
Progress on Ras/MAPK Signaling Research and Targeting in Blood and Solid Cancers


Abstract
:Simple Summary
Abstract
1. Introduction
2. Activation of Ras and Raf Proteins
2.1. Ras Activation by the Tyrosine Kinase, Interleukin (IL) Receptors, and G-Protein Coupled Receptors
2.2. Activation and Regulation of Raf Protein
2.3. Ras-Raf-MEK-ERK Pathway Interaction with p53
3. Ras-Raf Pathway Mutations
3.1. Ras Mutations
3.2. Raf Mutations
3.3. MEK and ERK Mutations
4. Ras-Raf Signaling in Various Types of Cancers
4.1. Leukemia/Lymphoma
4.2. Solid Tumors
5. Targeting of Ras-Raf Signaling in Cancers
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MAPK | Mitogen-activated protein kinase |
| ATLL | Adult T-cell leukemia/lymphoma |
| HTLV-1 | Human T-cell lymphotropic virus type 1 |
| ERK | Extracellular signal-regulated kinase |
| JNK | c-Jun N-terminal kinase |
| EBV | Epstein–Barr virus |
| HBV | Hepatitis B virus |
| HCV | Hepatitis C virus |
| HPV | Human papillomaviruses |
| GDP | Guanosine diphosphate |
| GTP | Guanosine triphosphate |
| Ras-GRF | Ras protein-specific guanine nucleotide releasing factor 1 |
| FTase | Farnesyl transferase |
| GGTase1 | Geranylgeranyl transferase 1 |
| Rce1 | Ras converting enzyme |
| Icmt | Isoprenylcysteine carboxyl methyltransferase |
| PDEδ | Phosphodiesterase delta |
| GPCR | G-protein-coupled receptors |
| IL | Interleukin |
| JAK2 | Janus kinase 2 |
| SH2 | Src homology 2 domain |
| Grb2 | Growth factor receptor bound protein 2 |
| SOS | Son of sevenless |
| GEF | Guanine nucleotide exchange factor |
| Gal-1 | Galectin-1 |
| EGFR | Epidermal growth factor receptor |
| PKA | Protein kinase A |
| PLCβ | Phospholipase C-β |
| RBD | Ras-binding domain |
| PAK | p21-activated protein kinase |
| PKC | Protein kinase C |
| BL | Burkitt’s lymphoma |
| NHL | non-Hodgkin’s lymphoma |
| HCC | Hepatocellular carcinoma |
| NSCLC | Non-small cell lung cancer |
| PAC | Pancreatic adenocarcinoma |
| CRC | Colorectal cancer |
| Shrp2 | Src homology region 2 domain-containing phosphatase 2 |
| Csk | C-terminal Src kinase |
| SFKs | Src family kinases (SFKs) |
| Epac-1 | Exchange protein directly activated by cAMP |
| PLCγ | Phospholipase C-γ |
| PAG | Phosphoprotein associated with glycosphingolipid-enriched microdomains |
| PP1 | Protein phosphatase 1 |
References
- Marshall, C.J. MAP kinase kinase kinase, MAP kinase kinase and MAP kinase. Curr. Opin. Genet. Dev. 1994, 4, 82–89. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, W.-Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.-F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. 2015, 35, 600–604. [Google Scholar] [CrossRef]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Mor, A.; Philips, M.R. Compartmentalized ras/mapk signaling. Annu. Rev. Immunol. 2006, 24, 771–800. [Google Scholar] [CrossRef]
- Weiss, R.A. A perspective on the early days of RAS research. Cancer Metastasis Rev. 2020, 39, 1023–1028. [Google Scholar] [CrossRef]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Noh, S.J.; Zhou, G.; Dixon, J.E.; Guan, K.-L. Selective Activation of MEK1 but Not MEK2 by A-Raf from Epidermal Growth Factor-stimulated Hela Cells. J. Biol. Chem. 1996, 271, 3265–3271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marais, R.; Light, Y.; Paterson, H.F.; Mason, C.S.; Marshall, C.J. Differential Regulation of Raf-1, A-Raf, and B-Raf by Oncogenic Ras and Tyrosine Kinases. J. Biol. Chem. 1997, 272, 4378–4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibney, G.T.; Messina, J.L.; Fedorenko, I.V.; Sondak, V.K.; Smalley, K.S.M. Paradoxical oncogenesis—The long-term effects of BRAF inhibition in melanoma. Nat. Rev. Clin. Oncol. 2013, 10, 390–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coles, L.C.; Shaw, P.E. PAK1 primes MEK1 for phosphorylation by Raf-1 kinase during cross-cascade activation of the ERK pathway. Oncogene 2002, 21, 2236–2244. [Google Scholar] [CrossRef] [Green Version]
- Payne, D.M.; Rossomando, A.J.; Martino, P.; Erickson, A.K.; Her, J.H.; Shabanowitz, J.; Hunt, D.F.; Weber, M.J.; Sturgill, T.W. Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase). EMBO J. 1991, 10, 885–892. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ebisuya, M.; Ashida, F.; Okamoto, K.; Yonehara, S.; Nishida, E. Continuous ERK Activation Downregulates Antiproliferative Genes throughout G1 Phase to Allow Cell-Cycle Progression. Curr. Biol. 2006, 16, 1171–1182. [Google Scholar] [CrossRef] [Green Version]
- Alao, J.P. The regulation of cyclin D1 degradation: Roles in cancer development and the potential for therapeutic invention. Mol. Cancer 2007, 6, 24–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sewing, A.; Wiseman, B.; Lloyd, A.C.; Land, H. High-intensity Raf signal causes cell cycle arrest mediated by p21Cip. Mol. Cell. Biol. 1997, 17, 5588–5597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, D.; Parry, D.; Cherwinski, H.; Bosch, E.; Lees, E.; McMahon, M. Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip. Mol. Cell. Biol. 1997, 17, 5598–5611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, A.M.; Gysin, S.; Malek, N.; Nakayama, K.-I.; Roberts, J.M.; McMahon, M. Cooperative Regulation of the Cell Division Cycle by the Protein Kinases RAF and AKT. Mol. Cell. Biol. 2004, 24, 10868–10881. [Google Scholar] [CrossRef] [Green Version]
- Bertics, P.J.; Gill, G.N. Self-phosphorylation enhances the protein-tyrosine kinase activity of the epidermal growth factor receptor. J. Biol. Chem. 1985, 260, 14642–14647. [Google Scholar] [CrossRef]
- Batzer, A.G.; Rotin, D.; Ureña, J.M.; Skolnik, E.Y.; Schlessinger, J. Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol. Cell. Biol. 1994, 14, 5192–5201. [Google Scholar] [CrossRef] [Green Version]
- Margolis, B.; Skolnik, E.Y. Activation of Ras by receptor tyrosine kinases. J. Am. Soc. Nephrol. 1994, 5, 1288–1299. [Google Scholar] [CrossRef]
- Blaževitš, O.; Mideksa, Y.G.; Šolman, M.; Ligabue, A.; Ariotti, N.; Nakhaeizadeh, H.; Fansa, E.K.; Papageorgiou, A.; Wittinghofer, A.; Ahmadian, M.R.; et al. Galectin-1 dimers can scaffold Raf-effectors to increase H-ras nanoclustering. Sci. Rep. 2016, 6, 24165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigil, D.; Cherfils, J.; Rossman, K.L.; Der, C.J. Ras superfamily GEFs and GAPs: Validated and tractable targets for cancer therapy? Nat. Rev. Cancer 2010, 10, 842–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoie, H.; Therrien, M. Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol. 2015, 16, 281–298. [Google Scholar] [CrossRef]
- James, B.P.; Bunch, T.A.; Krishnamoorthy, S.; Perkins, L.A.; Brower, D.L. Nuclear localization of the ERK MAP kinase mediated by Drosophila αPS2βPS integrin and importin-7. Mol. Biol. Cell 2007, 18, 4190–4199. [Google Scholar] [CrossRef] [Green Version]
- Lorenzen, J.; Baker, S.; Denhez, F.; Melnick, M.; Brower, D.; Perkins, L. Nuclear import of activated D-ERK by DIM-7, an importin family member encoded by the gene moleskin. Development 2001, 128, 1403–1414. [Google Scholar] [CrossRef]
- Chang, F.; Steelman, L.S.; Lee, J.T.; Shelton, J.G.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.A.; McCubrey, J. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: Potential targeting for therapeutic intervention. Leukemia 2003, 17, 1263–1293. [Google Scholar] [CrossRef]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human Viral Oncogenesis: A Cancer Hallmarks Analysis. Cell Host Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef] [Green Version]
- Olagnier, D.; Sze, A.; Hadj, S.B.; Chiang, C.; Steel, C.; Han, X.; Routy, J.-P.; Lin, R.; Hiscott, J.; Van Grevenynghe, J. HTLV-1 Tax-Mediated Inhibition of FOXO3a Activity Is Critical for the Persistence of Terminally Differentiated CD4+ T Cells. PLOS Pathog. 2014, 10, e1004575. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Longnecker, R. Latent membrane protein 2A inhibits transforming growth factor-beta 1-induced apoptosis through the phosphatidylinositol 3-kinase/Akt pathway. J. Virol. 2004, 78, 1697–1705. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wu, J.; Ling, M.T.; Zhao, L.; Zhao, K.-N. The role of the PI3K/Akt/mTOR signalling pathway in human cancers induced by infection with human papillomaviruses. Mol. Cancer 2015, 14, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, M.; Jain, P.; Tsukasaki, K.; Bangham, C. HTLV-1 Infection and Its Associated Diseases. Leuk. Res. Treat. 2012, 2012, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.; Khan, Z.K.; Wigdahl, B.; Jennings, S.R.; Tangy, F.; Jain, P. Murine FLT3 Ligand-Derived Dendritic Cell-Mediated Early Immune Responses Are Critical to Controlling Cell-Free Human T Cell Leukemia Virus Type 1 Infection. J. Immunol. 2010, 186, 390–402. [Google Scholar] [CrossRef] [Green Version]
- Mostoller, K.; Norbury, C.C.; Jain, P.; Wigdahl, B. Human T-cell leukemia virus type I Tax induces the expression of dendritic cell markers associated with maturation and activation. J. Neurovirol. 2004, 10, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Lavorgna, A.; Sehgal, M.; Gao, L.; Ginwala, R.; Sagar, D.; Harhaj, E.W.; Khan, Z.K. Myocyte enhancer factor (MEF)-2 plays essential roles in T-cell transformation associated with HTLV-1 infection by stabilizing complex between Tax and CREB. Retrovirology 2015, 12, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Jain, P.; Manuel, S.L.; Khan, Z.K.; Ahuja, J.; Quann, K.; Wigdahl, B. DC-SIGN Mediates Cell-Free Infection and Transmission of Human T-Cell Lymphotropic Virus Type 1 by Dendritic Cells. J. Virol. 2009, 83, 10908–10921. [Google Scholar] [CrossRef] [Green Version]
- Jain, P.; Ahuja, J.; Khan, Z.K.; Shimizu, S.; Meucci, O.; Jennings, S.R.; Wigdahl, B. Modulation of dendritic cell maturation and function by the Tax protein of human T cell leukemia virus type. J. Leukoc. Biol. 2007, 82, 44–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginwala, R.; Caruso, B.; Khan, Z.K.; Pattekar, A.; Chew, G.M.; Corley, M.J.; Loonawat, R.; Jacobson, S.; Sreedhar, S.; Ndhlovu, L.C.; et al. HTLV-1 Infection and Neuropathogenesis in the Context of Rag1(-/-)gammac(-/-) (RAG1-Hu) and BLT Mice. J. Neuroimmune Pharmacol. 2017, 12, 504–520. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589. [Google Scholar] [CrossRef] [Green Version]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Adachi, Y.; Ito, K.; Hayashi, Y.; Kimura, R.; Tan, T.Z.; Yamaguchi, R.; Ebi, H. Epithelial-to-Mesenchymal Transition is a Cause of Both Intrinsic and Acquired Resistance to KRAS G12C Inhibitor in KRAS G12C–Mutant Non–Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 5962–5973. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1268. [Google Scholar] [CrossRef] [Green Version]
- Brock, E.J.; Ji, K.; Reiners, J.J.; Mattingly, R.R. How to Target Activated Ras Proteins: Direct Inhibition vs. Induced Mislocalization. Mini-Rev. Med. Chem. 2016, 16, 358–369. [Google Scholar] [CrossRef]
- Wang, J.; Yao, X.; Huang, J. New tricks for human farnesyltransferase inhibitor: Cancer and beyond. MedChemComm 2017, 8, 841–854. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porru, M.; Pompili, L.; Caruso, C.; Biroccio, A.; Leonetti, C. Targeting KRAS in metastatic colorectal cancer: Current strategies and emerging opportunities. J. Exp. Clin. Cancer Res. 2018, 37, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dharmaiah, S.; Bindu, L.; Tran, T.H.; Gillette, W.K.; Frank, P.H.; Ghirlando, R.; Nissley, D.V.; Esposito, D.; McCormick, F.; Stephen, A.G.; et al. Structural basis of recognition of farnesylated and methylated KRAS4b by PDEdelta. Proc. Natl. Acad. Sci. USA 2016, 113, E6766–E6775. [Google Scholar] [CrossRef] [Green Version]
- Choy, E.; Chiu, V.K.; Silletti, J.; Feoktistov, M.; Morimoto, T.; Michaelson, D.; Ivanov, I.E.; Philips, M.R. Endomembrane Trafficking of Ras: The CAAX Motif Targets Proteins to the ER and Golgi. Cell 1999, 98, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Deschenes, R.J. Plasma Membrane Localization of Ras Requires Class C Vps Proteins and Functional Mitochondria in Saccharomyces cerevisiae. Mol. Cell. Biol. 2006, 26, 3243–3255. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.D.; Der, C.J.; Philips, M.R. Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery? Clin. Cancer Res. 2015, 21, 1819–1827. [Google Scholar] [CrossRef] [Green Version]
- Dougan, M.; Dranoff, G.; Dougan, S.K. GM-CSF, IL-3, and IL-5 Family of Cytokines: Regulators of Inflammation. Immunity 2019, 50, 796–811. [Google Scholar] [CrossRef]
- Broughton, S.; Dhagat, U.; Hercus, T.; Nero, T.; Grimbaldeston, M.A.; Bonder, C.S.; Lopez, A.F.; Parker, M.W. The GM-CSF/IL-3/IL-5 cytokine receptor family: From ligand recognition to initiation of signaling. Immunol. Rev. 2012, 250, 277–302. [Google Scholar] [CrossRef]
- Reddy, E.P.; Korapati, A.; Chaturvedi, P.; Rane, S. IL-3 signaling and the role of Src kinases, JAKs and STATs: A covert liaison unveiled. Oncogene 2000, 19, 2532–2547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.; Prignent, S. Insights into the Shc Family of Adaptor Proteins. J. Mol. Signal. 2017, 12, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, A.C.; Monkhouse, J.L.; Hamilton, J.A.; Csar, X.F. Direct binding of Shc, Grb2, SHP-2 and p40 to the murine granulocyte colony-stimulating factor receptor. Biochim. Biophys. Acta Bioenerg. 1998, 1448, 70–76. [Google Scholar] [CrossRef] [Green Version]
- Kerr, D.L.; Haderk, F.; Bivona, T.G. Allosteric SHP2 inhibitors in cancer: Targeting the intersection of RAS, resistance, and the immune microenvironment. Curr. Opin. Chem. Biol. 2021, 62, 1–12. [Google Scholar] [CrossRef]
- Hanafusa, H.; Torii, S.; Yasunaga, T.; Nishida, E. Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat. Cell Biol. 2002, 4, 850–858. [Google Scholar] [CrossRef]
- Jarvis, L.A.; Toering, S.J.; Simon, M.A.; Krasnow, M.A.; Smith-Bolton, R. Sprouty proteins are in vivo targets of Corkscrew/SHP-2 tyrosine phosphatases. Development 2006, 133, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- Montagner, A.; Yart, A.; Dance, M.; Perret, B.; Salles, J.-P.; Raynal, P. A Novel Role for Gab1 and SHP2 in Epidermal Growth Factor-induced Ras Activation. J. Biol. Chem. 2005, 280, 5350–5360. [Google Scholar] [CrossRef] [Green Version]
- Agazie, Y.M.; Hayman, M.J. Molecular Mechanism for a Role of SHP2 in Epidermal Growth Factor Receptor Signaling. Mol. Cell. Biol. 2003, 23, 7875–7886. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. Src protein–tyrosine kinase structure and regulation. Biochem. Biophys. Res. Commun. 2004, 324, 1155–1164. [Google Scholar] [CrossRef]
- Song, Y.; Zhao, M.; Zhang, H.; Yu, B. Double-edged roles of protein tyrosine phosphatase SHP2 in cancer and its inhibitors in clinical trials. Pharmacol. Ther. 2021, 107966. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.Q.; Feng, X.; Zhou, W.; Knyazev, P.G.; Ullrich, A.; Chen, Z. Csk-binding protein (Cbp) negatively regulates epidermal growth factor-induced cell transformation by controlling Src activation. Oncogene 2006, 25, 5495–5506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hrdinka, M.; Horejsi, V. PAG—A multipurpose transmembrane adaptor protein. Oncogene 2013, 33, 4881–4892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bivona, T.G.; De Castro, I.P.; Ahearn, I.M.; Grana, T.M.; Chiu, V.K.; Lockyer, P.J.; Cullen, P.J.; Pellicer, A.; Cox, A.D.; Philips, M.R. Phospholipase Cgamma activates Ras on the Golgi apparatus by means of RasGRP1. Nature 2003, 424, 694–698. [Google Scholar] [CrossRef]
- McKay, M.M.; Morrison, D.K. Integrating signals from RTKs to ERK/MAPK. Oncogene 2007, 26, 3113–3121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Medarde, A.; Santos, E. The RasGrf family of mammalian guanine nucleotide exchange factors. Biochim. Biophys. Acta Bioenerg. 2011, 1815, 170–188. [Google Scholar] [CrossRef]
- Le, D.T.; Shannon, K.M. Ras processing as a therapeutic target in hematologic malignancies. Curr. Opin. Hematol. 2002, 9, 308–315. [Google Scholar] [CrossRef] [PubMed]
- De Mora, J.F.; Esteban, L.M.; Burks, D.J.; Núñez, A.; Garcés, C.; García-Barrado, M.J.; Iglesias-Osma, M.C.; Moratinos, J.; Ward, J.M.; Santos, E. Ras-GRF1 signaling is required for normal beta-cell development and glucose homeostasis. EMBO J. 2003, 22, 3039–3049. [Google Scholar] [CrossRef]
- Ruiz, S.; Santos, E.; Bustelo, X.R. RasGRF2, a Guanosine Nucleotide Exchange Factor for Ras GTPases, Participates in T-Cell Signaling Responses. Mol. Cell. Biol. 2007, 27, 8127–8142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, J.C. Regulation and Function of the RasGRP Family of Ras Activators in Blood Cells. Genes Cancer 2011, 2, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Pan, C.; He, S.-M.; Lang, B.; Gao, G.-D.; Wang, X.-L.; Wang, Y. The Ras Superfamily of Small GTPases in Non-neoplastic Cerebral Diseases. Front. Mol. Neurosci. 2019, 12, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gideon, P.; John, J.; Frech, M.; Lautwein, A.; Clark, R.; Scheffler, J.E.; Wittinghofer, A. Mutational and kinetic analyses of the GTPase-activating protein (GAP)-p21 interaction: The C-terminal domain of GAP is not sufficient for full activity. Mol. Cell. Biol. 1992, 12, 2050–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudack, T.; Xia, F.; Schlitter, J.; Kötting, C.; Gerwert, K. Ras and GTPase-activating protein (GAP) drive GTP into a precatalytic state as revealed by combining FTIR and biomolecular simulations. Proc. Natl. Acad. Sci. USA 2012, 109, 15295–15300. [Google Scholar] [CrossRef] [Green Version]
- Maertens, O.; Cichowski, K. An expanding role for RAS GTPase activating proteins (RAS GAPs) in cancer. Adv. Biol. Regul. 2014, 55, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Trahey, M.; McCormick, F. A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science 1987, 238, 542–545. [Google Scholar] [CrossRef]
- Patil, S.; Chamberlain, R.S. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist 2012, 17, 101–116. [Google Scholar] [CrossRef] [Green Version]
- McGillicuddy, L.T.; Fromm, J.A.; Hollstein, P.E.; Kubek, S.; Beroukhim, R.; De Raedt, T.; Johnson, B.W.; Williams, S.M.; Nghiemphu, P.; Liau, L.M.; et al. Proteasomal and Genetic Inactivation of the NF1 Tumor Suppressor in Gliomagenesis. Cancer Cell 2009, 16, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Dote, H.; Toyooka, S.; Tsukuda, K.; Yano, M.; Ota, T.; Murakami, M.; Naito, M.; Toyota, M.; Gazdar, A.F.; Shimizu, N. Aberrant promoter methylation in human DAB2 interactive protein (hDAB2IP) gene in gastrointestinal tumour. Br. J. Cancer 2005, 92, 1117–1125. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Tu, S.-W.; Hsieh, J.-T. Down-regulation of Human DAB2IP Gene Expression Mediated by Polycomb Ezh2 Complex and Histone Deacetylase in Prostate Cancer. J. Biol. Chem. 2005, 280, 22437–22444. [Google Scholar] [CrossRef] [Green Version]
- Yano, M.; Toyooka, S.; Tsukuda, K.; Dote, H.; Ouchida, M.; Hanabata, T.; Aoe, M.; Date, H.; Gazdar, A.F.; Shimizu, N. Aberrant promoter methylation of human DAB2 interactive protein (hDAB2IP) gene in lung cancers. Int. J. Cancer 2005, 113, 59–66. [Google Scholar] [CrossRef]
- Xie, D.; Gore, C.; Liu, J.; Pong, R.-C.; Mason, R.; Hao, G.; Long, M.; Kabbani, W.; Yu, L.; Zhang, H.; et al. Role of DAB2IP in modulating epithelial-to-mesenchymal transition and prostate cancer metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 2485–2490. [Google Scholar] [CrossRef] [Green Version]
- Min, J.; Zaslavsky, A.; Fedele, G.; McLaughlin, S.K.; Reczek, E.E.; De Raedt, T.; Guney, I.; Strochlic, D.E.; MacConaill, L.E.; Beroukhim, R.; et al. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat. Med. 2010, 16, 286–294. [Google Scholar] [CrossRef]
- Zhang, X.; Li, N.; Li, X.; Zhao, W.; Qiao, Y.; Liang, L.; Ding, Y. Low expression of DAB2IP contributes to malignant development and poor prognosis in hepatocellular car-cinoma. J. Gastroenterol. Hepatol. 2012, 27, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, L.; Yao, B.; Liu, Q.; Guo, C. miR-1307-3p promotes tumor growth and metastasis of hepatocellular carcinoma by repressing DAB2 inter-acting protein. Biomed. Pharmacother. 2019, 117, 109055. [Google Scholar] [CrossRef]
- Prior, I.; Muncke, C.; Parton, R.G.; Hancock, J.F. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J. Cell Biol. 2003, 160, 165–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henis, Y.; Hancock, J.; Prior, I.A. Ras acylation, compartmentalization and signaling nanoclusters (Review). Mol. Membr. Biol. 2009, 26, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotblat, B.; Prior, I.A.; Muncke, C.; Parton, R.G.; Kloog, Y.; Henis, Y.; Hancock, J.F. Three Separable Domains Regulate GTP-Dependent Association of H-ras with the Plasma Membrane. Mol. Cell. Biol. 2004, 24, 6799–6810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, I.A.; Harding, A.; Yan, J.; Sluimer, J.; Parton, R.G.; Hancock, J.F. GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat. Cell Biol. 2001, 3, 368–375. [Google Scholar] [CrossRef]
- Weise, K.; Triola, G.; Brunsveld, L.; Waldmann, H.; Winter, R. Influence of the Lipidation Motif on the Partitioning and Association of N-Ras in Model Membrane Subdomains. J. Am. Chem. Soc. 2009, 131, 1557–1564. [Google Scholar] [CrossRef]
- Tebar, F.; Enrich, C.; Rentero, C.; Grewal, T. GTPases Rac1 and Ras Signaling from Endosomes. Mol. Evol. Evid. Monophyly Metazoa 2018, 57, 65–105. [Google Scholar] [CrossRef]
- Cristea, S.; Sage, J. Is the Canonical RAF/MEK/ERK Signaling Pathway a Therapeutic Target in SCLC? J. Thorac. Oncol. 2016, 11, 1233–1241. [Google Scholar] [CrossRef] [Green Version]
- Goldsmith, Z.G.; Dhanasekaran, D.N. G Protein regulation of MAPK networks. Oncogene 2007, 26, 3122–3142. [Google Scholar] [CrossRef] [Green Version]
- Dubrovska, A.; Cojoc, M.; Peitzsch, C.; Trautmann, F.; Polishchuk, L.; Telegeev, G.D. Emerging targets in cancer management: Role of the CXCL12/CXCR4 axis. OncoTargets Ther. 2013, 6, 1347–1361. [Google Scholar] [CrossRef] [Green Version]
- Gesty-Palmer, D.; Chen, M.; Reiter, E.; Ahn, S.; Nelson, C.D.; Wang, S.; Eckhardt, A.E.; Cowan, C.L.; Spurney, R.F.; Luttrell, L.M.; et al. Distinct beta-arrestin- and G protein-dependent pathways for parathyroid hormone recep-tor-stimulated ERK1/2 activation. J. Biol. Chem. 2006, 281, 10856–10864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vossler, M.R.; Yao, H.; York, R.D.; Pan, M.-G.; Rim, C.S.; Stork, P.J. cAMP Activates MAP Kinase and Elk-1 through a B-Raf- and Rap1-Dependent Pathway. Cell 1997, 89, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Winitz, S.; Russell, M.; Qian, N.X.; Gardner, A.; Dwyer, L.; Johnson, G.L. Involvement of Ras and Raf in the Gi-coupled acetylcholine muscarinic m2 receptor activation of mito-gen-activated protein (MAP) kinase kinase and MAP kinase. J. Biol. Chem. 1993, 268, 19196–19199. [Google Scholar] [CrossRef]
- Della Rocca, G.J.; van Biesen, T.; Daaka, Y.; Luttrell, D.K.; Luttrell, L.; Lefkowitz, R.J. Ras-dependent Mitogen-activated Protein Kinase Activation by G Protein-coupled Receptors. J. Biol. Chem. 1997, 272, 19125–19132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutkind, J.S. The Pathways Connecting G Protein-coupled Receptors to the Nucleus through Divergent Mitogen-activated Protein Kinase Cascades. J. Biol. Chem. 1998, 273, 1839–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan, D.H.; Yung, L.Y.; Ye, R.D.; Wong, Y.H. Activation of ras-dependent signaling pathways by G 14 -coupled receptors requires the adaptor protein TPR. J. Cell. Biochem. 2012, 113, 3486–3497. [Google Scholar] [CrossRef]
- Nugent, A.; Proia, R.L. The role of G protein-coupled receptors in lymphoid malignancies. Cell. Signal. 2017, 39, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Mashiko, M.; Kurosawa, A.; Tani, Y.; Tsuji, T.; Takeda, S. GPR31 and GPR151 are activated under acidic conditions. J. Biochem. 2019, 166, 317–322. [Google Scholar] [CrossRef]
- Fehrenbacher, N.; Philips, M.R. Targeting RAS—Will GPR31 deliver us a new path forward? Mol. Cell. Oncol. 2017, 4, e1359228. [Google Scholar] [CrossRef]
- Fehrenbacher, N.; Da Silva, I.T.; Ramirez, C.; Zhou, Y.; Cho, K.-J.; Kuchay, S.; Shi, J.; Thomas, S.; Pagano, M.; Hancock, J.; et al. The G protein–coupled receptor GPR31 promotes membrane association of KRAS. J. Cell Biol. 2017, 216, 2329–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Eishingdrelo, A.; Kongsamut, S.; Eishingdrelo, H. G-protein-coupled receptors mediate 14-3-3 signal transduction. Signal. Transduct. Target. Ther. 2016, 1, 16018. [Google Scholar] [CrossRef] [Green Version]
- McCudden, C.R.; Hains, M.D.; Kimple, R.J.; Siderovski, D.; Willard, F.S. G-protein signaling: Back to the future. Cell. Mol. Life Sci. 2005, 62, 551–577. [Google Scholar] [CrossRef] [Green Version]
- Ito, A.; Satoh, T.; Kaziro, Y.; Itoh, H. G protein βγ subunit activates Ras, Raf, and MAP kinase in HEK 293 cells. FEBS Lett. 1995, 368, 183–187. [Google Scholar] [CrossRef] [Green Version]
- Harden, T.K. G Protein-dependent Regulation of Phospholipase C by Cell Surface Receptors. Am. Rev. Respir. Dis. 1990, 141, 119–122. [Google Scholar] [CrossRef]
- Bivona, T.G.; Quatela, S.E.; Bodemann, B.O.; Ahearn, I.M.; Soskis, M.J.; Mor, A.; Miura, J.; Wiener, H.H.; Wright, L.; Saba, S.G.; et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mito-chondria and induces apoptosis. Mol. Cell 2006, 21, 481–493. [Google Scholar] [CrossRef]
- Alvarez-Moya, B.; López-Alcalá, C.; Drosten, M.; Bachs, O.; Agell, N. K-Ras4B phosphorylation at Ser181 is inhibited by calmodulin and modulates K-Ras activity and function. Oncogene 2010, 29, 5911–5922. [Google Scholar] [CrossRef] [Green Version]
- Kohno, T.; Matsuda, E.; Sasaki, H.; Sasaki, T. Protein-tyrosine kinase CAKbeta/PYK2 is activated by binding Ca2+/calmodulin to FERM F2 alpha2 helix and thus forming its dimer. Biochem. J. 2008, 410, 513–523. [Google Scholar] [CrossRef] [Green Version]
- Blaukat, A.; Ivankovic-Dikic, I.; Gronroos, E.; Dolfi, F.; Tokiwa, G.; Vuori, K.; Dikic, I. Adaptor Proteins Grb2 and Crk Couple Pyk2 with Activation of Specific Mitogen-activated Protein Kinase Cascades. J. Biol. Chem. 1999, 274, 14893–14901. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.F.; Kumahara, E.; Saffen, D. A CalDAG-GEFI/Rap1/B-Raf cassette couples M(1) muscarinic acetylcholine receptors to the activation of ERK1/2. J. Biol. Chem. 2001, 276, 25568–25581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.L.; Wang, R.C.; Cheng, K.; Ring, B.Z.; Su, L. Roles of Rap1 signaling in tumor cell migration and invasion. Cancer. Biol. Med. 2017, 14, 90–99. [Google Scholar]
- Feng, H.; Liu, K.W.; Guo, P.; Zhang, P.; Cheng, T.; McNiven, M.A.; Johnson, G.R.; Hu, B.; Cheng, S.Y. Dynamin 2 mediates PDGFRalpha-SHP-2-promoted glioblastoma growth and invasion. Oncogene 2012, 31, 2691–2702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wunderlich, L.; Faragó, A.; Buday, L. Characterization of Interactions of Nck with Sos and Dynamin. Cell. Signal. 1999, 11, 25–29. [Google Scholar] [CrossRef]
- Kranenburg, O.; Verlaan, I.; Hordijk, P.L.; Moolenaar, W.H. Gi-mediated activation of the Ras/MAP kinase pathway involves a 100 kDa tyrosine-phosphorylated Grb2 SH3 binding protein, but not Src nor Shc. EMBO J. 1997, 16, 3097–3105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kranenburg, O.; Verlaan, I.; Moolenaar, W.H. Gi-mediated tyrosine phosphorylation of Grb2 (growth-factor-receptor-bound protein 2)-bound dynamin-II by lysophosphatidic acid. Biochem. J. 1999, 339 Pt 1, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Sassone-Corsi, P. The cyclic AMP pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011148. [Google Scholar] [CrossRef]
- Ster, J.; De Bock, F.; Guerineau, N.; Janossy, A.; Barrere-Lemaire, S.; Bos, J.L.; Bockaert, J.; Fagni, L. Exchange protein activated by cAMP (Epac) mediates cAMP activation of p38 MAPK and modulation of Ca2+-dependent K+ channels in cerebellar neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 2519–2524. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, J.M.; Stork, P.J. PKA phosphorylation of Src mediates cAMP’s inhibition of cell growth via Rap1. Mol. Cell 2002, 9, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Schramm, K.; Niehof, M.; Radziwill, G.; Rommel, C.; Moelling, K. Phosphorylation of c-Raf-1 by Protein Kinase A Interferes with Activation. Biochem. Biophys. Res. Commun. 1994, 201, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Umeda, K.; Negishi, M.; Katoh, H. RasGRF1 mediates brain-derived neurotrophic factor-induced axonal growth in primary cultured cortical neurons. Biochem. Biophys. Rep. 2018, 17, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Nazmun, N.; Hassan, S.; Liu, X.; Yang, J. BRAF mutation and its inhibitors in sarcoma treatment. Cancer Med. 2020, 9, 4881–4896. [Google Scholar] [CrossRef]
- Park, E.; Rawson, S.; Li, K.; Kim, B.W.; Ficarro, S.B.; Gonzalez-Del Pino, G.; Sharif, H.; Marto, J.A.; Jeon, H.; Eck, M.J. Architecture of autoinhibited and active BRAF-MEK1-14-3-3 complexes. Nature 2019, 575, 545–550. [Google Scholar] [CrossRef]
- Cutler, R.E.; Stephens, R.M.; Saracino, M.R.; Morrison, D.K. Autoregulation of the Raf-1 serine/threonine kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 9214–9219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obsilova, V.; Obsil, T. The 14-3-3 Proteins as Important Allosteric Regulators of Protein Kinases. Int. J. Mol. Sci. 2020, 21, 8824. [Google Scholar] [CrossRef]
- Liau, N.P.D.; Venkatanarayan, A.; Quinn, J.G.; Phung, W.; Malek, S.; Hymowitz, S.G.; Sudhamsu, J. Dimerization Induced by C-Terminal 14–3–3 Binding Is Sufficient for BRAF Kinase Activation. Biochemistry 2020, 59, 3982–3992. [Google Scholar] [CrossRef] [PubMed]
- Pennington, K.L.; Chan, T.Y.; Torres, M.; Andersen, J.L. The dynamic and stress-adaptive signaling hub of 14-3-3: Emerging mechanisms of regulation and context-dependent protein–protein interactions. Oncogene 2018, 37, 5587–5604. [Google Scholar] [CrossRef] [Green Version]
- Yaffe, M.B. How do 14-3-3 proteins work?—Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett. 2002, 513, 53–57. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Pollock, C.; Steen, H.; Shaw, P.E.; Mischak, H.; Kolch, W. Cyclic AMP-Dependent Kinase Regulates Raf-1 Kinase Mainly by Phosphorylation of Serine. Mol. Cell. Biol. 2002, 22, 3237–3246. [Google Scholar] [CrossRef] [Green Version]
- Rommel, C.; Clarke, B.A.; Zimmermann, S.; Nuñez, L.; Rossman, R.; Reid, K.; Moelling, K.; Yancopoulos, G.D.; Glass, D.J. Differentiation Stage-Specific Inhibition of the Raf-MEK-ERK Pathway by Akt. Science 1999, 286, 1738–1741. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, S.; Moelling, K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 1999, 286, 1741–1744. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Ng, W.H.; Yap, J.; Chia, B.; Huang, X.; Wang, M.; Hu, J. The AMPK inhibitor overcomes the paradoxical effect of RAF inhibitors through blocking phospho–Ser-621 in the C terminus of CRAF. J. Biol. Chem. 2018, 293, 14276–14284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Dong, X.; Yap, J.; Hu, J. The MAPK and AMPK signalings: Interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 2020, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.-H.; Yuan, P.; Perez-Lorenzo, R.; Zhang, Y.; Lee, S.X.; Ou, Y.; Asara, J.M.; Cantley, L.; Zheng, B. Phosphorylation of BRAF by AMPK Impairs BRAF-KSR1 Association and Cell Proliferation. Mol. Cell 2013, 52, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, D.; Nguyen, L.; Matallanas, D.; Halasz, M.; Doherty, C.; Kholodenko, B.; Kolch, W. Protein interaction switches coordinate Raf-1 and MST2/Hippo signalling. Nat. Cell Biol. 2014, 16, 673–684. [Google Scholar] [CrossRef]
- Tran, T.H.; Chan, A.H.; Young, L.C.; Bindu, L.; Neale, C.; Messing, S.; Dharmaiah, S.; Taylor, T.; Denson, J.-P.; Esposito, D.; et al. KRAS interaction with RAF1 RAS-binding domain and cysteine-rich domain provides insights into RAS-mediated RAF activation. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Luo, Z.; Diaz, B.; Marshall, M.S.; Avruch, J. An intact Raf zinc finger is required for optimal binding to processed Ras and for ras-dependent Raf activation in situ. Mol. Cell. Biol. 1997, 17, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, D.; Podar, K.; Pacher, M.; Kubicek, M.; Welzel, N.; Hemmings, B.A.; Dilworth, S.M.; Mischak, H.; Kolch, W.; Baccarini, M. Raf-1-associated Protein Phosphatase 2A as a Positive Regulator of Kinase Activation. J. Biol. Chem. 2000, 275, 22300–22304. [Google Scholar] [CrossRef] [Green Version]
- Jaumot, M.; Hancock, J.F. Protein phosphatases 1 and 2A promote Raf-1 activation by regulating 14-3-3 interactions. Oncogene 2001, 20, 3949–3958. [Google Scholar] [CrossRef]
- Ory, S.; Zhou, M.; Conrads, T.P.; Veenstra, T.D.; Morrison, D.K. Protein Phosphatase 2A Positively Regulates Ras Signaling by Dephosphorylating KSR1 and Raf-1 on Critical 14-3-3 Binding Sites. Curr. Biol. 2003, 13, 1356–1364. [Google Scholar] [CrossRef] [Green Version]
- Young, L.C.; Hartig, N.; del Río, I.B.; Sari, S.; Ringham-Terry, B.; Wainwright, J.R.; Jones, G.G.; McCormick, F.; Rodriguez-Viciana, P. SHOC2–MRAS–PP1 complex positively regulates RAF activity and contributes to Noonan syndrome pathogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, E10576–E10585. [Google Scholar] [CrossRef] [Green Version]
- Yin, Q.; Han, T.; Fang, B.; Zhang, G.; Zhang, C.; Roberts, E.R.; Izumi, V.; Zheng, M.; Jiang, S.; Yin, X.; et al. K27-linked ubiquitination of BRAF by ITCH engages cytokine response to maintain MEK-ERK signaling. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Abankwa, D.; Gorfe, A. Mechanisms of Ras Membrane Organization and Signaling: Ras Rocks Again. Biomolecules 2020, 10, 1522. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Stites, E.C.; Yu, H.; Germino, E.A.; Meharena, H.S.; Stork, P.J.; Kornev, A.; Taylor, S.S.; Shaw, A.S. Allosteric Activation of Functionally Asymmetric RAF Kinase Dimers. Cell 2013, 154, 1036–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drosten, M.; Sum, E.Y.M.; Lechuga, C.G.; Simón-Carrasco, L.; Jacob, H.K.C.; García-Medina, R.; Huang, S.; Beijersbergen, R.; Bernards, R.; Barbacid, M. Loss of p53 induces cell proliferation via Ras-independent activation of the Raf/Mek/Erk signaling pathway. Proc. Natl. Acad. Sci. USA 2014, 111, 15155–15160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drosten, M.; Barbacid, M. Ras and p53: An unsuspected liaison. Mol. Cell. Oncol. 2015, 3, e996001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras Provokes Premature Cell Senescence Associated with Accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Qi, Z.; Yin, H.; Yang, G. Interaction between p53 and Ras signaling controls cisplatin resistance via HDAC4- and HIF-1alpha-mediated reg-ulation of apoptosis and autophagy. Theranostics 2019, 9, 1096–1114. [Google Scholar] [CrossRef]
- Lu, L.; Zeng, J. Evaluation of K-ras and p53 expression in pancreatic adenocarcinoma using the cancer genome atlas. PLoS ONE 2017, 12, e0181532. [Google Scholar] [CrossRef] [Green Version]
- Ries, S.; Biederer, C.; Woods, D.; Shifman, O.; Shirasawa, S.; Sasazuki, T.; McMahon, M.; Oren, M.; McCormick, F. Opposing Effects of Ras on p53: Transcriptional Activation of mdm2 and Induction of p19ARF. Cell 2000, 103, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Shin, S.; Zhang, R.; Firozi, P.F.; Yang, L.; Abbruzzese, J.L.; Reddy, S.A.G. Hdm2 is regulated by K-Ras and mediates p53-independent functions in pancreatic cancer cells. Oncogene 2008, 28, 709–720. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M.; Tortola, S.; Toyota, M.; Capella, G.; Peinado, M.A.; Baylin, S.B.; Herman, J.G. Hypermethylation-associated inactivation of p14(ARF) is independent of p16(INK4a) methylation and p53 mutational status. Cancer Res. 2000, 60, 129–133. [Google Scholar]
- Li, L.; Zhao, G.-D.; Shi, Z.; Qi, L.-L.; Zhou, L.-Y.; Fu, Z.-X. The Ras/Raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC. Oncol. Lett. 2016, 12, 3045–3050. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, M.R.M.; Baig, M.; Mohamoud, H.S.A.; Ul-Haq, Z.; Hoessli, D.C.; Khogeer, G.S.; Al-Sayed, R.R.; Al-Aama, J.Y. BRAF gene: From human cancers to developmental syndromes. Saudi J. Biol. Sci. 2015, 22, 359–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, Y.; Niihori, T.; Inoue, S.-I.; Matsubara, Y. Recent advances in RASopathies. J. Hum. Genet. 2016, 61, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khrenova, M.; Mironov, V.; Grigorenko, B.; Nemukhin, A.V. Modeling the Role of G12V and G13V Ras Mutations in the Ras-GAP-Catalyzed Hydrolysis Reaction of Guanosine Triphosphate. Biochemistry 2014, 53, 7093–7099. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P.L.; Stone, R.M.; Al-Kali, A.; Barta, S.K.; Bejar, R.; Bennett, J.M.; Carraway, H.; De Castro, C.M.; Deeg, H.J.; DeZern, A.E.; et al. Myelodysplastic Syndromes, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2016, 15, 60–87. [Google Scholar] [CrossRef] [Green Version]
- Steensma, D.P. Myelodysplastic syndromes current treatment algorithm. Blood Cancer J. 2018, 8, 1–7. [Google Scholar] [CrossRef]
- Ma, X. Epidemiology of Myelodysplastic Syndromes. Am. J. Med. 2012, 125, S2–S5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, J.L. ras oncogenes in human cancer: A review. Cancer Res. 1989, 49, 4682–4689. [Google Scholar]
- Kadia, T.M.; Kantarjian, H.; Kornblau, S.; Borthakur, G.; Faderl, S.; Freireich, E.J.; Luthra, R.; Garcia-Manero, G.; Pierce, S.; Cortes, J.; et al. Clinical and proteomic characterization of acute myeloid leukemia with mutated RAS. Cancer 2012, 118, 5550–5559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tallman, M.S.; Wang, E.S.; Altman, J.K.; Appelbaum, F.R.; Bhatt, V.R.; Bixby, D.; Coutre, S.E.; De Lima, M.; Fathi, A.T.; Fiorella, M.; et al. Acute Myeloid Leukemia, Version 3.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2019, 17, 721–749. [Google Scholar] [CrossRef] [Green Version]
- DeSantis, C.E.; Lin, C.C.; Mariotto, A.B.; Siegel, R.L.; Stein, K.D.; Kramer, J.L.; Alteri, R.; Robbins, A.S.; Jemal, A. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 2014, 64, 252–271. [Google Scholar] [CrossRef]
- Neubauer, A.; Maharry, K.; Mrózek, K.; Thiede, C.; Marcucci, G.; Paschka, P.; Mayer, R.J.; Larson, R.A.; Liu, E.T.; Bloomfield, C.D. Patients with Acute Myeloid Leukemia and RAS Mutations Benefit Most From Postremission High-Dose Cytarabine: A Cancer and Leukemia Group B Study. J. Clin. Oncol. 2008, 26, 4603–4609. [Google Scholar] [CrossRef] [Green Version]
- Brown, P.; Inaba, H.; Annesley, C.; Beck, J.; Colace, S.; Dallas, M.; DeSantes, K.; Kelly, K.; Kitko, C.; Lacayo, N.; et al. Pediatric Acute Lymphoblastic Leukemia, Version 2.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2020, 18, 81–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaime-Pérez, J.C.; Jiménez-Castillo, R.A.; Herrera-Garza, J.L.; Gutiérrez-Aguirre, H.; Marfil-Rivera, L.J.; Gómez-Almaguer, D. Survival Rates of Adults with Acute Lymphoblastic Leukemia in a Low-Income Population: A Decade of Experience at a Single Institution in Mexico. Clin. Lymphoma Myeloma Leuk. 2017, 17, 60–68. [Google Scholar] [CrossRef]
- Knight, T.; Irving, J.A. Ras/Raf/MEK/ERK Pathway Activation in Childhood Acute Lymphoblastic Leukemia and Its Therapeutic Targeting. Front. Oncol. 2014, 4, 160. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, B.; Angelini, S.; Sander, B.; Christensson, B.; Hemminki, K.; Kumar, R. Mutations in the BRAF and N-ras genes in childhood acute lymphoblastic leukaemia. Leukemia 2004, 19, 310–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pleyer, L.; Germing, U.; Sperr, W.R.; Linkesch, W.; Burgstaller, S.; Stauder, R.; Girschikofsky, M.; Schreder, M.; Pfeilstocker, M.; Lang, A.H.; et al. Azacitidine in CMML: Matched-pair analyses of daily-life patients reveal modest effects on clinical course and survival. Leuk. Res. 2014, 38, 475–483. [Google Scholar] [CrossRef] [Green Version]
- Gelsi-Boyer, V.; Trouplin, V.; Adélaïde, J.; Aceto, N.; Remy, V.; Pinson, S.; Houdayer, C.; Arnoulet, C.; Sainty, D.; Bentires-Alj, M.; et al. Genome profiling of chronic myelomonocytic leukemia: Frequent alterations of RAS and RUNX1genes. BMC Cancer 2008, 8, 299. [Google Scholar] [CrossRef] [Green Version]
- Onida, F.; Kantarjian, H.M.; Smith, T.L.; Ball, G.; Keating, M.J.; Estey, E.H.; Glassman, A.B.; Albitar, M.; Kwari, M.I.; Beran, M. Prognostic factors and scoring systems in chronic myelomonocytic leukemia: A retrospective analysis of 213 patients. Blood 2002, 99, 840–849. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Tefferi, A. Cytogenetic and molecular abnormalities in chronic myelomonocytic leukemia. Blood Cancer J. 2016, 6, e393. [Google Scholar] [CrossRef]
- Zhang, L.; Singh, R.R.; Patel, K.P.; Stingo, F.; Routbort, M.; You, M.J.; Miranda, R.N.; Garcia-Manero, G.; Kantarjian, H.M.; Medeiros, L.J.; et al. BRAF kinase domain mutations are present in a subset of chronic myelomonocytic leukemia with wild-type RAS. Am. J. Hematol. 2014, 89, 499–504. [Google Scholar] [CrossRef] [Green Version]
- Radich, J.P.; Deininger, M.; Abboud, C.N.; Altman, J.K.; Berman, E.; Bhatia, R.; Bhatnagar, B.; Curtin, P.; DeAngelo, D.J.; Gotlib, J.; et al. Chronic Myeloid Leukemia, Version 1.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2018, 16, 1108–1135. [Google Scholar] [CrossRef]
- Liu, E.; Hjelle, B.; Bishop, J.M. Transforming genes in chronic myelogenous leukemia. Proc. Natl. Acad. Sci. USA 1988, 85, 1952–1956. [Google Scholar] [CrossRef] [Green Version]
- Tempero, M.A.; Malafa, M.P.; Chiorean, E.G.; Czito, B.; Scaife, C.; Narang, A.K.; Fountzilas, C.; Wolpin, B.M.; Al-Hawary, M.; Asbun, H.; et al. Pancreatic Adenocarcinoma, Version 1.2019. J. Natl. Compr. Cancer Netw. 2019, 17, 202–210. [Google Scholar] [CrossRef] [Green Version]
- Le, N.; Sund, M.; Vinci, A.; Beyer, G.; Javed, M.A.; Krug, S.; Neessee, A.; Schober, M. Prognostic and predictive markers in pancreatic adenocarcinoma. Dig. Liver Dis. 2016, 48, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Caldas, C.; Kern, S.E. K-ras mutation and pancreatic adenocarcinoma. Int. J. Pancreatol. 1995, 18, 1–6. [Google Scholar] [CrossRef]
- Schultz, N.A.; Roslind, A.; Christensen, I.J.; Horn, T.; Høgdall, E.; Pedersen, L.N.; Kruhøffer, M.; Burcharth, F.; Wøjdemann, M.; Johansen, J.S. Frequencies and Prognostic Role of KRAS and BRAF Mutations in Patients with Localized Pancreatic and Ampullary Adenocarcinomas. Pancreas 2012, 41, 759–766. [Google Scholar] [CrossRef]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Garnett, M.J.; Marais, R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell 2004, 6, 313–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ball, N.J.; Yohn, J.J.; Morelli, J.G.; Norris, D.A.; Golitz, L.E.; Hoeffler, J.P. RAS Mutations in Human Melanoma: A Marker of Malignant Progression. J. Investig. Dermatol. 1994, 102, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non-Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. [Google Scholar] [CrossRef]
- Ettinger, D.S.; Wood, D.E.; Aggarwal, C.; Aisner, D.L.; Akerley, W.; Bauman, J.R.; Bharat, A.; Bruno, D.S.; Chang, J.Y.; Chirieac, L.R.; et al. NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 1.2020. J. Natl. Compr. Cancer Netw. 2019, 17, 1464–1472. [Google Scholar] [CrossRef] [Green Version]
- Riely, G.J.; Marks, J.; Pao, W. KRAS Mutations in Non-Small Cell Lung Cancer. Proc. Am. Thorac. Soc. 2009, 6, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Torres, J.M.; Viteri, S.; Molina, M.A.; Rosell, R. BRAF mutant non-small cell lung cancer and treatment with BRAF inhibitors. Transl. Lung Cancer Res. 2013, 2, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Mitsudomi, T.; Viallet, J.; Mulshine, J.; Linnoila, R.I.; Minna, J.D.; Gazdar, A.F. Mutations of ras genes distinguish a subset of non-small-cell lung cancer cell lines from small-cell lung cancer cell lines. Oncogene 1991, 6, 1353–1362. [Google Scholar] [PubMed]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Gustavsson, B.; Carlsson, G.; Machover, D.; Petrelli, N.; Roth, A.; Schmoll, H.-J.; Tveit, K.-M.; Gibson, F. A Review of the Evolution of Systemic Chemotherapy in the Management of Colorectal Cancer. Clin. Color. Cancer 2015, 14, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breivik, J.; Meling, G.I.; Spurkland, A.; Rognum, T.O.; Gaudernack, G. K-ras mutation in colorectal cancer: Relations to patient age, sex and tumour location. Br. J. Cancer 1994, 69, 367–371. [Google Scholar] [CrossRef] [Green Version]
- Oldenburg, J.; Fosså, S.D.; Nuver, J.; Heidenreich, A.; Schmoll, H.-J.; Bokemeyer, C.; Horwich, A.; Beyer, J.; Kataja, V. Testicular seminoma and non-seminoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2013, 24, vi125–vi132. [Google Scholar] [CrossRef]
- Dong, W.; Gang, W.; Liu, M.; Zhang, H. Analysis of the prognosis of patients with testicular seminoma. Oncol. Lett. 2016, 11, 1361–1366. [Google Scholar] [CrossRef] [Green Version]
- Mulder, M.P.; Keijzer, W.; Verkerk, A.; Boot, A.J.; Prins, M.E.; Splinter, T.A.; Bos, J.L. Activated ras genes in human seminoma: Evidence for tumor heterogeneity. Oncogene 1989, 4, 1345–1351. [Google Scholar]
- DeGeorge, K.C.; Holt, H.R.; Hodges, S.C. Bladder Cancer: Diagnosis and Treatment. Am. Fam. Physician 2017, 96, 507–514. [Google Scholar]
- Abdollah, F.; Gandaglia, G.; Thuret, R.; Schmitges, J.; Tian, Z.; Jeldres, C.; Passoni, N.M.; Briganti, A.; Shariat, S.F.; Perrotte, P.; et al. Incidence, survival and mortality rates of stage-specific bladder cancer in United States: A trend analysis. Cancer Epidemiol. 2013, 37, 219–225. [Google Scholar] [CrossRef]
- Oxford, G.; Theodorescu, D. Review Article: The Role of Ras Superfamily Proteins in Bladder Cancer Progression. J. Urol. 2003, 170, 1987–1993. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K. Bladder Cancer: New Insights into Its Molecular Pathology. Cancers 2018, 10, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosetti, C.; Turati, F.; La Vecchia, C. Hepatocellular carcinoma epidemiology. Best Pr. Res. Clin. Gastroenterol. 2014, 28, 753–770. [Google Scholar] [CrossRef] [PubMed]
- Taketomi, A.; Shirabe, K.; Muto, J.; Yoshiya, S.; Motomura, T.; Mano, Y.; Ikegami, T.; Yoshizumi, T.; Sugio, K.; Maehara, Y. A rare point mutation in the Ras oncogene in hepatocellular carcinoma. Surg. Today 2013, 43, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Orr, B.; Edwards, R.P. Diagnosis and Treatment of Ovarian Cancer. Hematol. Clin. North. Am. 2018, 32, 943–964. [Google Scholar] [CrossRef]
- Protani, M.M.; Nagle, C.M.; Webb, P. Obesity and Ovarian Cancer Survival: A Systematic Review and Meta-analysis. Cancer Prev. Res. 2012, 5, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Alsharedi, M.; Katz, H. Check point inhibitors a new era in renal cell carcinoma treatment. Med. Oncol. 2018, 35, 85. [Google Scholar] [CrossRef]
- Motzer, R.J.; Jonasch, E.; Michaelson, M.D.; Nandagopal, L.; Gore, J.L.; George, S.; Alva, A.; Haas, N.; Harrison, M.R.; Plimack, E.R.; et al. NCCN Guidelines Insights: Kidney Cancer, Version 2.2020. J. Natl. Compr. Cancer Netw. 2019, 17, 1278–1285. [Google Scholar] [CrossRef] [Green Version]
- Bayrak, O.; Sen, H.; Bulut, E.; Cengiz, B.; Karakok, M.; Erturhan, S.; Seckiner, I. Evaluation of EGFR, KRAS and BRAF gene mutations in renal cell carcinoma. J. Kidney Cancer VHL 2014, 1, 40–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesweg, M.; Kasper, S.; Worm, K.; Herold, T.; Reis, H.; Sara, L.; Metzenmacher, M.; Abendroth, A.; Darwiche, K.; Aigner, C.; et al. Impact of RAS mutation subtype on clinical outcome—A cross-entity comparison of patients with advanced non-small cell lung cancer and colorectal cancer. Oncogene 2019, 38, 2953–2966. [Google Scholar] [CrossRef] [PubMed]
- Terrell, E.M.; Durrant, D.E.; Ritt, D.A.; Sealover, N.E.; Sheffels, E.; Spencer-Smith, R.; Esposito, D.; Zhou, Y.; Hancock, J.F.; Kortum, R.; et al. Distinct Binding Preferences between Ras and Raf Family Members and the Impact on Oncogenic Ras Signaling. Mol. Cell 2019, 76, 872–884.e5. [Google Scholar] [CrossRef] [PubMed]
- Barbacid, M. ras oncogenes: Their role in neoplasia. Eur. J. Clin. Investig. 2008, 20, 225–235. [Google Scholar] [CrossRef]
- Cook, J.H.; Melloni, G.E.M.; Gulhan, D.C.; Park, P.J.; Haigis, K.M. The origins and genetic interactions of KRAS mutations are allele- and tissue-specific. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Nair, S.G.; Loppnow, G.R. Comparison of K-Ras and N-Ras Mutagenic Hot Spots for UVC Damage. ACS Omega 2019, 4, 3469–3475. [Google Scholar] [CrossRef] [Green Version]
- Holderfield, M.; Deuker, M.M.; McCormick, F.; McMahon, M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 2014, 14, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Cancer Genome Project; Jones, C.M.; Marshall, C.J.; Springer, C.J.; et al. Mechanical acts RAF-Erk signal pathway by oncological mutations B-Raf. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Hanrahan, A.J.; Sylvester, B.E.; Chang, M.T.; ElZein, A.; Gao, J.; Han, W.; Liu, Y.; Xu, D.; Gao, S.P.; Gorelick, A.N.; et al. Leveraging Systematic Functional Analysis to Benchmark an In Silico Framework Distinguishes Driver from Passenger MEK Mutants in Cancer. Cancer Res. 2020, 80, 4233–4243. [Google Scholar] [CrossRef]
- Greger, J.G.; Eastman, S.D.; Zhang, V.; Bleam, M.R.; Hughes, A.M.; Smitheman, K.N.; Dickerson, S.H.; Laquerre, S.G.; Liu, L.; Gilmer, T.M. Combinations of BRAF, MEK, and PI3K/mTOR Inhibitors Overcome Acquired Resistance to the BRAF Inhibitor GSK2118436 Dabrafenib, Mediated by NRAS or MEK Mutations. Mol. Cancer Ther. 2012, 11, 909–920. [Google Scholar] [CrossRef] [Green Version]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Viciana, P.; Rauen, K.A. Biochemical characterization of novel germline BRAF and MEK mutations in car-dio-facio-cutaneous syndrome. Methods Enzymol. 2008, 438, 277–289. [Google Scholar]
- Smorodinsky-Atias, K.; Soudah, N.; Engelberg, D. Mutations That Confer Drug-Resistance, Oncogenicity and Intrinsic Activity on the ERK MAP Kinases—Current State of the Art. Cells 2020, 9, 129. [Google Scholar] [CrossRef] [Green Version]
- Emery, C.M.; Vijayendran, K.G.; Zipser, M.C.; Sawyer, A.M.; Niu, L.; Kim, J.J.; Hatton, C.; Chopra, R.; Oberholzer, P.A.; Karpova, M.B.; et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 20411–20416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, E.M.; Ghandi, M.; Treacy, D.J.; Wagle, N.; Garraway, L.A. ERK Mutations Confer Resistance to Mitogen-Activated Protein Kinase Pathway Inhibitors. Cancer Res. 2014, 74, 7079–7089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, B.S.; Durinck, S.; Stawiski, E.W.; Yin, J.; Wang, W.; Lin, E.; Moffat, J.G.; Martin, S.E.; Modrusan, Z.; Seshagiri, S. ERK Mutations and Amplification Confer Resistance to ERK-Inhibitor Therapy. Clin. Cancer Res. 2018, 24, 4044–4055. [Google Scholar] [CrossRef] [Green Version]
- Phillips, A.A.; Harewood, J.C.K. Adult T Cell Leukemia-Lymphoma (ATL): State of the Art. Curr. Hematol. Malign. Rep. 2018, 13, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Mehta-Shah, N.; Ratner, L.; Horwitz, S.M. Adult T-Cell Leukemia/Lymphoma. J. Oncol. Pract. 2017, 13, 487–492. [Google Scholar] [CrossRef]
- Poiesz, B.J.; Ruscetti, F.W.; Gazdar, A.F.; Bunn, P.A.; Minna, J.D.; Gallo, R.C. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci. USA 1980, 77, 7415–7419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, R.C. History of the discoveries of the first human retroviruses: HTLV-1 and HTLV-2. Oncogene 2005, 24, 5926–5930. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, M.; Watanabe, T. Molecular Hallmarks of Adult T Cell Leukemia. Front. Microbiol. 2012, 3, 334. [Google Scholar] [CrossRef] [Green Version]
- Krump, N.A.; You, J. Molecular mechanisms of viral oncogenesis in humans. Nat. Rev. Genet. 2018, 16, 684–698. [Google Scholar] [CrossRef]
- Bangham, C.R.; Ratner, L. How does HTLV-1 cause adult T-cell leukaemia/lymphoma (ATL)? Curr. Opin. Virol. 2015, 14, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasunaga, J.-I. Strategies of Human T-Cell Leukemia Virus Type 1 for Persistent Infection: Implications for Leukemogenesis of Adult T-Cell Leukemia-Lymphoma. Front. Microbiol. 2020, 11, 979. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Wang, W.; Li, M.; Liu, Y.; Zheng, D. Tax1 enhances cancer cell proliferation via Ras-Raf-MEK-ERK signaling pathway. IUBMB Life 2009, 61, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Nagata, Y.; Kitanaka, A.; Shiraishi, Y.; Shimamura, T.; Yasunaga, J.-I.; Totoki, Y.; Chiba, K.; Sato-Otsubo, A.; Nagae, G.; et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat. Genet. 2015, 47, 1304–1315. [Google Scholar] [CrossRef]
- Stoppa, G.; Rumiato, E.; Saggioro, D. Ras signaling contributes to survival of human T-cell leukemia/lymphoma virus type 1 (HTLV-1) Tax-positive T-cells. Apoptosis 2011, 17, 219–228. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, J.L. Burkitt’s Lymphoma. N. Engl. J. Med. 1981, 305, 735–745. [Google Scholar] [CrossRef]
- Dunleavy, K.; Little, R.F.; Wilson, W.H. Update on Burkitt Lymphoma. Hematol. Clin. North. Am. 2016, 30, 1333–1343. [Google Scholar] [CrossRef]
- De-Thé, G.; Geser, A.; Day, N.E.; Tukei, P.M.; Williams, E.H.; Beri, D.P.; Smith, P.G.; Dean, A.G.; Bornkamm, G.W.; Feorino, P.; et al. Epidemiological evidence for causal relationship between Epstein-Barr virus and Burkitt’s lymphoma from Ugandan prospective study. Nature 1978, 274, 756–761. [Google Scholar] [CrossRef]
- Casulo, C.; Friedberg, J. Treating Burkitt Lymphoma in Adults. Curr. Hematol. Malign. Rep. 2015, 10, 266–271. [Google Scholar] [CrossRef]
- Dalla-Favera, R.; Bregni, M.; Erikson, J.; Patterson, D.; Gallo, R.C.; Croce, C.M. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7824–7827. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Meng, L.; Jiang, W.; Zhang, H.; Zhou, A.; Zeng, N. Identification of clinical molecular targets for childhood Burkitt lymphoma. Transl. Oncol. 2020, 13, 100855. [Google Scholar] [CrossRef]
- Strati, P.; Nastoupil, L.J.; Davis, R.E.; Fayad, L.E.; Fowler, N.; Hagemeister, F.B.; Kwak, L.; Oki, Y.; Wang, M.; Westin, J.; et al. A phase 1 trial of alisertib and romidepsin for relapsed/refractory aggressive B-cell and T-cell lymphomas. Haematologica 2019, 105, e26–e28. [Google Scholar] [CrossRef] [PubMed]
- Wever, C.M.; Geoffrion, D.; Grande, B.M.; Yu, S.; Alcaide, M.; Lemaire, M.; Riazalhosseini, Y.; Hébert, J.; Gavino, C.; Vinh, D.; et al. The genomic landscape of two Burkitt lymphoma cases and derived cell lines: Comparison between primary and relapse samples. Leuk. Lymphoma 2018, 59, 2159–2174. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Viatour, P. Hepatocellular carcinoma: Old friends and new tricks. Exp. Mol. Med. 2020, 52, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Balogh, J.; Victor, D., III; Asham, E.H.; Burroughs, S.G.; Boktour, M.; Saharia, A.; Li, X.; Ghobrial, R.M.; Monsour, H.P., Jr. Hepatocellular carcinoma: A review. J. Hepatocell. Carcinoma 2016, 3, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.-N.; Bai, T.; Chen, Z.-S.; Wu, F.-X.; Chen, Y.-Y.; De Xiang, B.; Peng, T.; Han, Z.-G.; Li, L.-Q. The p53 mutation spectrum in hepatocellular carcinoma from Guangxi, China: Role of chronic hepatitis B virus infection and aflatoxin B1 exposure. Liver Int. 2014, 35, 999–1009. [Google Scholar] [CrossRef]
- Hou, W.; Liu, J.; Chen, P.; Wang, H.; Ye, B.C.; Qiang, F. Mutation analysis of key genes in RAS/RAF and PI3K/PTEN pathways in Chinese patients with hepatocellular carcinoma. Oncol. Lett. 2014, 8, 1249–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, Y.H.; Choi, J.Y.; Kim, S.; Chung, E.S.; Kim, T.; Koh, S.S.; Lee, B.; Bae, S.H.; Kim, J.; Park, Y.M. Over-expression of c-raf-1 proto-oncogene in liver cirrhosis and hepatocellular carcinoma. Hepatol. Res. 2004, 29, 113–121. [Google Scholar] [CrossRef]
- Chen, L.; Shi, Y.; Jiang, C.Y.; Wei, L.X.; Wang, Y.L.; Dai, G.H. Expression and prognostic role of pan-Ras, Raf-1, pMEK1 and pERK1/2 in patients with hepatocellular carcinoma. Eur. J. Surg. Oncol. 2011, 37, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Nguyen, T.T.T.; Chow, K.-H.K.-P.; Tan, P.H.; Soo, K.C.; Tran, E. Over-expression of the mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK in hepatocellular carcinoma: Its role in tumor progression and apoptosis. BMC Gastroenterol. 2003, 3, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, Y.; Sasaki, Y.; Horimoto, M.; Wada, S.; Tanaka, Y.; Kasahara, A.; Ueki, T.; Hirano, T.; Yamamoto, H.; Fujimoto, J.; et al. Activation of mitogen-activated protein kinases/extracellular signal-regulated kinases in human hepatocellular carcinoma. Hepatology 1998, 27, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Hisamoto, T.; Akiba, J.; Koga, H.; Nakamura, K.; Tokunaga, Y.; Hanada, S.; Kumemura, H.; Maeyama, M.; Harada, M.; et al. Spreds, inhibitors of the Ras/ERK signal transduction, are dysregulated in human hepatocellular carcinoma and linked to the malignant phenotype of tumors. Oncogene 2006, 25, 6056–6066. [Google Scholar] [CrossRef] [Green Version]
- Vareedayah, A.A.; Alkaade, S.; Taylor, J.R. Pancreatic Adenocarcinoma. MO. Med. 2018, 115, 230–235. [Google Scholar]
- Jonckheere, N.; Vasseur, R.; Van Seuningen, I. The cornerstone K-RAS mutation in pancreatic adenocarcinoma: From cell sig-naling network, target genes, biological processes to therapeutic targeting. Crit. Rev. Oncol. Hematol. 2017, 111, 7–19. [Google Scholar] [CrossRef]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 2140–2141. [Google Scholar] [CrossRef]
- Neuzillet, C.; Hammel, P.; Tijeras-Raballand, A.; Couvelard, A.; Raymond, E. Targeting the Ras–ERK pathway in pancreatic adenocarcinoma. Cancer Metastasis Rev. 2012, 32, 147–162. [Google Scholar] [CrossRef]
- Drosten, M.; Barbacid, M. Targeting the MAPK Pathway in KRAS-Driven Tumors. Cancer Cell 2020, 37, 543–550. [Google Scholar] [CrossRef]
- Goebel, C.; Louden, C.L.; McKenna, R.; Onugha, O.; Wachtel, A.; Long, T. Diagnosis of Non-small Cell Lung Cancer for Early Stage Asymptomatic Patients. Cancer Genom. Proteom. 2019, 16, 229–244. [Google Scholar] [CrossRef] [Green Version]
- Kerr, E.M.; Gaude, E.; Turrell, F.K.; Frezza, C.; Martins, C.P. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nat. Cell Biol. 2016, 531, 110–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ricciuti, B.; Nguyen, T.; Li, X.; Rabin, M.S.; Awad, M.M.; Lin, X.; Johnson, B.E.; Christiani, D.C. Association between Smoking History and Tumor Mutation Burden in Advanced Non–Small Cell Lung Cancer. Cancer Res. 2021, 81, 2566–2573. [Google Scholar] [CrossRef] [PubMed]
- Dogan, S.; Shen, R.; Ang, D.C.; Johnson, M.L.; D’Angelo, S.P.; Paik, P.K.; Brzostowski, E.B.; Riely, G.J.; Kris, M.; Zakowski, M.F.; et al. Molecular Epidemiology of EGFR and KRAS Mutations in 3,026 Lung Adenocarcinomas: Higher Susceptibility of Women to Smoking-Related KRAS-Mutant Cancers. Clin. Cancer Res. 2012, 18, 6169–6177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihle, N.T.; Byers, L.A.; Kim, E.S.; Saintigny, P.; Lee, J.J.; Blumenschein, G.R.; Tsao, A.; Liu, S.; Larsen, J.; Wang, J.; et al. Effect of KRAS Oncogene Substitutions on Protein Behavior: Implications for Signaling and Clinical Outcome. J. Natl. Cancer Inst. 2012, 104, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Yuan, M.; Li, X.; Chen, L.; Yang, J.; Zhao, X.; Ma, W.; Xin, J. Prognostic value of K-RAS mutations in patients with non-small cell lung cancer: A systematic review with meta-analysis. Lung Cancer 2013, 81, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Sima, C.S.; Shen, R.; Kass, S.; Gainor, J.; Shaw, A.; Hames, M.; Iams, W.; Aston, J.; Lovly, C.; et al. Prognostic Impact of KRAS Mutation Subtypes in 677 Patients with Metastatic Lung Adenocarcinomas. J. Thorac. Oncol. 2015, 10, 431–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepherd, F.A.; Domerg, C.; Hainaut, P.; Jänne, P.A.; Pignon, J.-P.; Graziano, S.; Douillard, J.-Y.; Brambilla, E.; Le Chevalier, T.; Seymour, L.; et al. Pooled Analysis of the Prognostic and Predictive Effects of KRAS Mutation Status and KRAS Mutation Subtype in Early-Stage Resected Non–Small-Cell Lung Cancer in Four Trials of Adjuvant Chemotherapy. J. Clin. Oncol. 2013, 31, 2173–2181. [Google Scholar] [CrossRef] [Green Version]
- Glodde, N.; Hölzel, M. RAS and PD-L1: A Masters’ Liaison in Cancer Immune Evasion. Immunity 2017, 47, 1007–1009. [Google Scholar] [CrossRef] [Green Version]
- Coelho, M.A.; Trécesson, S.D.C.; Rana, S.; Zecchin, D.; Moore, C.; Molina-Arcas, M.; East, P.; Spencer-Dene, B.; Nye, E.; Barnouin, K.; et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity 2017, 47, 1083–1099.e6. [Google Scholar] [CrossRef] [Green Version]
- Zdanov, S.; Mandapathil, M.; Abu Eid, R.; Adamson-Fadeyi, S.; Wilson, W.; Qian, J.; Carnie, A.; Tarasova, N.; Mkrtichyan, M.; Berzofsky, J.A.; et al. Mutant KRAS Conversion of Conventional T Cells into Regulatory T Cells. Cancer Immunol. Res. 2016, 4, 354–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF–MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalili, J.S.; Liu, S.; Rodríguez-Cruz, T.G.; Whittington, M.; Wardell, S.; Liu, C.; Zhang, M.; Cooper, Z.; Frederick, D.T.; Li, Y.; et al. Oncogenic BRAF(V600E) Promotes Stromal Cell-Mediated Immunosuppression Via Induction of Interleukin-1 in Melanoma. Clin. Cancer Res. 2012, 18, 5329–5340. [Google Scholar] [CrossRef] [Green Version]
- Boni, A.; Cogdill, A.; Dang, P.; Udayakumar, D.; Njauw, C.-N.J.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E Inhibition Enhances T-Cell Recognition of Melanoma without Affecting Lymphocyte Function. Cancer Res. 2010, 70, 5213–5219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, S.; Melendez, B.; Talukder, A.; Lizée, G. Trouble at the core: BRAF(V600E) drives multiple modes of T-cell suppression in melanoma. Oncoimmunology 2015, 5, e1078966. [Google Scholar] [CrossRef] [Green Version]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Miller, W.H.; Carlino, M.S.; Fisher, R.; Long, G.V.; Hodi, F.S.; Tsoi, J.; et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat. Med. 2019, 25, 936–940. [Google Scholar] [CrossRef]
- Pratilas, C.A.; Hanrahan, A.J.; Halilovic, E.; Persaud, Y.; Soh, J.; Chitale, D.; Shigematsu, H.; Yamamoto, H.; Sawai, A.; Janakiraman, M.; et al. Genetic Predictors of MEK Dependence in Non–Small Cell Lung Cancer. Cancer Res. 2008, 68, 9375–9383. [Google Scholar] [CrossRef] [Green Version]
- Karreth, F.A.; Frese, K.K.; DeNicola, G.; Baccarini, M.; Tuveson, D.A. C-Raf Is Required for the Initiation of Lung Cancer by K-RasG12D. Cancer Discov. 2011, 1, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Blasco, R.B.; Francoz, S.; Santamaría, D.; Cañamero, M.; Dubus, P.; Charron, J.; Baccarini, M.; Barbacid, M. c-Raf, but Not B-Raf, Is Essential for Development of K-Ras Oncogene-Driven Non-Small Cell Lung Carcinoma. Cancer Cell 2011, 19, 652–663. [Google Scholar] [CrossRef] [Green Version]
- Takezawa, K.; Okamoto, I.; Yonesaka, K.; Hatashita, E.; Yamada, Y.; Fukuoka, M.; Nakagawa, K. Sorafenib Inhibits Non–Small Cell Lung Cancer Cell Growth by Targeting B-RAF in KRAS Wild-Type Cells and C-RAF in KRAS Mutant Cells. Cancer Res. 2009, 69, 6515–6521. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, M.H.; Gmachl, M.; Ramharter, J.; Savarese, F.; Gerlach, D.; Marszalek, J.R.; Sanderson, M.P.; Kessler, D.; Trapani, F.; Arnhof, H.; et al. BI-3406, a Potent and Selective SOS1–KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers through Combined MEK Inhibition. Cancer Discov. 2021, 11, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Koczywas, M.; Haura, E.; Janne, P.A.; Pacheco, J.M.; Ulahannan, S.; Wang, J.S.; Burris, H.A.; Riess, J.W.; McCoach, C.; Gordon, M.S.; et al. Abstract LB001: Anti-tumor activity and tolerability of the SHP2 inhibitor RMC-4630 as a single agent in patients with RAS-addicted solid cancers. Cancer Res. 2021, 81. [Google Scholar] [CrossRef]
- Karp, J.E.; Lancet, J.E. Development of the farnesyltransferase inhibitor tipifarnib for therapy of hematologic malignancies. Futur. Oncol. 2005, 1, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Hidalgo, M.; Canon, J.; Macarulla, T.; Bazin, I.; Poddubskaya, E.; Manojlovic, N.; Radenkovic, D.; Verslype, C.; Raymond, E.; et al. Phase I/II trial of pimasertib plus gemcitabine in patients with metastatic pancreatic cancer. Int. J. Cancer 2018, 143, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.L.; Brana, I.; Haddad, R.; Bauman, J.; Bible, K.; Oosting, S.; Wong, D.J.; Ahn, M.-J.; Boni, V.; Even, C.; et al. Tipifarnib in Head and Neck Squamous Cell Carcinoma with HRAS Mutations. J. Clin. Oncol. 2021, 39, 1856–1864. [Google Scholar] [CrossRef]
- Untch, B.R.; Dos Anjos, V.; Garcia-Rendueles, M.E.R.; Knauf, J.A.; Krishnamoorthy, G.P.; Saqcena, M.; Bhanot, U.K.; Socci, N.D.; Ho, A.L.; Ghossein, R.; et al. Tipifarnib Inhibits HRAS-Driven Dedifferentiated Thyroid Cancers. Cancer Res. 2018, 78, 4642–4657. [Google Scholar] [CrossRef] [Green Version]
- Dunnett-Kane, V.; Nicola, P.; Blackhall, F.; Lindsay, C. Mechanisms of Resistance to KRAS(G12C) Inhibitors. Cancers 2021, 13, 151. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.-C.; Mansour, J.C.; Mollaee, M.; Wagner, K.-U.; Koduru, P.; Yopp, A.C.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Sakamoto, K.; Kamada, Y.; Sameshima, T.; Yaguchi, M.; Niida, A.; Sasaki, S.; Miwa, M.; Ohkubo, S.; Sakamoto, J.-I.; Kamaura, M.; et al. K-Ras(G12D)-selective inhibitory peptides generated by random peptide T7 phage display technology. Biochem. Biophys. Res. Commun. 2017, 484, 605–611. [Google Scholar] [CrossRef]
- Carlino, M.S.; Kwan, V.; Miller, D.K.; Saunders, C.A.; Yip, D.; Nagrial, A.M.; Tomlinson, J.; Grimmond, S.M.; Scolyer, R.A.; Kefford, R.F.; et al. New RAS-Mutant Pancreatic Adenocarcinoma with Combined BRAF and MEK Inhibition for Metastatic Melanoma. J. Clin. Oncol. 2015, 33, e52–e56. [Google Scholar] [CrossRef] [Green Version]
- Boussemart, L.; Girault, I.; Malka-Mahieu, H.; Mateus, C.; Routier, E.; Rubington, M.; Kamsu-Kom, N.; Thomas, M.; Tomasic, G.; Agoussi, S.; et al. Secondary Tumors Arising in Patients Undergoing BRAF Inhibitor Therapy Exhibit Increased BRAF–CRAF Heterodimerization. Cancer Res. 2016, 76, 1476–1484. [Google Scholar] [CrossRef] [Green Version]
- Mellema, W.W.; Burgers, S.A.; Smit, E.F. Tumor flare after start of RAF inhibition in KRAS mutated NSCLC: A case report. Lung Cancer 2015, 87, 201–203. [Google Scholar] [CrossRef]
- Miyauchi, S.; Shien, K.; Takeda, T.; Araki, K.; Nakata, K.; Miura, A.; Takahashi, Y.; Kurihara, E.; Ogoshi, Y.; Namba, K.; et al. Antitumor Effects of Pan-RAF Inhibitor LY3009120 Against Lung Cancer Cells Harboring Oncogenic BRAF Mutation. Anticancer Res. 2020, 40, 2667–2673. [Google Scholar] [CrossRef]
- Semrad, T.J.; Gandara, D.R.; Lara, P.N. Enhancing the clinical activity of sorafenib through dose escalation: Rationale and current experience. Ther. Adv. Med. Oncol. 2011, 3, 95–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.; Liu, H.; Ming, L. Multiple Roles of Autophagy in the Sorafenib Resistance of Hepatocellular Carcinoma. Cell. Physiol. Biochem. 2017, 44, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Choi, G.H.; Jung, Y.; Kim, K.M.; Jang, S.-J.; Yu, E.S.; Lee, H.C. Downregulation of Raf-1 kinase inhibitory protein as a sorafenib resistance mechanism in hepatocellular carcinoma cell lines. J. Cancer Res. Clin. Oncol. 2018, 144, 1487–1501. [Google Scholar] [CrossRef]
- Ye, L.; Mayerle, J.; Ziesch, A.; Reiter, F.P.; Gerbes, A.L.; De Toni, E.N. The PI3K inhibitor copanlisib synergizes with sorafenib to induce cell death in hepatocellular carcinoma. Cell Death Discov. 2019, 5, 86. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Zhao, C.-R.; Yin, H.; Wang, K.; Gao, J.-J. Synergistic antitumor activity of sorafenib and artesunate in hepatocellular carcinoma cells. Acta Pharmacol. Sin. 2020, 41, 1609–1620. [Google Scholar] [CrossRef]
- Gedaly, R.; Angulo, P.; Hundley, J.; Daily, M.; Chen, C.; Koch, A.; Evers, B.M. PI-103 and sorafenib inhibit hepatocellular carcinoma cell proliferation by blocking Ras/Raf/MAPK and PI3K/AKT/mTOR pathways. Anticancer Res. 2010, 30, 4951–4958. [Google Scholar]
- Gedaly, R.; Angulo, P.; Hundley, J.; Daily, M.; Chen, C.; Evers, B.M. PKI-587 and Sorafenib Targeting PI3K/AKT/mTOR and Ras/Raf/MAPK Pathways Synergistically Inhibit HCC Cell Proliferation. J. Surg. Res. 2012, 176, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Daouti, S.; Li, W.H.; Wen, Y.; Rizzo, C.; Higgins, B.; Packman, K.; Rosen, N.; Boylan, J.F.; Heimbrook, D.; et al. Identification of the MEK1(F129L) activating mutation as a potential mechanism of acquired resistance to MEK inhi-bition in human cancers carrying the B-RafV600E mutation. Cancer Res. 2011, 71, 5535–5545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, K.; Bilal, F.; Morales, C.B.; Salcedo, M.T.; Macarulla, T.; Massó-Vallés, D.; Mohan, V.; Vivancos, A.; Carreras, M.-J.; Serres-Créixams, X.; et al. Pancreatic cancer heterogeneity and response to Mek inhibition. Oncogene 2017, 36, 5639–5647. [Google Scholar] [CrossRef] [PubMed]
- Kun, E.; Tsang, Y.; Ng, C.; Gershenson, D.; Wong, K. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat. Rev. 2021, 92, 102137. [Google Scholar] [CrossRef] [PubMed]
- Hayes, T.K.; Neel, N.F.; Hu, C.; Gautam, P.; Chenard, M.; Long, B.; Aziz, M.; Kassner, M.; Bryant, K.L.; Pierobon, M.; et al. Long-Term ERK Inhibition in KRAS-Mutant Pancreatic Cancer Is Associated with MYC Degradation and Senes-cence-like Growth Suppression. Cancer Cell 2016, 29, 75–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lake, D.; Correa, S.A.; Muller, J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell. Mol. Life Sci. 2016, 73, 4397–4413. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Ohuchida, K.; Fei, S.; Zheng, B.; Guan, W.; Feng, H.; Kibe, S.; Ando, Y.; Koikawa, K.; Abe, T.; et al. Inhibition of ERK1/2 in cancer-associated pancreatic stellate cells suppresses cancer-stromal interaction and metastasis. J. Exp. Clin. Cancer Res. 2019, 38, 221. [Google Scholar] [CrossRef]
- Bhagwat, S.V.; McMillen, W.T.; Cai, S.; Zhao, B.; Whitesell, M.; Shen, W.; Kindler, L.; Flack, R.S.; Wu, W.; Anderson, B.; et al. ERK Inhibitor LY3214996 Targets ERK Pathway–Driven Cancers: A Therapeutic Approach Toward Precision Medicine. Mol. Cancer Ther. 2019, 19, 325–336. [Google Scholar] [CrossRef] [Green Version]
- Hatzivassiliou, G.; Liu, B.; O’Brien, C.; Spoerke, J.M.; Hoeflich, K.P.; Haverty, P.M.; Soriano, R.; Forrest, W.F.; Heldens, S.; Chen, H.; et al. ERK Inhibition Overcomes Acquired Resistance to MEK Inhibitors. Mol. Cancer Ther. 2012, 11, 1143–1154. [Google Scholar] [CrossRef] [Green Version]
- Ning, C.; Liang, M.; Liu, S.; Wang, G.; Edwards, H.; Xia, Y.; Polin, L.; Dyson, G.; Taub, J.W.; Mohammad, R.M.; et al. Targeting ERK enhances the cytotoxic effect of the novel PI3K and mTOR dual inhibitor VS-5584 in preclinical models of pancreatic cancer. Oncotarget 2017, 8, 44295–44311. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Kong, X.; Ribas, A.; Lo, R.S. Combinatorial treatments that overcome PDGFRbeta-driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer Res. 2011, 71, 5067–5074. [Google Scholar] [CrossRef] [Green Version]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Dudek-Perić, A.M.; Maes, H.; Garg, A.D.; Gabrysiak, M.; Demirsoy, S.; Swinnen, J.V.; Agostinis, P. Concurrent MEK and autophagy inhibition is required to restore cell death associated danger-signalling in Vemuraf-enib-resistant melanoma cells. Biochem. Pharmacol. 2015, 93, 290–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Zhou, S.; Xia, H.; Yu, H.; Tang, Q.; Bi, F. MEK nuclear localization promotes YAP stability via sequestering beta-TrCP in KRAS mutant cancer cells. Cell Death Differ. 2019, 26, 2400–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, J.H.; Gao, Y.; Soucheray, M.; Pulido, I.; Kikuchi, E.; Rodríguez, M.L.; Gandhi, R.; Lafuente-Sanchis, A.; Aupí, M.; Fernández-Coronado, J.A.; et al. CXCR7 Reactivates ERK Signaling to Promote Resistance to EGFR Kinase Inhibitors in NSCLC. Cancer Res. 2019, 79, 4439–4452. [Google Scholar] [CrossRef]
- Neto, J.M.F.; Nadal, E.; Bosdriesz, E.; Ooft, S.N.; Farre, L.; McLean, C.; Klarenbeek, S.; Jurgens, A.; Hagen, H.; Wang, L.; et al. Multiple low dose therapy as an effective strategy to treat EGFR inhibitor-resistant NSCLC tumours. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Ray, P.; Dutta, D.; Haque, I.; Nair, G.; Mohammed, J.; Parmer, M.; Kale, N.; Orr, M.; Jain, P.; Banerjee, S.; et al. pH-Sensitive Nanodrug Carriers for Codelivery of ERK Inhibitor and Gemcitabine Enhance the Inhibition of Tumor Growth in Pancreatic Cancer. Mol. Pharm. 2021, 18, 87–100. [Google Scholar] [CrossRef]
- Mao, J.; Yang, H.; Cui, T.; Pan, P.; Kabir, N.; Chen, D.; Ma, J.; Chen, X.; Chen, Y.; Yang, Y. Combined treatment with sorafenib and silibinin synergistically targets both HCC cells and cancer stem cells by enhanced inhibition of the phosphorylation of STAT3/ERK/AKT. Eur. J. Pharmacol. 2018, 832, 39–49. [Google Scholar] [CrossRef]
- Bian, Y.; Guo, D. Targeted Therapy for Hepatocellular Carcinoma: Co-Delivery of Sorafenib and Curcumin Using Lactosylated pH-Responsive Nanoparticles. Drug Des. Dev. Ther. 2020, 14, 647–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawazoe, T.; Taniguchi, K. The Sprouty/Spred family as tumor suppressors: Coming of age. Cancer Sci. 2019, 110, 1525–1535. [Google Scholar] [CrossRef] [Green Version]
- Momeny, M.; Khorramizadeh, M.R.; Ghaffari, S.H.; Yousefi, M.; Yekaninejad, M.S.; Esmaeili, R.; Jahanshiri, Z.; Nooridaloii, M.R. Effects of silibinin on cell growth and invasive properties of a human hepatocellular carcinoma cell line, HepG-2, through inhibition of extracellular signal-regulated kinase 1/2 phosphorylation. Eur. J. Pharmacol. 2008, 591, 13–20. [Google Scholar] [CrossRef]
- Varghese, L.; Agarwal, C.; Tyagi, A.; Singh, R.P.; Agarwal, R. Silibinin Efficacy against Human Hepatocellular Carcinoma. Clin. Cancer Res. 2005, 11, 8441–8448. [Google Scholar] [CrossRef] [Green Version]
- Ailioaie, L.M.; Litscher, G. Curcumin and Photobiomodulation in Chronic Viral Hepatitis and Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 7150. [Google Scholar] [CrossRef] [PubMed]
- Abdelmoaty, A.A.A.; Zhang, P.; Lin, W.; Fan, Y.J.; Ye, S.N.; Xu, J.H. C0818, a novel curcumin derivative, induces ROS-dependent cytotoxicity in human hepatocellular car-cinoma cells in vitro via disruption of Hsp90 function. Acta Pharmacol. Sin. 2021. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Brentjens, R.; Rivière, I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Miao, L.; Zhang, Y.; Huang, L. mRNA vaccine for cancer immunotherapy. Mol. Cancer 2021, 20, 1–23. [Google Scholar] [CrossRef]
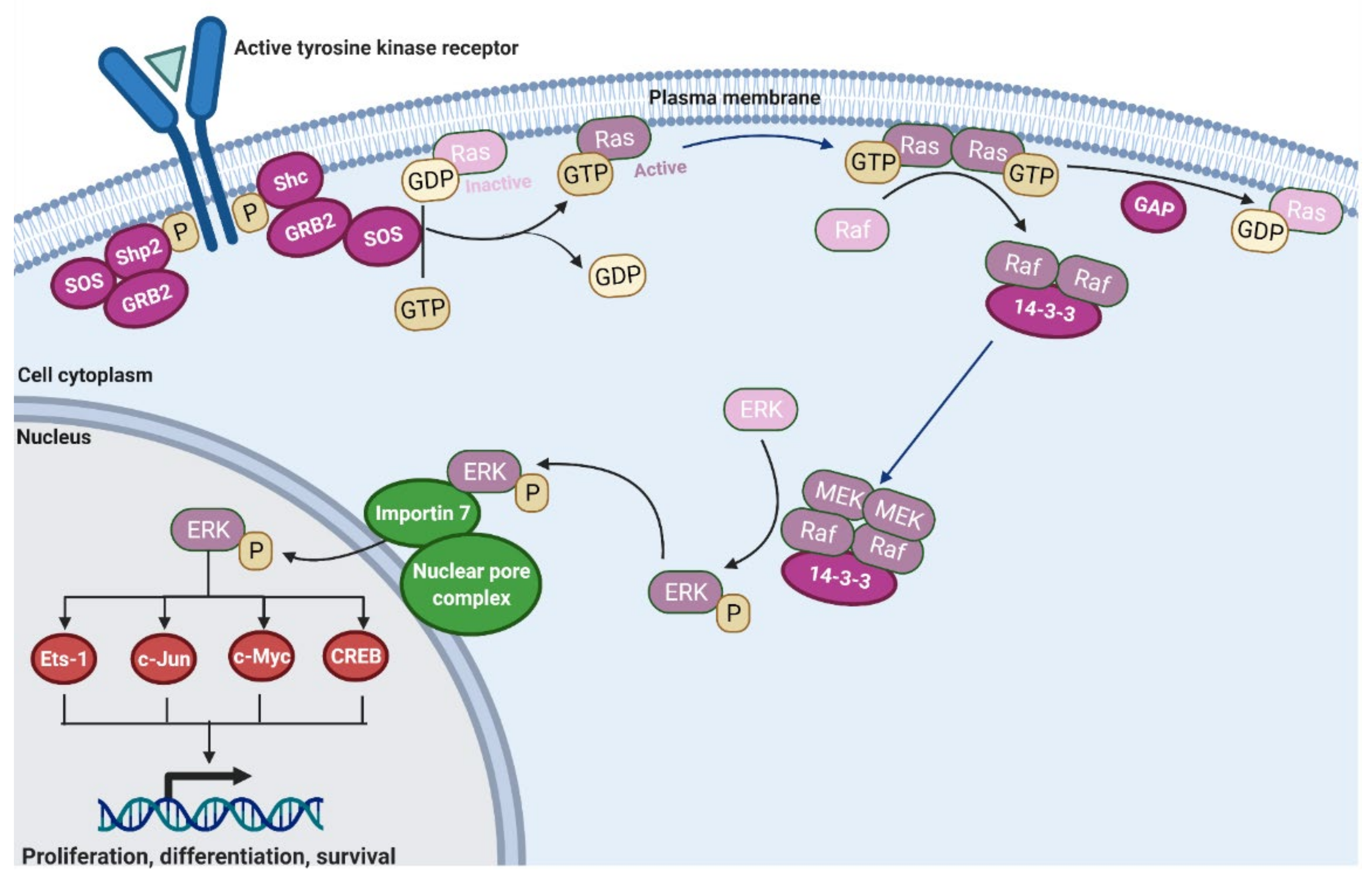


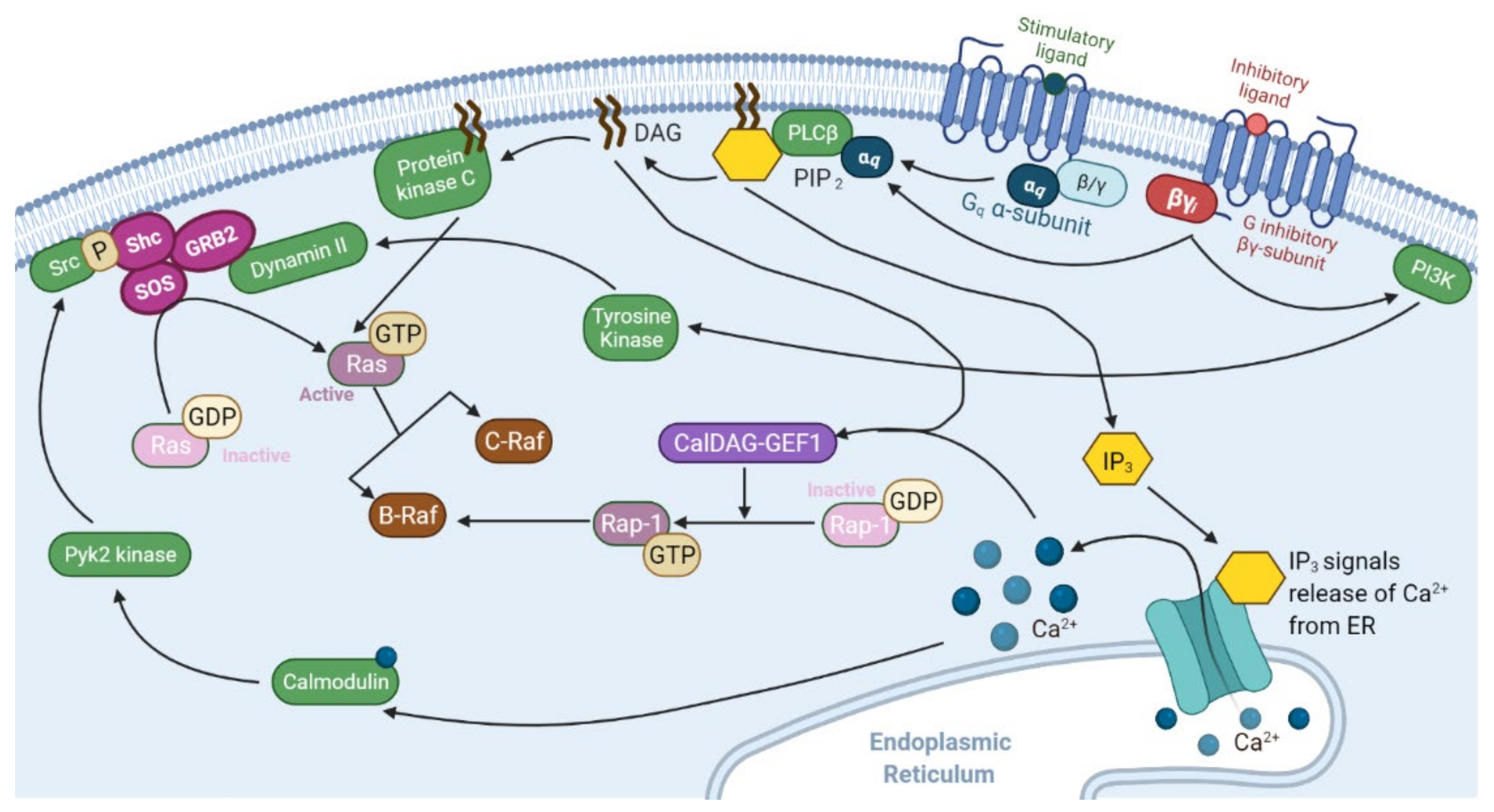
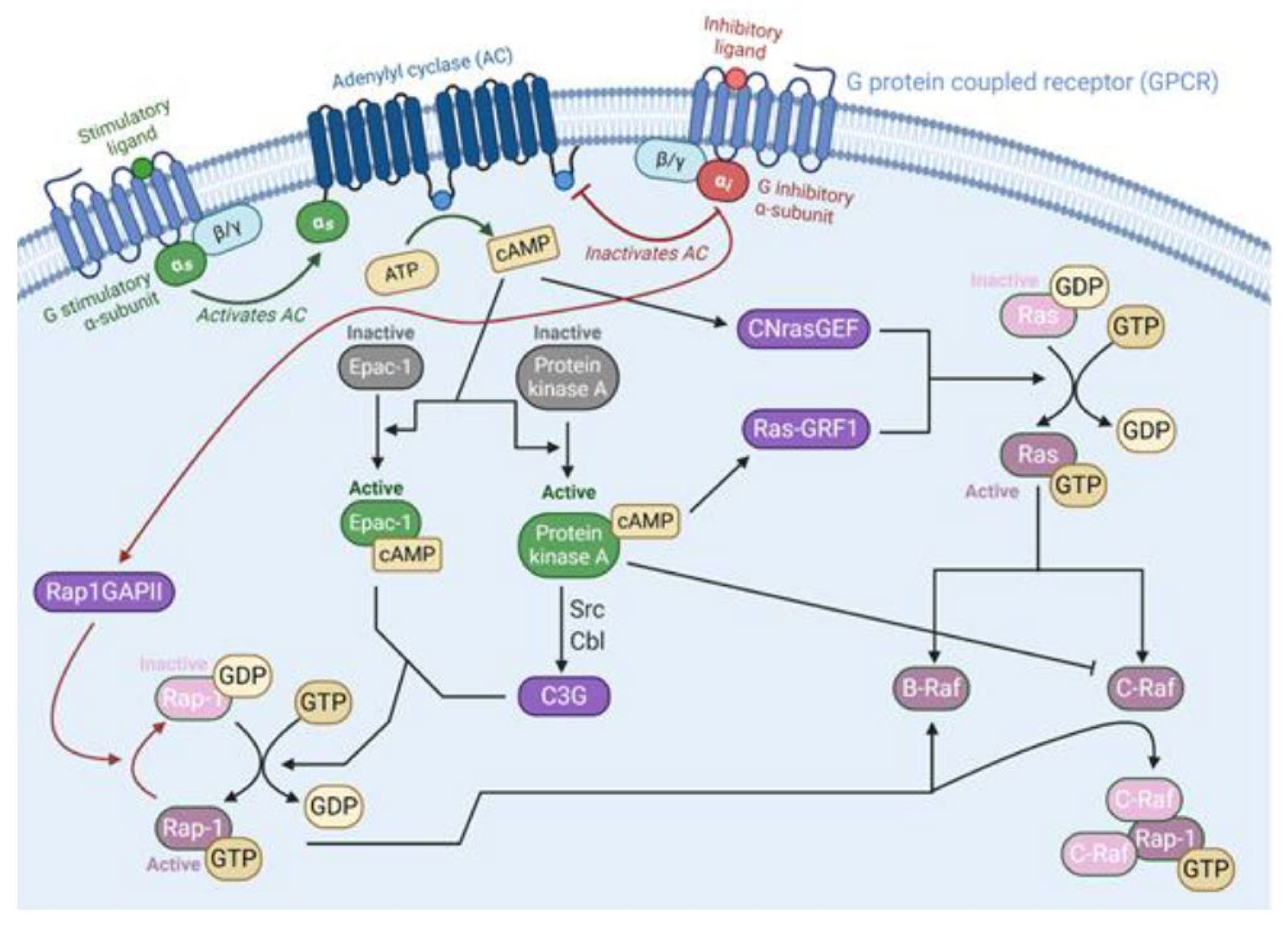

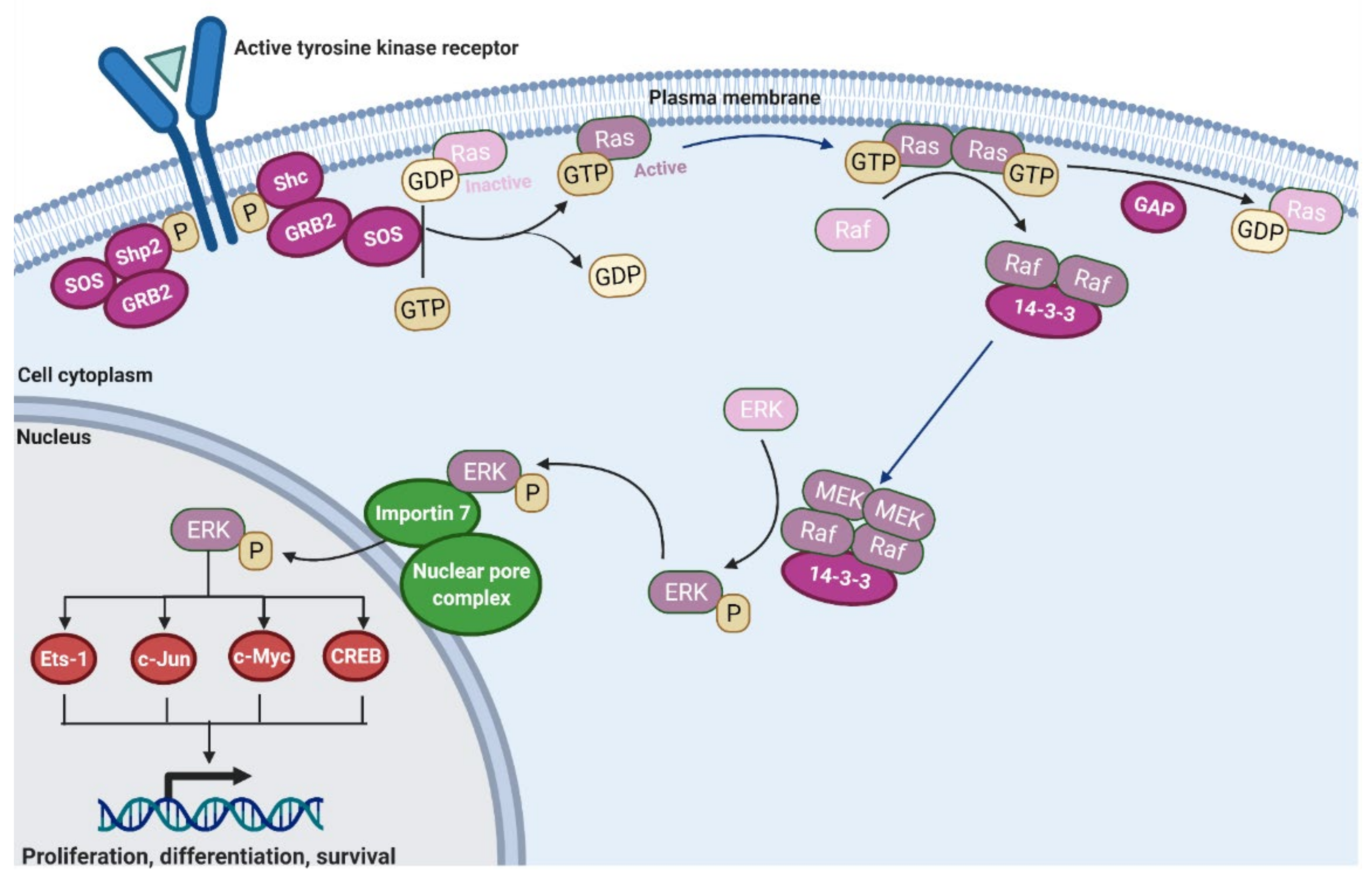{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Blood Cancer | Ras-Raf Pathology | General Treatment Protocol | 5-Year Survival | References |
|---|---|---|---|---|
| Myelodysplastic Syndromes | NRas, KRas mutation in 7–48% of patients. | Use of erythropoietin stimulating agents to mitigate symptoms. Allogeneic stem cell transplant for higher risk patients. | 29% | [163,164,165,166,167] |
| Acute Myeloid Leukemia | NRas, KRas mutations in 10–27% of de novo patients. | Induction via cytarabine and the addition of an anthracycline for patients followed by consolidation. | 24% | [167,168,169,170] |
| Acute Lymphoblastic Leukemia | NRas, KRas mutations in 5–22% of patients. BRaf mutations have been found in infants and children with acute B and T-cell lymphoblastic leukemia. Small sample found ~21% of ALL patients with BRaf mutations. | Induction using vincristine, corticosteroids, L asparaginase, and an anthracycline for patients followed by consolidation. | Between 30 and 45% | [171,172,173,174] |
| Chronic Myelomonocytic Leukemia | NRas, KRas mutations in 30–50% with CMML. Subset of patients with CMML-1 presented BRaf mutations (~7%). | Lack of consensus about a treatment that markedly expands overall survival rate. Hypomethylating agents have shown some promise and are approved by the FDA for CMML. Allogeneic stem cell transplantation regarded as only curative treatment. | 18.5% | [175,176,177,178,179] |
| Chronic Myeloid Leukemia | NRas mutations in up to 1/3 of atypical CML. Chronic CML patients present up to 17% of Ras mutations, up to 58% of acute CML patients have Ras mutations. | Standard treatment involves the use of tyrosine kinase inhibitors in sequence. | 61% | [166,169,180,181] |
| Solid Cancer | Ras-Raf Pathology | General Treatment Protocol | 5-Year Survival Rate | References |
|---|---|---|---|---|
| Pancreatic Adenocarcinoma | KRas mutations in up to 90% of patients. BRaf mutation in 14% of patients. | Surgical resection and/or chemotherapy. | less than 5%. | [166,182,183,184,185] |
| Melanoma | Ras mutations in up to 36% of patients. BRaf mutations in 27–70% of patients. | Surgical resection. Chemotherapy and novel targeted therapies, including BRaf inhibitors, may be used when surgical resection is not possible. | 91%. | [169,186,187,188,189] |
| Non-Small Cell Lung Cancer | KRas mutations in 22–36% of patients. BRaf in ~2 to 5% of patients. | Surgical resection and/or chemotherapy. | 25% | [190,191,192,193,194,195] |
| Colorectal Cancer | KRas mutations in 40–60% of patients. BRaf mutation in 18% of patients. | Surgical resection and/or chemotherapy. | 65% | [169,187,196,197] |
| Seminoma | KRas, NRas mutations in 7–40% of patients. | Radical orchiectomy with subsequent chemotherapy. | 86.4% | [166,198,199,200] |
| Bladder Cancer | HRas, NRas mutations shown in up to 80% of patients, although Ras mutations generally considered present in 10% of patients. | Immunotherapy and/or chemotherapy, with radical cystectomy after invasion of muscle. | 80.8% | [201,202,203,204] |
| Hepatocellular Carcinoma | NRas mutations in 30%, KRas mutations in 1.6% of patients. BRaf mutations in 14% of patients. | Surgical resection, liver transplantation, and/or chemotherapy. | 15% | [156,166,187,205,206] |
| Ovarian Cancer | KRas mutation in 13.7% of patients. BRaf mutations in 4–30% of patients | Cytoreductive surgery followed by chemotherapy. | 40% | [187,188,207,208] |
| Renal Cell Carcinoma | Ras mutations in up to 13% of patients. | Tyrosine kinase, m-TOR, and VEGF inhibitors. Other targeting therapies including immunotherapy are used as well. | Between 92.5 and 12% depending on localization. | [166,209,210,211] |
| Inhibitor | Notes | Clinical Trials | Targeted Cancers |
|---|---|---|---|
| BI 1701963 | Binds SOS1 protein, inhibiting its activation of KRas | NCT04111458, NCT04975256, NCT04835714 | Lung, colon, and lung cancer |
| RMC-4630 | Selective inhibitor of Shp2, indirectly inhibiting KRas | NCT03634982, NCT03989115, NCT04916236 | Pancreatic, colorectal, and non-small cell lung cancer |
| TNO155 | Selective inhibitor of Shp2, indirectly inhibiting KRas | NCT04330664, NCT04292119, NCT04000529 | Lung, head and neck, esophageal, gastrointestinal and colorectal cancer |
| BBP-398 | Selective inhibitor of Shp2, indirectly inhibiting KRas | NCT04528836 | Advanced solid tumors |
| Sorafenib | Inhibitor of multiple intracellular and cell surface kinases such as CRaf and BRaf that are involved in tumor cell signaling, angiogenesis, and apoptosis | NCT01730937, NCT03518502, NCT01371981 | Liver and thyroid cancer, leukemias |
| Sotorasib (AMG 510) | KRas inhibition specific to G12C mutation | NCT03600883, NCT04303780, NCT04933695 | Non-small cell lung cancer |
| MRTX849 | KRas inhibition selective to the G12C mutation | NCT04793958, NCT04685135 | Non-small cell lung and colorectal cancer |
| JAB-21822 | KRas inhibition selective to the G12C mutation | NCT05009329, NCT05002270 | Non-small cell lung and colorectal cancer |
| GFH925 | KRas inhibition selective to the G12C mutation | NCT05005234 | Advanced solid tumors |
| LY3537982 | KRas inhibition selective to the G12C mutation | NCT04956640 | Non-small cell lung, colorectal, endometrial, ovarian, and pancreatic cancer |
| Tipifarnib (R115777) | Farnesyltransferase inhibitor that prevents post-translational processing of Ras proteins. Prevents Ras from membrane binding | NCT03496766, NCT04997902, NCT03155620 | Non-small cell lung and head and neck cancer, non-Hodgkin lymphoma |
| Rigosertib | Targets mutated Ras pathway in leukemia by interacting with effectors proteins containing Ras binding domains | NCT04263090, NCT04263090, NCT03786237 | Myelodysplastic syndrome, non-small cell lung cancer, and squamous cell carcinoma |
| Trametinib (GSK1120212) | Non-ATP-competitive inhibitor of MEK1/2. FDA approved, suggested to use in combination with BRaf inhibitors due to resistance. | NCT03340506, NCT04940052, NCT02101788 | Non-small cell lung, thyroid, ovarian and peritoneal cancer, melanoma |
| Mirdametinib (PD0325901) | MEK1/2 inhibitor (derivative of CI-1040) | NCT02022982, NCT03905148, NCT04923126 | Ovarian, endometrial, pancreatic, thyroid, and non-small cell lung cancer, melanoma, glioma |
| Selumetinib (AZD6244) | Highly selective, non-ATP-competitive MEK1 inhibitor. | NCT04576117, NCT03705507, NCT03705507 | Non-small cell lung cancer, glioma, leukemia |
| Binimetinib (MEKTOVI; MEK162) | Highly selective, non-ATP-competitive MEK1/2 inhibitor. | NCT04657991, NCT02928224, NCT03843775 | Colorectal cancer, melanoma, and other BRaf mutant malignancies |
| RO5126766 (CH5127566) | Selective Raf/MEK1/2 inhibitor | NCT02407509, NCT03875820, NCT04720417 | Non-small cell lung, ovarian, and colorectal cancer, multiple myeloma, melanoma |
| HL-085 | Selective MEK1 inhibitor | NCT03973151, NCT03990077, NCT03781219 | Non-small cell lung cancer, melanoma, and BRaf mutant solid cancers |
| Ulixertinib (BVD-523) | ATP-competitive, reversible inhibitor of ERK1/2 | NCT04145297, NCT03417739, NCT04488003 | Gastrointestinal cancers, melanoma, and BRaf mutant solid cancer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dillon, M.; Lopez, A.; Lin, E.; Sales, D.; Perets, R.; Jain, P. Progress on Ras/MAPK Signaling Research and Targeting in Blood and Solid Cancers. Cancers 2021, 13, 5059. https://doi.org/10.3390/cancers13205059
Dillon M, Lopez A, Lin E, Sales D, Perets R, Jain P. Progress on Ras/MAPK Signaling Research and Targeting in Blood and Solid Cancers. Cancers. 2021; 13(20):5059. https://doi.org/10.3390/cancers13205059
Chicago/Turabian StyleDillon, Martha, Antonio Lopez, Edward Lin, Dominic Sales, Ron Perets, and Pooja Jain. 2021. "Progress on Ras/MAPK Signaling Research and Targeting in Blood and Solid Cancers" Cancers 13, no. 20: 5059. https://doi.org/10.3390/cancers13205059
APA StyleDillon, M., Lopez, A., Lin, E., Sales, D., Perets, R., & Jain, P. (2021). Progress on Ras/MAPK Signaling Research and Targeting in Blood and Solid Cancers. Cancers, 13(20), 5059. https://doi.org/10.3390/cancers13205059