
Hepatocyte Polyploidy: Driver or Gatekeeper of Chronic Liver Diseases

 , and
, and 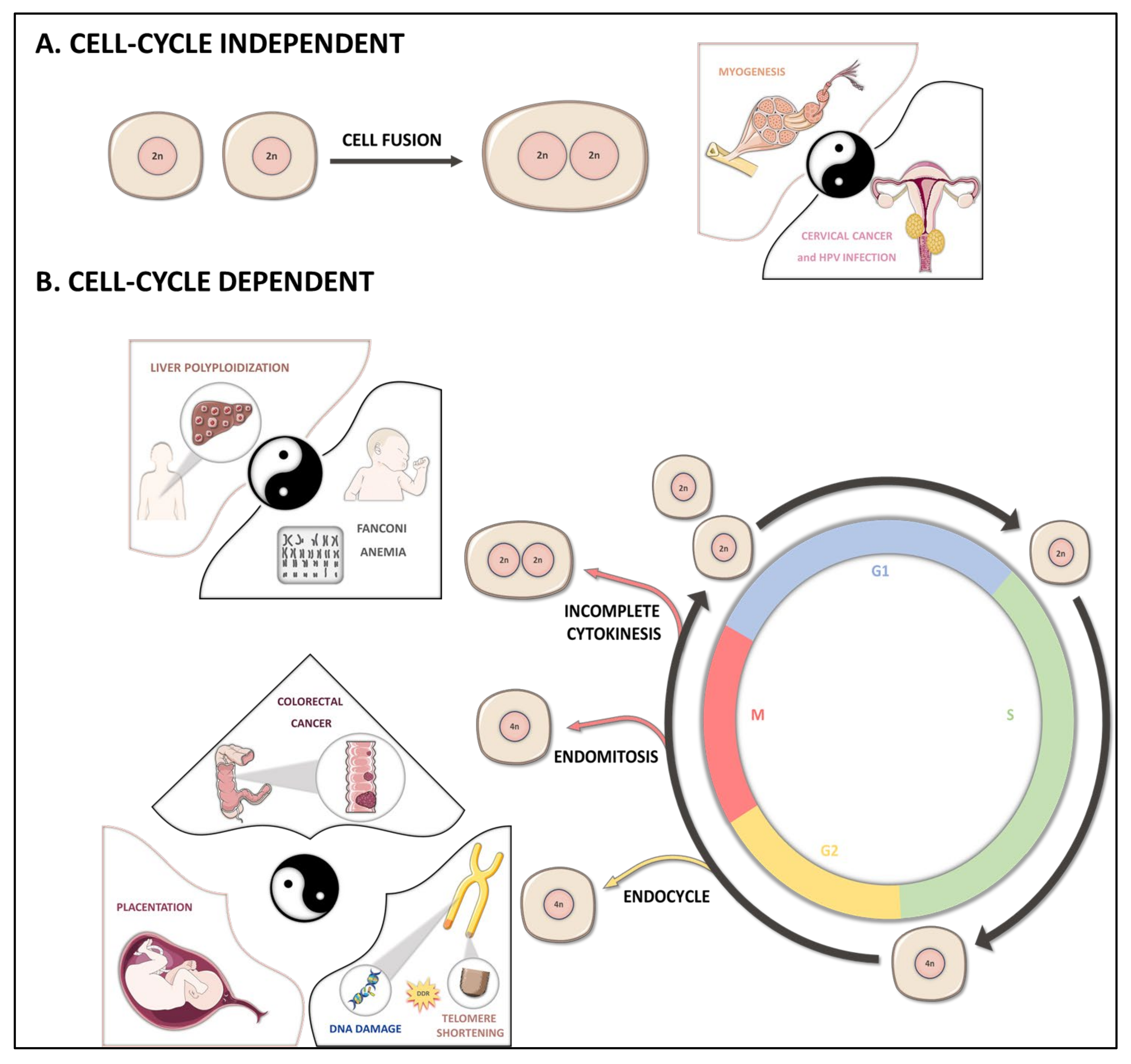{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Polyploidization Mechanisms
2.1. Cell Fusion
2.2. Endoreplication
2.3. Cytokinesis Failure
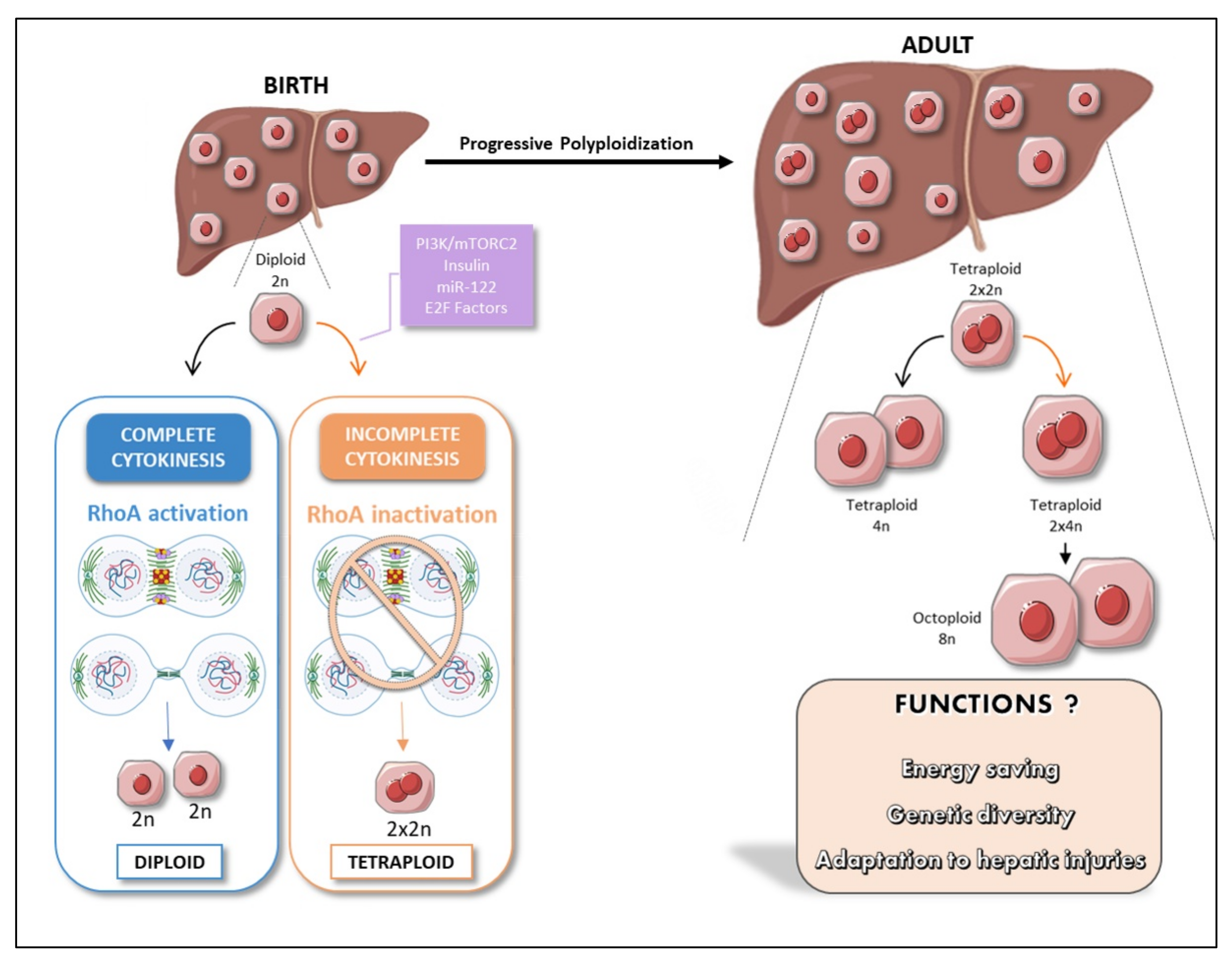
3. The Liver: A Singular Polyploid Organ
4. Polyploidy, an Alternative Cell Cycle Program during Development
5. Does Liver Polypaloidy Boost Hepatic Function?
6. Physiological Polyploidy in the Liver: A Strategy for Cellular Senescence?
7. Liver Polyploidy and Chronic Liver Disease
8. Polyploidy and Centrosome Amplification
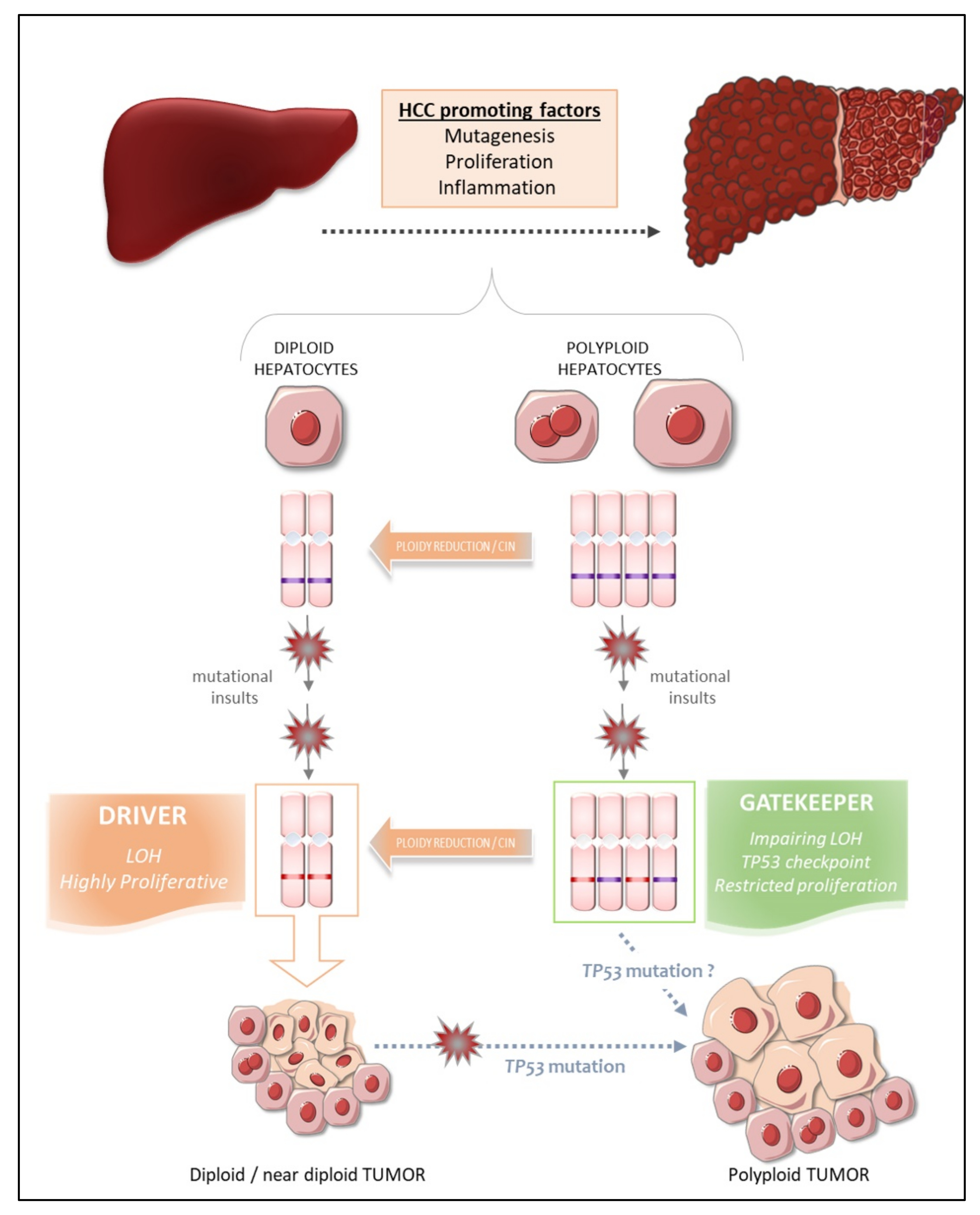
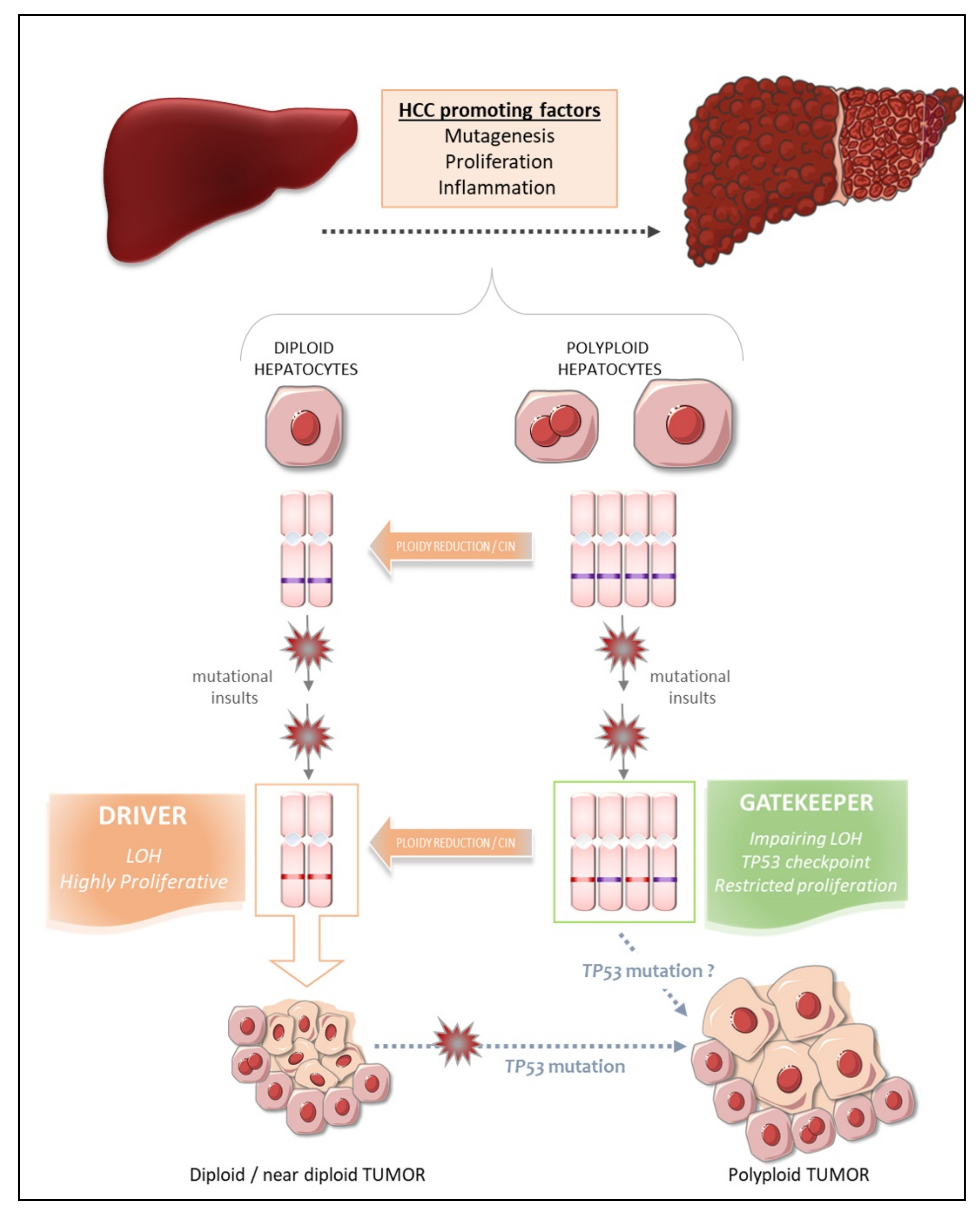
9. The Fate of Polyploid Hepatocytes during Liver Tumorigenesis
10. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Comai, L. The Advantages and Disadvantages of Being Polyploid. Nat. Rev. Genet. 2005, 6, 836–846. [Google Scholar] [CrossRef]
- Otto, S.P.; Whitton, J. Polyploid Incidence and Evolution. Annu. Rev. Genet. 2000, 34, 401–437. [Google Scholar] [CrossRef] [Green Version]
- Soltis, D.E.; Bell, C.D.; Kim, S.; Soltis, P.S. Origin and Early Evolution of Angiosperms. Ann. N. Y. Acad. Sci. 2008, 1133, 3–25. [Google Scholar] [CrossRef]
- Mable, B.; Alexandrou, M.A.; Taylor, M.I. Genome Duplication in Amphibians and Fish: An Extended Synthesis. J. Zool. 2011, 284, 151–182. [Google Scholar] [CrossRef]
- Van de Peer, Y.; Maere, S.; Meyer, A. The Evolutionary Significance of Ancient Genome Duplications. Nat. Rev. Genet. 2009, 10, 725–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The Evolutionary Significance of Polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Creasy, M.R.; Alberman, E.D. Congenital Malformations of the Central Nervous System in Spontaneous Abortions. J. Med. Genet. 1976, 13, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, M.H. Histochemical Identification of Primordial Germ Cells and Differentiation of the Gonads in Homozygous Tetraploid Mouse Embryos. J. Anat. 1991, 179, 169–181. [Google Scholar] [PubMed]
- Gallardo, M.H.; Bickham, J.W.; Honeycutt, R.L.; Ojeda, R.A.; Köhler, N. Discovery of Tetraploidy in a Mammal. Nature 1999, 401, 341. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; de Lange, T. The Causes and Consequences of Polyploidy in Normal Development and Cancer. Annu. Rev. Cell Dev. Biol. 2011, 27, 585–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, D.T.; Duronio, R.J. Endoreplication and Polyploidy: Insights into Development and Disease. Development 2013, 140, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovrebo, J.I.; Edgar, B.A. Polyploidy in Tissue Homeostasis and Regeneration. Development 2018, 145, dev156034. [Google Scholar] [CrossRef] [Green Version]
- Gjelsvik, K.J.; Besen-McNally, R.; Losick, V.P. Solving the Polyploid Mystery in Health and Disease. Trends Genet. 2019, 35, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Pandit, S.K.; Westendorp, B.; de Bruin, A. Physiological Significance of Polyploidization in Mammalian Cells. Trends Cell Biol. 2013, 23, 556–566. [Google Scholar] [CrossRef]
- Losick, V.P. Wound-Induced Polyploidy Is Required for Tissue Repair. Adv. Wound Care 2016, 5, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Losick, V.P.; Fox, D.T.; Spradling, A.C. Polyploidization and Cell Fusion Contribute to Wound Healing in the Adult Drosophila Epithelium. Curr. Biol. 2013, 23, 2224–2232. [Google Scholar] [CrossRef] [Green Version]
- Ganem, N.J.; Storchova, Z.; Pellman, D. Tetraploidy, Aneuploidy and Cancer. Curr. Opin. Genet. Dev. 2007, 17, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Lens, S.M.A.; Medema, R.H. Cytokinesis Defects and Cancer. Nat. Rev. Cancer 2019, 19, 32–45. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [Green Version]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhsng, C.-Z.; Wala, J.; Mermel, C.H.; et al. Pan-Cancer Patterns of Somatic Copy Number Alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [Green Version]
- Storchova, Z.; Pellman, D. From Polyploidy to Aneuploidy, Genome Instability and Cancer. Nat. Rev. Mol. Cell Biol. 2004, 5, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.G.; Ravid, K. Polyploidy: Mechanisms and cancer promotion in hematopoietic and other cells. In Polyploidization and Cancer; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2010; pp. 105–122. ISBN 978-1-4419-6198-3. [Google Scholar]
- Wangsa, D.; Braun, R.; Schiefer, M.; Gertz, E.M.; Bronder, D.; Quintanilla, I.; Padilla-Nash, H.M.; Torres, I.; Hunn, C.; Warner, L.; et al. The Evolution of Single Cell-Derived Colorectal Cancer Cell Lines Is Dominated by the Continued Selection of Tumor Specific Genomic Imbalances, Despite Random Chromosomal Instability. Carcinogenesis 2018, 39, 993–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donne, R.; Saroul-Aïnama, M.; Cordier, P.; Celton-Morizur, S.; Desdouets, C. Polyploidy in Liver Development, Homeostasis and Disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 1–15. [Google Scholar] [CrossRef]
- Duelli, D.; Lazebnik, Y. Cell-to-Cell Fusion as a Link between Viruses and Cancer. Nat. Rev. Cancer 2007, 7, 968–976. [Google Scholar] [CrossRef]
- Olaharski, A.J.; Sotelo, R.; Solorza-Luna, G.; Gonsebatt, M.E.; Guzman, P.; Mohar, A.; Eastmond, D.A. Tetraploidy and Chromosomal Instability Are Early Events during Cervical Carcinogenesis. Carcinogenesis 2006, 27, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Edgar, B.A.; Zielke, N.; Gutierrez, C. Endocycles: A Recurrent Evolutionary Innovation for Post-Mitotic Cell Growth. Nat. Rev. Mol. Cell Biol. 2014, 15, 197–210. [Google Scholar] [CrossRef]
- Lee, H.O.; Davidson, J.M.; Duronio, R.J. Endoreplication: Polyploidy with Purpose. Genes Dev. 2009, 23, 2461–2477. [Google Scholar] [CrossRef] [Green Version]
- Lazzeri, E.; Angelotti, M.L.; Peired, A.; Conte, C.; Marschner, J.A.; Maggi, L.; Mazzinghi, B.; Lombardi, D.; Melica, M.E.; Nardi, S.; et al. Endocycle-Related Tubular Cell Hypertrophy and Progenitor Proliferation Recover Renal Function after Acute Kidney Injury. Nat. Commun. 2018, 9, 1344. [Google Scholar] [CrossRef]
- Adachi, S.; Minamisawa, K.; Okushima, Y.; Inagaki, S.; Yoshiyama, K.; Kondou, Y.; Kaminuma, E.; Kawashima, M.; Toyoda, T.; Matsui, M.; et al. Programmed Induction of Endoreduplication by DNA Double-Strand Breaks in Arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 10004–10009. [Google Scholar] [CrossRef] [Green Version]
- Davoli, T.; Denchi, E.L.; de Lange, T. Persistent Telomere Damage Induces Bypass of Mitosis and Tetraploidy. Cell 2010, 141, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Davoli, T.; de Lange, T. Telomere-Driven Tetraploidization Occurs in Human Cells Undergoing Crisis and Promotes Transformation of Mouse Cells. Cancer Cell 2012, 21, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Vitale, I.; Galluzzi, L.; Castedo, M.; Kroemer, G. Mitotic Catastrophe: A Mechanism for Avoiding Genomic Instability. Nat. Rev. Mol. Cell Biol. 2011, 12, 385–392. [Google Scholar] [CrossRef]
- Sinha, D.; Duijf, P.H.G.; Khanna, K.K. Mitotic Slippage: An Old Tale with a New Twist. Cell Cycle 2019, 18, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Brito, D.A.; Rieder, C.L. Mitotic Checkpoint Slippage in Humans Occurs via Cyclin B Destruction in the Presence of an Active Checkpoint. Curr. Biol. 2006, 16, 1194–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikovskaya, D.; Schiffmann, D.; Newton, I.P.; Oakley, A.; Kroboth, K.; Sansom, O.; Jamieson, T.J.; Meniel, V.; Clarke, A.; Näthke, I.S. Loss of APC Induces Polyploidy as a Result of a Combination of Defects in Mitosis and Apoptosis. J. Cell Biol. 2007, 176, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Balachandran, R.S.; Kipreos, E.T. Addressing a Weakness of Anticancer Therapy with Mitosis Inhibitors: Mitotic Slippage. Mol. Cell. Oncol. 2017, 4, e1277293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuda, Y.; Iimori, M.; Nakashima, Y.; Nakanishi, R.; Ando, K.; Ohgaki, K.; Kitao, H.; Saeki, H.; Oki, E.; Maehara, Y. Mitotic Slippage and the Subsequent Cell Fates after Inhibition of Aurora B during Tubulin-Binding Agent-Induced Mitotic Arrest. Sci. Rep. 2017, 7, 16762. [Google Scholar] [CrossRef] [Green Version]
- Cheng, B.; Crasta, K. Consequences of Mitotic Slippage for Antimicrotubule Drug Therapy. Endocr. Relat. Cancer 2017, 24, T97–T106. [Google Scholar] [CrossRef] [Green Version]
- Green, R.A.; Paluch, E.; Oegema, K. Cytokinesis in Animal Cells. Annu. Rev. Cell Dev. Biol. 2012, 28, 29–58. [Google Scholar] [CrossRef] [Green Version]
- Lacroix, B.; Maddox, A.S. Cytokinesis, Ploidy and Aneuploidy. J. Pathol. 2012, 226, 338–351. [Google Scholar] [CrossRef]
- Rios, A.C.; Fu, N.Y.; Jamieson, P.R.; Pal, B.; Whitehead, L.; Nicholas, K.R.; Lindeman, G.J.; Visvader, J.E. Essential Role for a Novel Population of Binucleated Mammary Epithelial Cells in Lactation. Nat. Commun. 2016, 7, 11400. [Google Scholar] [CrossRef]
- Rappaport, A.M.; Borowy, Z.J.; Lougheed, W.M.; Lotto, W.N. Subdivision of Hexagonal Liver Lobules into a Structural and Functional Unit; Role in Hepatic Physiology and Pathology. Anat. Rec. 1954, 119, 11–33. [Google Scholar] [CrossRef] [PubMed]
- Jungermann, K.; Kietzmann, T. Zonation of Parenchymal and Nonparenchymal Metabolism in Liver. Annu. Rev. Nutr. 1996, 16, 179–203. [Google Scholar] [CrossRef] [PubMed]
- Benhamouche, S.; Decaens, T.; Perret, C.; Colnot, S. Wnt/beta-catenin pathway and liver metabolic zonation: A new player for an old concept. Med. Sci. 2006, 22, 904–906. [Google Scholar] [CrossRef] [Green Version]
- Halpern, K.B.; Shenhav, R.; Matcovitch-Natan, O.; Toth, B.; Lemze, D.; Golan, M.; Massasa, E.E.; Baydatch, S.; Landen, S.; Moor, A.E.; et al. Single-Cell Spatial Reconstruction Reveals Global Division of Labour in the Mammalian Liver. Nature 2017, 542, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.A. Experimental Carcinoma of the Liver. “Regeneration” of Liver Cells in Premalignant Stages. Am. J. Pathol. 1961, 39, 209–220. [Google Scholar]
- Bucher, N.L. Liver Regeneration: An Overview. J. Gastroenterol. Hepatol. 1991, 6, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Gentric, G.; Desdouets, C. Polyploidization in Liver Tissue. Am. J. Pathol. 2014, 184, 322–331. [Google Scholar] [CrossRef]
- Duncan, A.W.; Taylor, M.H.; Hickey, R.D.; Hanlon Newell, A.E.; Lenzi, M.L.; Olson, S.B.; Finegold, M.J.; Grompe, M. The Ploidy Conveyor of Mature Hepatocytes as a Source of Genetic Variation. Nature 2010, 467, 707–710. [Google Scholar] [CrossRef] [Green Version]
- Guidotti, J.-E.; Brégerie, O.; Robert, A.; Debey, P.; Brechot, C.; Desdouets, C. Liver Cell Polyploidization: A Pivotal Role for Binuclear Hepatocytes. J. Biol. Chem. 2003, 278, 19095–19101. [Google Scholar] [CrossRef] [Green Version]
- Toyoda, H.; Bregerie, O.; Vallet, A.; Nalpas, B.; Pivert, G.; Brechot, C.; Desdouets, C. Changes to Hepatocyte Ploidy and Binuclearity Profiles during Human Chronic Viral Hepatitis. Gut 2005, 54, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Bou-Nader, M.; Caruso, S.; Donne, R.; Celton-Morizur, S.; Calderaro, J.; Gentric, G.; Cadoux, M.; L’Hermitte, A.; Klein, C.; Guilbert, T.; et al. Polyploidy Spectrum: A New Marker in HCC Classification. Gut 2019, 69, 355–364. [Google Scholar] [CrossRef]
- Styles, J.A.; Kelly, M.; Pritchard, N.R.; Elcombe, C.R. A Species Comparison of Acute Hyperplasia Induced by the Peroxisome Proliferator Methylclofenapate: Involvement of the Binucleated Hepatocyte. Carcinogenesis 1988, 9, 1647–1655. [Google Scholar] [CrossRef]
- Cullen, J.M.; Linzey, D.W.; Gebhard, D.H. Nuclear Ploidy of Normal and Neoplastic Hepatocytes from Woodchuck Hepatitis Virus-Infected and Uninfected Woodchucks. Hepatology 1994, 19, 1072–1078. [Google Scholar] [PubMed]
- Gupta, S. Hepatic Polyploidy and Liver Growth Control. Semin. Cancer Biol. 2000, 10, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Margall-Ducos, G.; Celton-Morizur, S.; Couton, D.; Brégerie, O.; Desdouets, C. Liver Tetraploidization Is Controlled by a New Process of Incomplete Cytokinesis. J. Cell Sci. 2007, 120, 3633–3639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, S.; Delgado, E.R.; Otero, P.A.; Teng, K.; Kutay, H.; Meehan, K.M.; Moroney, J.B.; Monga, J.K.; Hand, N.J.; Friedman, J.R.; et al. MicroRNA-122 Regulates Polyploidization in the Murine Liver. Hepatology 2016, 64, 599–615. [Google Scholar] [CrossRef] [Green Version]
- Celton-Morizur, S.; Merlen, G.; Couton, D.; Margall-Ducos, G.; Desdouets, C. The Insulin/Akt Pathway Controls a Specific Cell Division Program That Leads to Generation of Binucleated Tetraploid Liver Cells in Rodents. J. Clin. Investig. 2009, 119, 1880–1887. [Google Scholar] [CrossRef] [Green Version]
- Celton-Morizur, S.; Merlen, G.; Couton, D.; Desdouets, C. Polyploidy and Liver Proliferation: Central Role of Insulin Signaling. Cell Cycle 2010, 9, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Pandit, S.K.; Westendorp, B.; Nantasanti, S.; van Liere, E.; Tooten, P.C.J.; Cornelissen, P.W.A.; Toussaint, M.J.M.; Lamers, W.H.; de Bruin, A. E2F8 Is Essential for Polyploidization in Mammalian Cells. Nat. Cell Biol. 2012, 14, 1181–1191. [Google Scholar] [CrossRef]
- Chen, H.-Z.; Ouseph, M.M.; Li, J.; Pécot, T.; Chokshi, V.; Kent, L.; Bae, S.; Byrne, M.; Duran, C.; Comstock, G.; et al. Canonical and Atypical E2Fs Regulate the Mammalian Endocycle. Nat. Cell Biol. 2012, 14, 1192–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conner, E.A.; Lemmer, E.R.; Sanchez, A.; Factor, V.M.; Thorgeirsson, S.S. E2F1 Blocks and C-Myc Accelerates Hepatic Ploidy in Transgenic Mouse Models. Biochem. Biophys. Res. Commun. 2003, 302, 114–120. [Google Scholar] [CrossRef]
- Lammens, T.; Li, J.; Leone, G.; de Veylder, L. Atypical E2Fs: New Players in the E2F Transcription Factor Family. Trends Cell Biol. 2009, 19, 111–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diril, M.K.; Ratnacaram, C.K.; Padmakumar, V.C.; Du, T.; Wasser, M.; Coppola, V.; Tessarollo, L.; Kaldis, P. Cyclin-Dependent Kinase 1 (Cdk1) Is Essential for Cell Division and Suppression of DNA Re-Replication but Not for Liver Regeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 3826–3831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miettinen, T.P.; Pessa, H.K.J.; Caldez, M.J.; Fuhrer, T.; Diril, M.K.; Sauer, U.; Kaldis, P.; Björklund, M. Identification of Transcriptional and Metabolic Programs Related to Mammalian Cell Size. Curr. Biol. 2014, 24, 598–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selmecki, A.M.; Maruvka, Y.E.; Richmond, P.A.; Guillet, M.; Shoresh, N.; Sorenson, A.L.; De, S.; Kishony, R.; Michor, F.; Dowell, R.; et al. Polyploidy Can Drive Rapid Adaptation in Yeast. Nature 2015, 519, 349–352. [Google Scholar] [CrossRef]
- Gandillet, A.; Alexandre, E.; Holl, V.; Royer, C.; Bischoff, P.; Cinqualbre, J.; Wolf, P.; Jaeck, D.; Richert, L. Hepatocyte Ploidy in Normal Young Rat. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2003, 134, 665–673. [Google Scholar] [CrossRef]
- Asahina, K.; Shiokawa, M.; Ueki, T.; Yamasaki, C.; Aratani, A.; Tateno, C.; Yoshizato, K. Multiplicative Mononuclear Small Hepatocytes in Adult Rat Liver: Their Isolation as a Homogeneous Population and Localization to Periportal Zone. Biochem. Biophys. Res. Commun. 2006, 342, 1160–1167. [Google Scholar] [CrossRef] [Green Version]
- Morales-Navarrete, H.; Segovia-Miranda, F.; Klukowski, P.; Meyer, K.; Nonaka, H.; Marsico, G.; Chernykh, M.; Kalaidzidis, A.; Zerial, M.; Kalaidzidis, Y. A Versatile Pipeline for the Multi-Scale Digital Reconstruction and Quantitative Analysis of 3D Tissue Architecture. eLife 2015, 4, e11214. [Google Scholar] [CrossRef]
- Tanami, S.; Ben-Moshe, S.; Elkayam, A.; Mayo, A.; Bahar Halpern, K.; Itzkovitz, S. Dynamic Zonation of Liver Polyploidy. Cell Tissue Res. 2017, 368, 405–410. [Google Scholar] [CrossRef]
- Matsumoto, T.; Wakefield, L.; Tarlow, B.D.; Grompe, M. In Vivo Lineage Tracing of Polyploid Hepatocytes Reveals Extensive Proliferation during Liver Regeneration. Cell Stem Cell 2020, 26, 34–47. [Google Scholar] [CrossRef]
- Katsuda, T.; Hosaka, K.; Matsuzaki, J.; Usuba, W.; Prieto-Vila, M.; Yamaguchi, T.; Tsuchiya, A.; Terai, S.; Ochiya, T. Transcriptomic Dissection of Hepatocyte Heterogeneity: Linking Ploidy, Zonation, and Stem/Progenitor Cell Characteristics. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 161–183. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.L.; Deligiannis, I.K.; Yin, K.; Danese, A.; Lleshi, E.; Coupland, P.; Vallejos, C.A.; Matchett, K.P.; Henderson, N.C.; Colome-Tatche, M.; et al. Single-Nucleus RNA-Seq2 Reveals Functional Crosstalk between Liver Zonation and Ploidy. Nat. Commun. 2021, 12, 4264. [Google Scholar] [CrossRef] [PubMed]
- Sigal, S.H.; Gupta, S.; Gebhard, D.F., Jr.; Holst, P.; Neufeld, D.; Reid, L.M. Evidence for a Terminal Differentiation Process in the Rat Liver. Differentiation 1995, 59, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Ganem, N.J.; Cornils, H.; Chiu, S.-Y.; O’Rourke, K.P.; Arnaud, J.; Yimlamai, D.; Théry, M.; Camargo, F.D.; Pellman, D. Cytokinesis Failure Triggers Hippo Tumor Suppressor Pathway Activation. Cell 2014, 158, 833–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overturf, K.; Al-Dhalimy, M.; Finegold, M.; Grompe, M. The Repopulation Potential of Hepatocyte Populations Differing in Size and Prior Mitotic Expansion. Am. J. Pathol. 1999, 155, 2135–2143. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, P.D.; Alencastro, F.; Delgado, E.R.; Leek, M.P.; Weirich, M.P.; Otero, P.A.; Roy, N.; Brown, W.K.; Oertel, M.; Duncan, A.W. Polyploid Hepatocytes Facilitate Adaptation and Regeneration to Chronic Liver Injury. Am. J. Pathol. 2019, 189, 1241–1255. [Google Scholar] [CrossRef]
- Wang, M.-J.; Chen, F.; Li, J.-X.; Liu, C.-C.; Zhang, H.-B.; Xia, Y.; Yu, B.; You, P.; Xiang, D.; Lu, L.; et al. Reversal of Hepatocyte Senescence after Continuous in Vivo Cell Proliferation. Hepatolology 2014, 60, 349–361. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Afendy, M.; Fang, Y.; Younossi, Y.; Mir, H.; Srishord, M. Changes in the Prevalence of the Most Common Causes of Chronic Liver Diseases in the United States from 1988 to 2008. Clin. Gastroenterol. Hepatol. 2011, 9, 524–530. [Google Scholar] [CrossRef]
- Magami, Y.; Azuma, T.; Inokuchi, H.; Kokuno, S.; Moriyasu, F.; Kawai, K.; Hattori, T. Cell Proliferation and Renewal of Normal Hepatocytes and Bile Duct Cells in Adult Mouse Liver. Liver 2002, 22, 419–425. [Google Scholar] [CrossRef]
- Toyoda, H.; Kumada, T.; Bregerie, O.; Brechot, C.; Desdouets, C. Conserved Balance of Hepatocyte Nuclear DNA Content in Mononuclear and Binuclear Hepatocyte Populations during the Course of Chronic Viral Hepatitis. World J. Gastroenterol. 2006, 12, 4546–4548. [Google Scholar] [CrossRef]
- Rakotomalala, L.; Studach, L.; Wang, W.-H.; Gregori, G.; Hullinger, R.L.; Andrisani, O. Hepatitis B Virus X Protein Increases the Cdt1-to-Geminin Ratio Inducing DNA Re-Replication and Polyploidy. J. Biol. Chem. 2008, 283, 28729–28740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studach, L.; Wang, W.-H.; Weber, G.; Tang, J.; Hullinger, R.L.; Malbrue, R.; Liu, X.; Andrisani, O. Polo-like Kinase 1 Activated by the Hepatitis B Virus X Protein Attenuates Both the DNA Damage Checkpoint and DNA Repair Resulting in Partial Polyploidy. J. Biol. Chem. 2010, 285, 30282–30293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahodantin, J.; Bou-Nader, M.; Cordier, C.; Mégret, J.; Soussan, P.; Desdouets, C.; Kremsdorf, D. Hepatitis B Virus X Protein Promotes DNA Damage Propagation through Disruption of Liver Polyploidization and Enhances Hepatocellular Carcinoma Initiation. Oncogene 2019, 38, 2645–2657. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Liu, J.-C.; McNamara, G.; Levine, A.; Duan, L.; Lai, M.M.C. Hepatitis C Virus Causes Uncoupling of Mitotic Checkpoint and Chromosomal Polyploidy through the Rb Pathway. J. Virol. 2009, 83, 12590–12600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current Concepts and Future Challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Younossi, Z.M. The Epidemiology of Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2018, 11, 92–94. [Google Scholar] [CrossRef] [Green Version]
- Gentric, G.; Desdouets, C. Liver Polyploidy: Dr Jekyll or Mr Hide? Oncotarget 2015, 6, 8430–8431. [Google Scholar] [CrossRef]
- Gentric, G.; Maillet, V.; Paradis, V.; Couton, D.; L’Hermitte, A.; Panasyuk, G.; Fromenty, B.; Celton-Morizur, S.; Desdouets, C. Oxidative Stress Promotes Pathologic Polyploidization in Nonalcoholic Fatty Liver Disease. J. Clin. Investig. 2015, 125, 981–992. [Google Scholar] [CrossRef] [Green Version]
- Herrtwich, L.; Nanda, I.; Evangelou, K.; Nikolova, T.; Horn, V.; Erny, D.; Stefanowski, J.; Rogell, L.; Klein, C.; Gharun, K.; et al. DNA Damage Signaling Instructs Polyploid Macrophage Fate in Granulomas. Cell 2016, 167, 1264–1280. [Google Scholar] [CrossRef]
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A.; et al. The Oxygen-Rich Postnatal Environment Induces Cardiomyocyte Cell-Cycle Arrest through DNA Damage Response. Cell 2014, 157, 565–579. [Google Scholar] [CrossRef] [Green Version]
- Bettencourt-Dias, M.; Glover, D.M. Centrosome Biogenesis and Function: Centrosomics Brings New Understanding. Nat. Rev. Mol. Cell Biol. 2007, 8, 451–463. [Google Scholar] [CrossRef]
- Nigg, E.A. Centrosome Duplication: Of Rules and Licenses. Trends Cell Biol. 2007, 17, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.S.; Holland, A.J. The Impact of Mitotic Errors on Cell Proliferation and Tumorigenesis. Genes Dev. 2018, 32, 620–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A Mechanism Linking Extra Centrosomes to Chromosomal Instability. Nature 2009, 460, 278–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godinho, S.A.; Pellman, D. Causes and Consequences of Centrosome Abnormalities in Cancer. Philos. Trans. R. Soc. B. 2014, 369, 20130467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duensing, A.; Duensing, S. Centrosomes, Polyploidy and Cancer. Adv. Exp. Med. Biol. 2010, 676, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.W.; Hickey, R.D.; Paulk, N.K.; Culberson, A.J.; Olson, S.B.; Finegold, M.J.; Grompe, M. Ploidy Reductions in Murine Fusion-Derived Hepatocytes. PLoS Genet. 2009, 5, e1000385. [Google Scholar] [CrossRef] [Green Version]
- Duncan, A.W. Aneuploidy, Polyploidy and Ploidy Reversal in the Liver. Semin. Cell Dev. Biol. 2013, 24, 347–356. [Google Scholar] [CrossRef]
- Knouse, K.A.; Lopez, K.E.; Bachofner, M.; Amon, A. Chromosome Segregation Fidelity in Epithelia Requires Tissue Architecture. Cell 2018, 175, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Duncan, A.W.; Hanlon Newell, A.E.; Smith, L.; Wilson, E.M.; Olson, S.B.; Thayer, M.J.; Strom, S.C.; Grompe, M. Frequent Aneuploidy among Normal Human Hepatocytes. Gastroenterology 2012, 142, 25–28. [Google Scholar] [CrossRef] [Green Version]
- Andreassen, P.R.; Lohez, O.D.; Lacroix, F.B.; Margolis, R.L. Tetraploid State Induces P53-Dependent Arrest of Nontransformed Mammalian Cells in G1. Mol. Biol. Cell 2001, 12, 1315–1328. [Google Scholar] [CrossRef] [Green Version]
- Meraldi, P.; Honda, R.; Nigg, E.A. Aurora-A Overexpression Reveals Tetraploidization as a Major Route to Centrosome Amplification in P53-/- Cells. EMBO J. 2002, 21, 483–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuffer, C.; Kuznetsova, A.Y.; Storchová, Z. Abnormal Mitosis Triggers P53-Dependent Cell Cycle Arrest in Human Tetraploid Cells. Chromosoma 2013, 122, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis Failure Generating Tetraploids Promotes Tumorigenesis in P53-Null Cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Michael, D.; Shmueli, A.; Yabuta, N.; Nojima, H.; Oren, M. A Positive Feedback Loop between the P53 and Lats2 Tumor Suppressors Prevents Tetraploidization. Genes Dev. 2006, 20, 2687–2700. [Google Scholar] [CrossRef] [Green Version]
- Tinel, A.; Tschopp, J. The PIDDosome, a Protein Complex Implicated in Activation of Caspase-2 in Response to Genotoxic Stress. Science 2004, 304, 843–846. [Google Scholar] [CrossRef] [Green Version]
- Fava, L.L.; Schuler, F.; Sladky, V.; Haschka, M.D.; Soratroi, C.; Eiterer, L.; Demetz, E.; Weiss, G.; Geley, S.; Nigg, E.A.; et al. The PIDDosome Activates P53 in Response to Supernumerary Centrosomes. Genes Dev. 2017, 31, 34–45. [Google Scholar] [CrossRef]
- Sladky, V.C.; Knapp, K.; Soratroi, C.; Heppke, J.; Eichin, F.; Rocamora-Reverte, L.; Szabo, T.G.; Bongiovanni, L.; Westendorp, B.; Moreno, E.; et al. E2F-Family Members Engage the PIDDosome to Limit Hepatocyte Ploidy in Liver Development and Regeneration. Dev. Cell 2020, 52, 335–349. [Google Scholar] [CrossRef]
- Zasadil, L.M.; Britigan, E.M.C.; Weaver, B.A. 2n or Not 2n: Aneuploidy, Polyploidy and Chromosomal Instability in Primary and Tumor Cells. Semin. Cell Dev. Biol. 2013, 24, 370–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K.; Goto, H.; Nishimura, Y.; Kasahara, K.; Mizoguchi, A.; Inagaki, M. Tetraploidy in Cancer and Its Possible Link to Aging. Cancer Sci. 2018, 109, 2632–2640. [Google Scholar] [CrossRef] [PubMed]
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.A.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome Doubling Shapes the Evolution and Prognosis of Advanced Cancers. Nat. Genet. 2018, 50, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Maley, C.C.; Galipeau, P.C.; Li, X.; Sanchez, C.A.; Paulson, T.G.; Blount, P.L.; Reid, B.J. The Combination of Genetic Instability and Clonal Expansion Predicts Progression to Esophageal Adenocarcinoma. Cancer Res. 2004, 64, 7629–7633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anti, M.; Marra, G.; Rapaccini, G.L.; Rumi, C.; Bussa, S.; Fadda, G.; Vecchio, F.M.; Valenti, A.; Percesepe, A.; Pompili, M.; et al. DNA Ploidy Pattern in Human Chronic Liver Diseases and Hepatic Nodular Lesions. Flow Cytometric Analysis on Echo-Guided Needle Liver Biopsy. Cancer 1994, 73, 281–288. [Google Scholar] [CrossRef]
- Fujimoto, J.; Okamoto, E.; Yamanaka, N.; Toyosaka, A.; Mitsunobu, M. Flow Cytometric DNA Analysis of Hepatocellular Carcinoma. Cancer 1991, 67, 939–944. [Google Scholar] [CrossRef]
- Nagasue, N.; Kohno, H.; Hayashi, T.; Yamanoi, A.; Uchida, M.; Takemoto, Y.; Makino, Y.; Ono, T.; Hayashi, J.; Nakamura, T. Lack of Intratumoral Heterogeneity in DNA Ploidy Pattern of Hepatocellular Carcinoma. Gastroenterology 1993, 105, 1449–1454. [Google Scholar] [CrossRef]
- Kent, L.N.; Rakijas, J.B.; Pandit, S.K.; Westendorp, B.; Chen, H.-Z.; Huntington, J.T.; Tang, X.; Bae, S.; Srivastava, A.; Senapati, S.; et al. E2f8 Mediates Tumor Suppression in Postnatal Liver Development. J. Clin. Investig. 2016, 126, 2955–2969. [Google Scholar] [CrossRef]
- Sladky, V.C.; Knapp, K.; Szabo, T.G.; Braun, V.Z.; Bongiovanni, L.; van den Bos, H.; Spierings, D.C.; Westendorp, B.; Curinha, A.; Stojakovic, T.; et al. PIDDosome-Induced P53-Dependent Ploidy Restriction Facilitates Hepatocarcinogenesis. EMBO Rep. 2020, 21, e50893. [Google Scholar] [CrossRef]
- Moreno, E.; Toussaint, M.J.M.; van Essen, S.C.; Bongiovanni, L.; van Liere, E.A.; Koster, M.H.; Yuan, R.; van Deursen, J.M.; Westendorp, B.; de Bruin, A. E2F7 Is a Potent Inhibitor of Liver Tumor Growth in Adult Mice. Hepatology 2021, 73, 303–317. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, P.D.; Delgado, E.R.; Alencastro, F.; Leek, M.P.; Roy, N.; Weirich, M.P.; Stahl, E.C.; Otero, P.A.; Chen, M.I.; Brown, W.K.; et al. The Polyploid State Restricts Hepatocyte Proliferation and Liver Regeneration in Mice. Hepatolology 2019, 69, 1242–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zhou, K.; Luo, X.; Li, L.; Tu, H.-C.; Sehgal, A.; Nguyen, L.H.; Zhang, Y.; Gopal, P.; Tarlow, B.D.; et al. The Polyploid State Plays a Tumor-Suppressive Role in the Liver. Dev. Cell 2018, 44, 447–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Nguyen, L.H.; Zhou, K.; Tiu, H.C.; Sehgal, A.; Nassour, I.; Li, L.; Gopal, P.; Goodman, J.; Singal, A.G.; et al. Knockdown of Anillin Actin Binding Protein Blocks Cytokinesis in Hepatocytes and Reduces Liver Tumor Development in Mice without Affecting Regeneration. Gastroenterology 2018, 154, 1421–1434. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-H.; Zhang, S.; Zhu, M.; Lu, T.; Chen, K.; Wen, Z.; Wang, S.; Xiao, G.; Luo, D.; Jia, Y.; et al. Mice With Increased Numbers of Polyploid Hepatocytes Maintain Regenerative Capacity but Develop Fewer Tumors Following Chronic Liver Injury. Gastroenterology 2020, 158, 1698–1712. [Google Scholar] [CrossRef]
- Piekny, A.J.; Maddox, A.S. The Myriad Roles of Anillin during Cytokinesis. Semin. Cell Dev. Biol. 2010, 21, 881–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, M.; May, S.; Bird, T.G. Ploidy Dynamics Increase the Risk of Liver Cancer Initiation. Nat. Commun. 2021, 12, 1896. [Google Scholar] [CrossRef]
- Matsumoto, T.; Wakefield, L.; Peters, A.; Peto, M.; Spellman, P.; Grompe, M. Proliferative Polyploid Cells Give Rise to Tumors via Ploidy Reduction. Nat. Commun. 2021, 12, 646. [Google Scholar] [CrossRef]
- Vargas-Rondón, N.; Villegas, V.E.; Rondón-Lagos, M. The Role of Chromosomal Instability in Cancer and Therapeutic Responses. Cancers 2017, 10, 4. [Google Scholar] [CrossRef] [Green Version]
- Bach, D.-H.; Zhang, W.; Sood, A.K. Chromosomal Instability in Tumor Initiation and Development. Cancer Res. 2019, 79, 3995–4002. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Huang, Y.-S.; Fustin, J.-M.; Doi, M.; Chen, H.; Lai, H.-H.; Lin, S.-H.; Lee, Y.-L.; King, P.-C.; Hou, H.-S.; et al. Hyperpolyploidization of Hepatocyte Initiates Preneoplastic Lesion Formation in the Liver. Nat. Commun. 2021, 12, 645. [Google Scholar] [CrossRef]
- Carmena, M.; Wheelock, M.; Funabiki, H.; Earnshaw, W.C. The Chromosomal Passenger Complex (CPC): From Easy Rider to the Godfather of Mitosis. Nat. Rev. Mol. Cell Biol. 2012, 13, 789–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, A.W. Hepatocyte Ploidy Modulation in Liver Cancer. EMBO Rep. 2020, 21, e51922. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular Carcinoma. Nat. Rev. Dis. Primer 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donne, R.; Sangouard, F.; Celton-Morizur, S.; Desdouets, C. Hepatocyte Polyploidy: Driver or Gatekeeper of Chronic Liver Diseases. Cancers 2021, 13, 5151. https://doi.org/10.3390/cancers13205151
Donne R, Sangouard F, Celton-Morizur S, Desdouets C. Hepatocyte Polyploidy: Driver or Gatekeeper of Chronic Liver Diseases. Cancers. 2021; 13(20):5151. https://doi.org/10.3390/cancers13205151
Chicago/Turabian StyleDonne, Romain, Flora Sangouard, Séverine Celton-Morizur, and Chantal Desdouets. 2021. "Hepatocyte Polyploidy: Driver or Gatekeeper of Chronic Liver Diseases" Cancers 13, no. 20: 5151. https://doi.org/10.3390/cancers13205151
APA StyleDonne, R., Sangouard, F., Celton-Morizur, S., & Desdouets, C. (2021). Hepatocyte Polyploidy: Driver or Gatekeeper of Chronic Liver Diseases. Cancers, 13(20), 5151. https://doi.org/10.3390/cancers13205151