epigenetic regulation of ZEB1-RAB25/ESRP1 axis plays a critical role in phenylbutyrate treatment-resistant breast cancer. Oncotarget 2015, 7, 1741–1753. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, D.; Bhargava, D.K.; Dixit, A.; Sahoo, B.; Biswas, S.K.; Biswas, G.; Mishra, S.K. ERRβ signalling through FST and BCAS2 inhibits cellular proliferation in breast cancer cells. Br. J. Cancer 2014, 110, 2144–2158. [Google Scholar] [CrossRef] [Green Version]
- Bouin, A.-P.; Kyurmurkov, A.; Régent-Kloeckner, M.; Ribba, A.-S.; Faurobert, E.; Fournier, H.-N.; Bourrin-Reynard, I.; Manet-Dupé, S.; Oddou, C.; Balland, M.; et al. ICAP-1 monoubiquitination coordinates matrix density and rigidity sensing for cell migration through ROCK2- MRCKα balance. J. Cell Sci. 2017, 130, 626–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.K.; Gopalan, G. Aurora-A kinase interacting protein 1 (AURKAIP1) promotes Aurora-A degradation through an alternative ubiquitin-independent pathway. Biochem. J. 2007, 403, 119–127. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Xiang, H.; Zong, X.; Yan, X.; Yu, Y.; Liu, G.; Zou, D.; Yang, H. CDK2-AP1 inhibits growth of breast cancer cells by regulating cell cycle and increasing docetaxel sensitivity in vivo and in vitro. Cancer Cell Int. 2014, 14, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucharaba, A.; Serre, C.-M.; Guglielmi, J.; Bordet, J.-C.; Clezardin, P.; Peyruchaud, O. The type 1 lysophosphatidic acid receptor is a target for therapy in bone metastases. Proc. Natl. Acad. Sci. 2006, 103, 9643–9648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.W.; Herr, D.R.; Noguchi, K.; Yung, Y.C.; Lee, C.-W.; Mutoh, T. LPA Receptors: Subtypes and Biological Actions. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 157–186. [Google Scholar] [CrossRef] [Green Version]
- Sahay, D.; Leblanc, R.; Grunewald, T.G.P.; Ambatipudi, S.; Ribeiro, J.; Clézardin, P.; Peyruchaud, O. The LPA1/ZEB1/miR-21-activation pathway regulates metastasis in basal breast cancer. Oncotarget 2015, 6, 20604–20620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scalise, M.; Galluccio, M.; Console, L.; Pochini, L.; Indiveri, C. The Human SLC7A5 (LAT1): The Intriguing Histidine/Large Neutral Amino Acid Transporter and Its Relevance to Human Health. Front. Chem. 2018, 6, 243. [Google Scholar] [CrossRef]
- Fuchs, B.C.; Bode, B.P. Amino acid transporters ASCT2 and LAT1 in cancer: Partners in crime? Semin. Cancer Biol. 2005, 15, 254–266. [Google Scholar] [CrossRef]
- Jose, C.; Bellance, N.; Rossignol, R. Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochim. Biochim. et Biophys. Acta (BBA)-Bioenerg. 2011, 1807, 552–561. [Google Scholar] [CrossRef] [Green Version]
- Al-Masri, M.; Paliotti, K.; Tran, R.; Halaoui, R.; Lelarge, V.; Chatterjee, S.; Wang, L.-T.; Moraes, C.; McCaffrey, L. Architectural control of metabolic plasticity in epithelial cancer cells. Commun. Biol. 2021, 4, 1–16. [Google Scholar] [CrossRef]
- Damiani, C.; Gaglio, D.; Sacco, E.; Alberghina, L.; Vanoni, M. Systems metabolomics: From metabolomic snapshots to design principles. Curr. Opin. Biotechnol. 2020, 63, 190–199. [Google Scholar] [CrossRef]
- Terunuma, A.; Putluri, N.; Mishra, P.; Mathe, E.A.; Dorsey, T.H.; Yi, M.; Wallace, T.A.; Issaq, H.J.; Zhou, M.; Killian, J.K.; et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J. Clin. Investig. 2014, 124, 398–412. [Google Scholar] [CrossRef] [Green Version]
- Keating, S.; El-Osta, A. Epigenetics and Metabolism. Circ. Res. 2015, 116, 715–736. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Zhang, H.; Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 2021, 1–43. [Google Scholar] [CrossRef]
- Xiao, Z.; Locasale, J.W. Epigenomic links from metabolism—Methionine and chromatin architecture. Curr. Opin. Chem. Biol. 2021, 63, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Montellier, E.; Gaucher, J. Targeting the interplay between metabolism and epigenetics in cancer. Curr. Opin. Oncol. 2019, 31, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Desbats, M.A.; Giacomini, I.; Prayer-Galetti, T.; Montopoli, M. Metabolic Plasticity in Chemotherapy Resistance. Front. Oncol. 2020, 10, 281. [Google Scholar] [CrossRef]
- Chen, Z. The transrepression and transactivation roles of CtBPs in the pathogenesis of different diseases. J. Mol. Med. 2021, 1–13. [Google Scholar] [CrossRef]
- Birts, C.N.; Harding, R.; Soosaipillai, G.; Halder, T.; Azim-Araghi, A.; Darley, M.; Cutress, R.I.; Bateman, A.C.; Blaydes, J.P. Expression of CtBP family protein isoforms in breast cancer and their role in chemoresistance. Biol. Cell 2011, 103, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonanomi, M.; Salmistraro, N.; Fiscon, G.; Conte, F.; Paci, P.; Bravatà, V.; Forte, G.I.; Volpari, T.; Scorza, M.; Mastroianni, F.; et al. Transcriptomics and Metabolomics Integration Reveals Redox-Dependent Metabolic Rewiring in Breast Cancer Cells. Cancers 2021, 13, 5058. https://doi.org/10.3390/cancers13205058
Bonanomi M, Salmistraro N, Fiscon G, Conte F, Paci P, Bravatà V, Forte GI, Volpari T, Scorza M, Mastroianni F, et al. Transcriptomics and Metabolomics Integration Reveals Redox-Dependent Metabolic Rewiring in Breast Cancer Cells. Cancers. 2021; 13(20):5058. https://doi.org/10.3390/cancers13205058
Chicago/Turabian StyleBonanomi, Marcella, Noemi Salmistraro, Giulia Fiscon, Federica Conte, Paola Paci, Valentina Bravatà, Giusi Irma Forte, Tatiana Volpari, Manuela Scorza, Fabrizia Mastroianni, and et al. 2021. "Transcriptomics and Metabolomics Integration Reveals Redox-Dependent Metabolic Rewiring in Breast Cancer Cells" Cancers 13, no. 20: 5058. https://doi.org/10.3390/cancers13205058