
TPH1 and 5-HT7 Receptor Overexpression Leading to Gemcitabine-Resistance Requires Non-Canonical Permissive Action of EZH2 in Pancreatic Ductal Adenocarcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture and Viability Assay
2.3. Protein Extraction and Western Blotting
2.4. siRNA Transfection
2.5. Flow Cytometry Analysis
2.6. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.7. Sphere Formation Assay
2.8. Immunoprecipitation
2.9. Anti-Tumor Activity Measurement Using a Xenograft Tumor Model
2.10. Statistical Analyses
3. Results
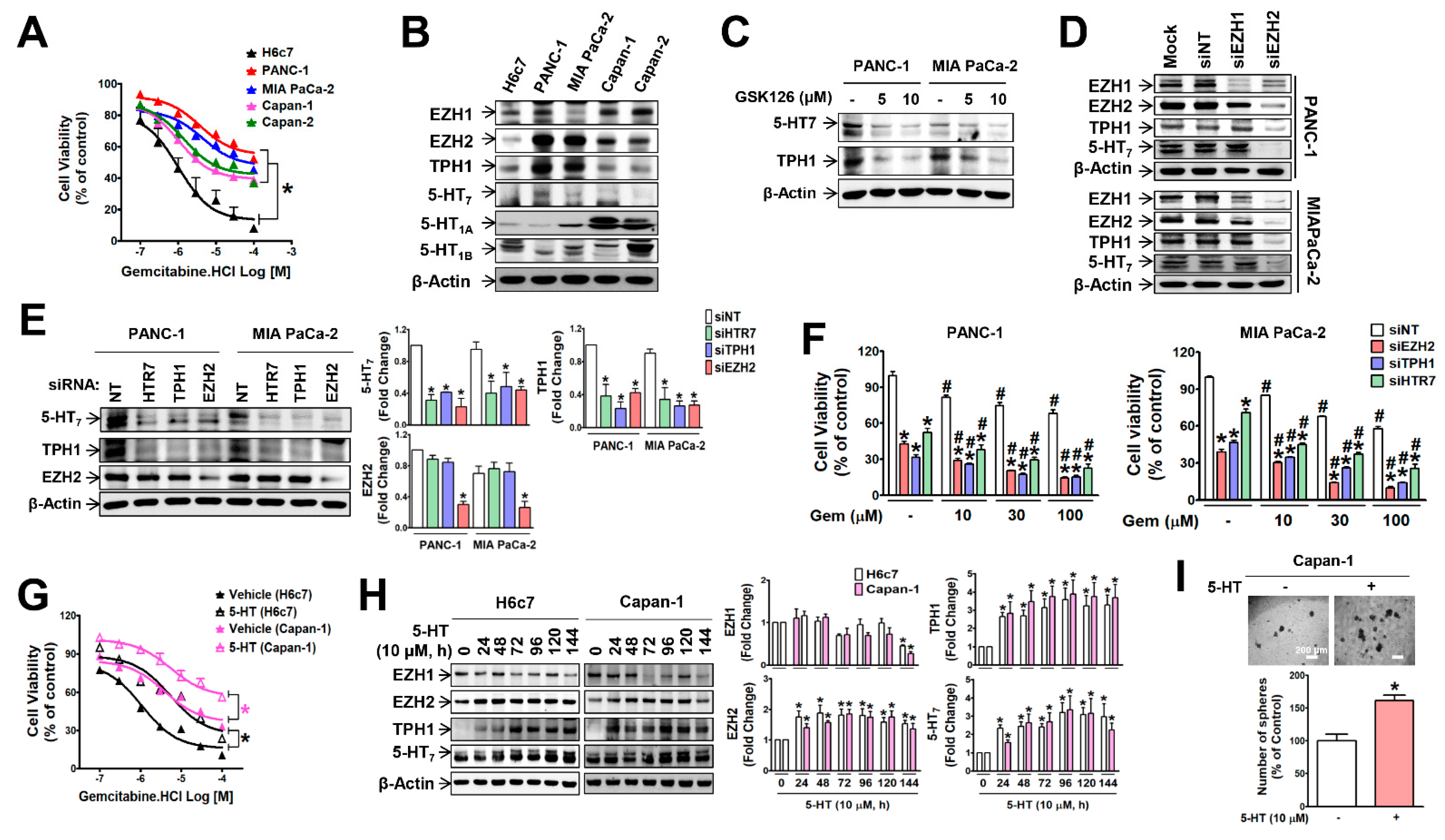
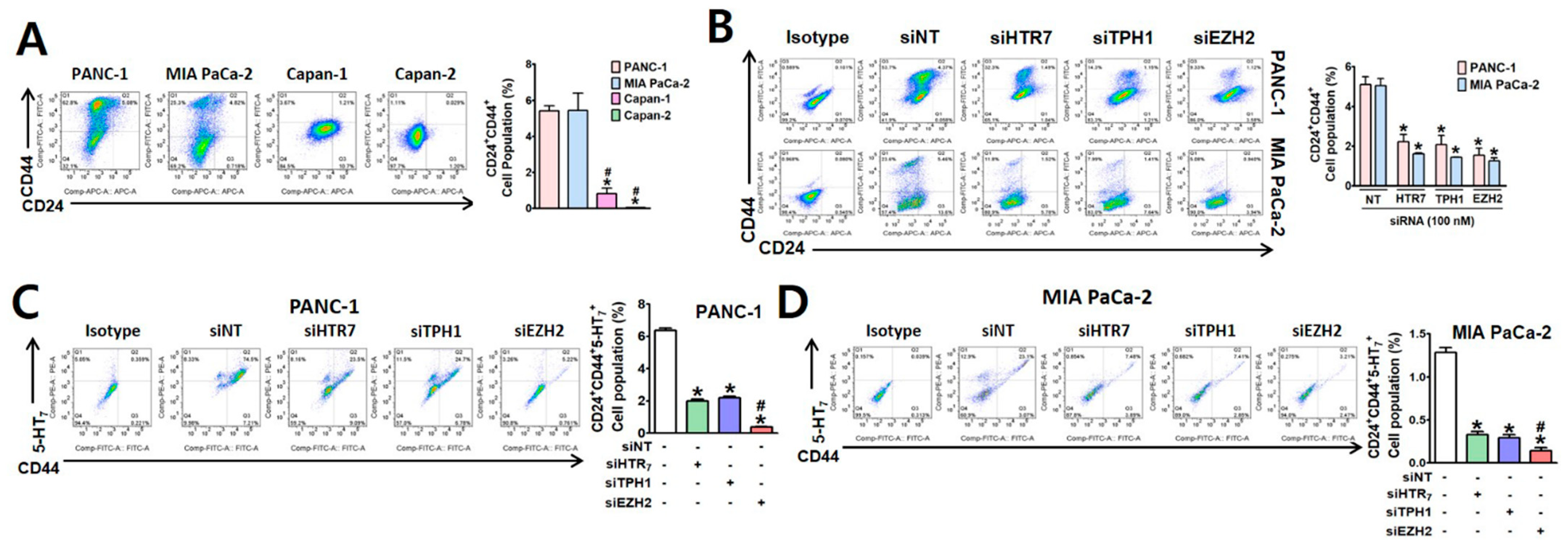
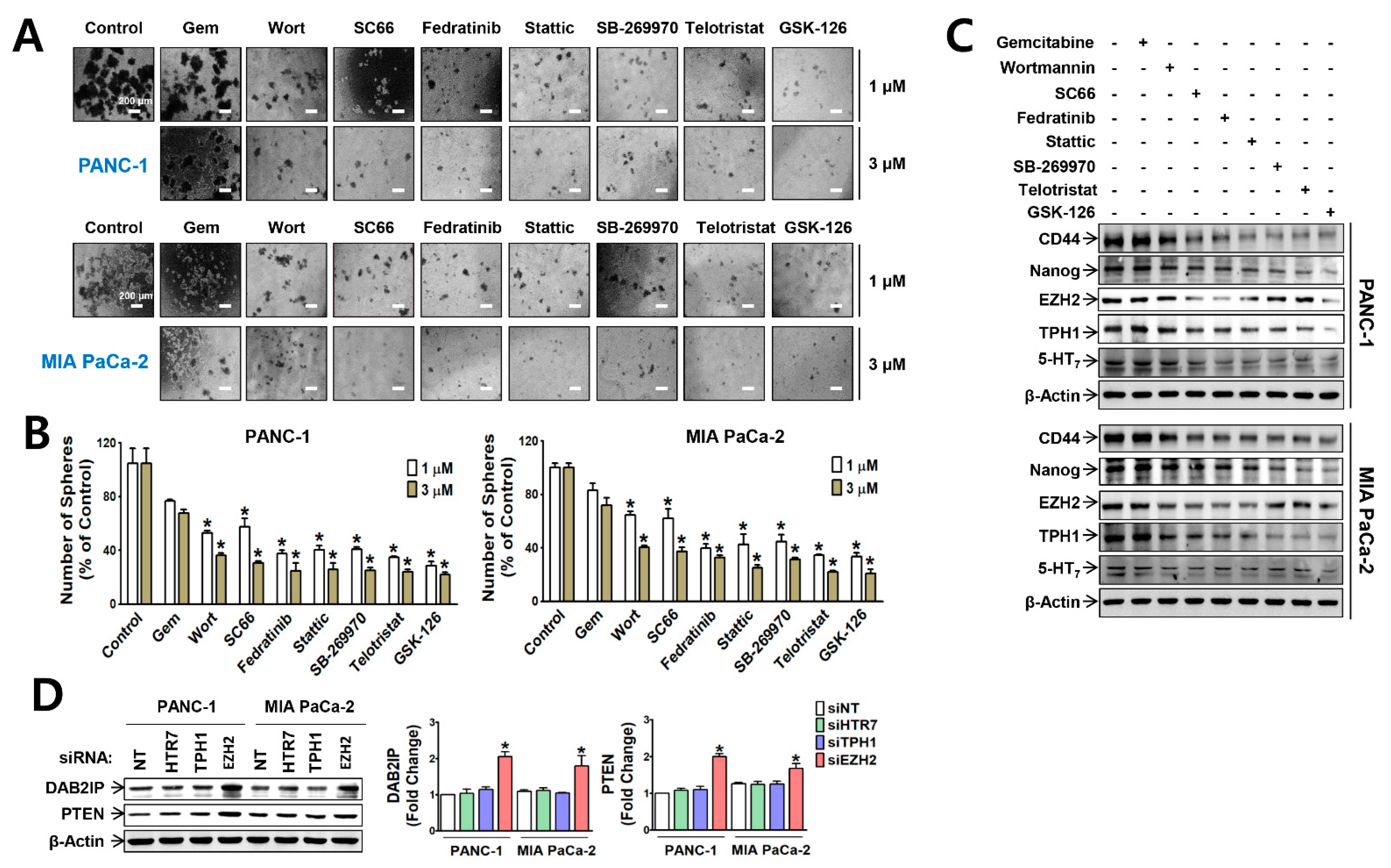
3.1. EZH2 Supports TPH1-5-HT7 Axis to Regulate Gemcitabine Resistance and Cancer Stem Cell Population in Pancreatic Cancer Cells
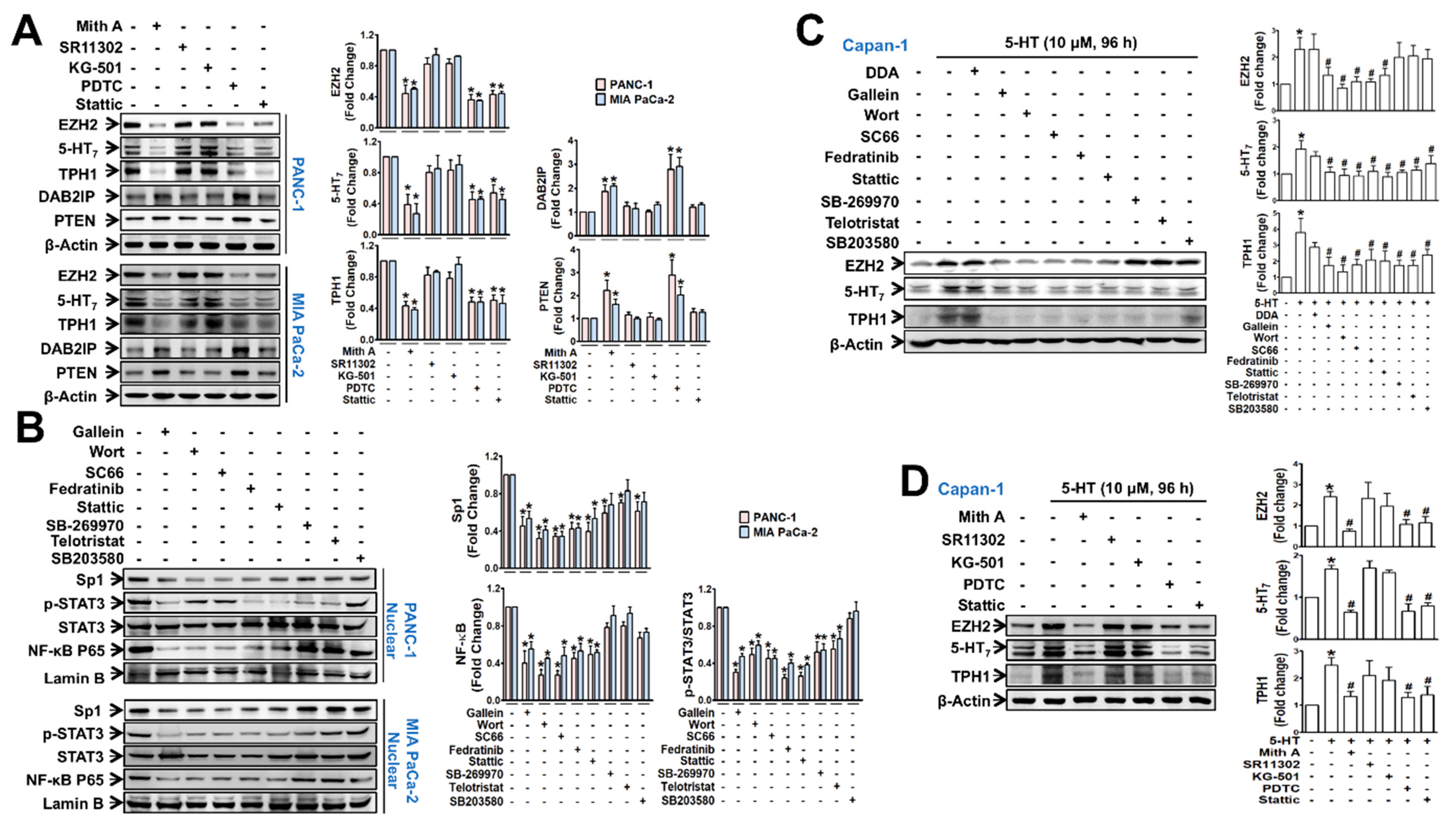
3.2. EZH2-Regulated Signaling Pathways Potentiate Nuclear Translocation of TFs Linked to TPH1-5-HT7 Axis in Pancreatic Cancer Cells
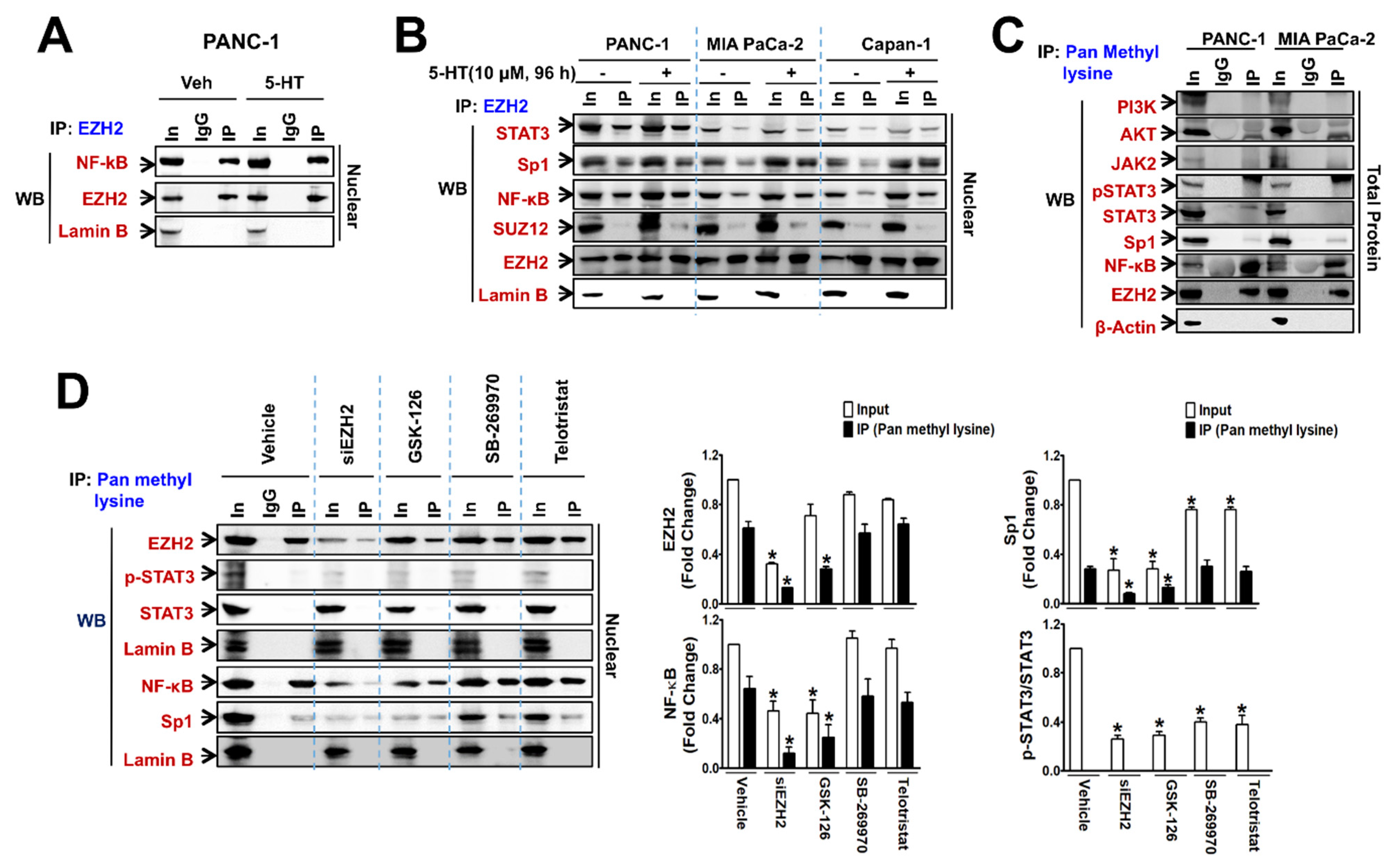
3.3. Automethylated EZH2 Serves as a Binding Scaffold for Methylated NF-κB and Sp1, and Unmethylated p-STAT3, in a PRC2-Independent Manner
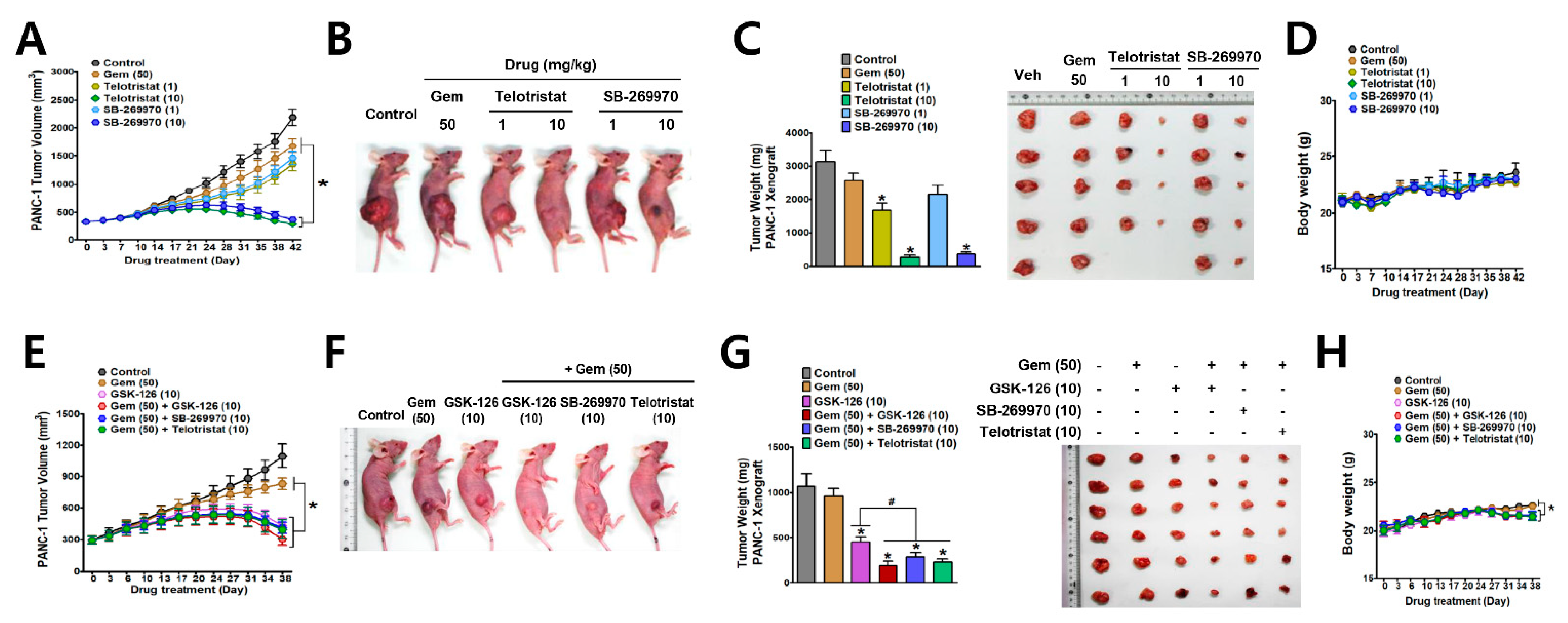
3.4. Antitumor Effects of EZH2-TPH1-5-HT7 Axis Inhibition in PANC-1 Xenograft Tumor Model in Mice
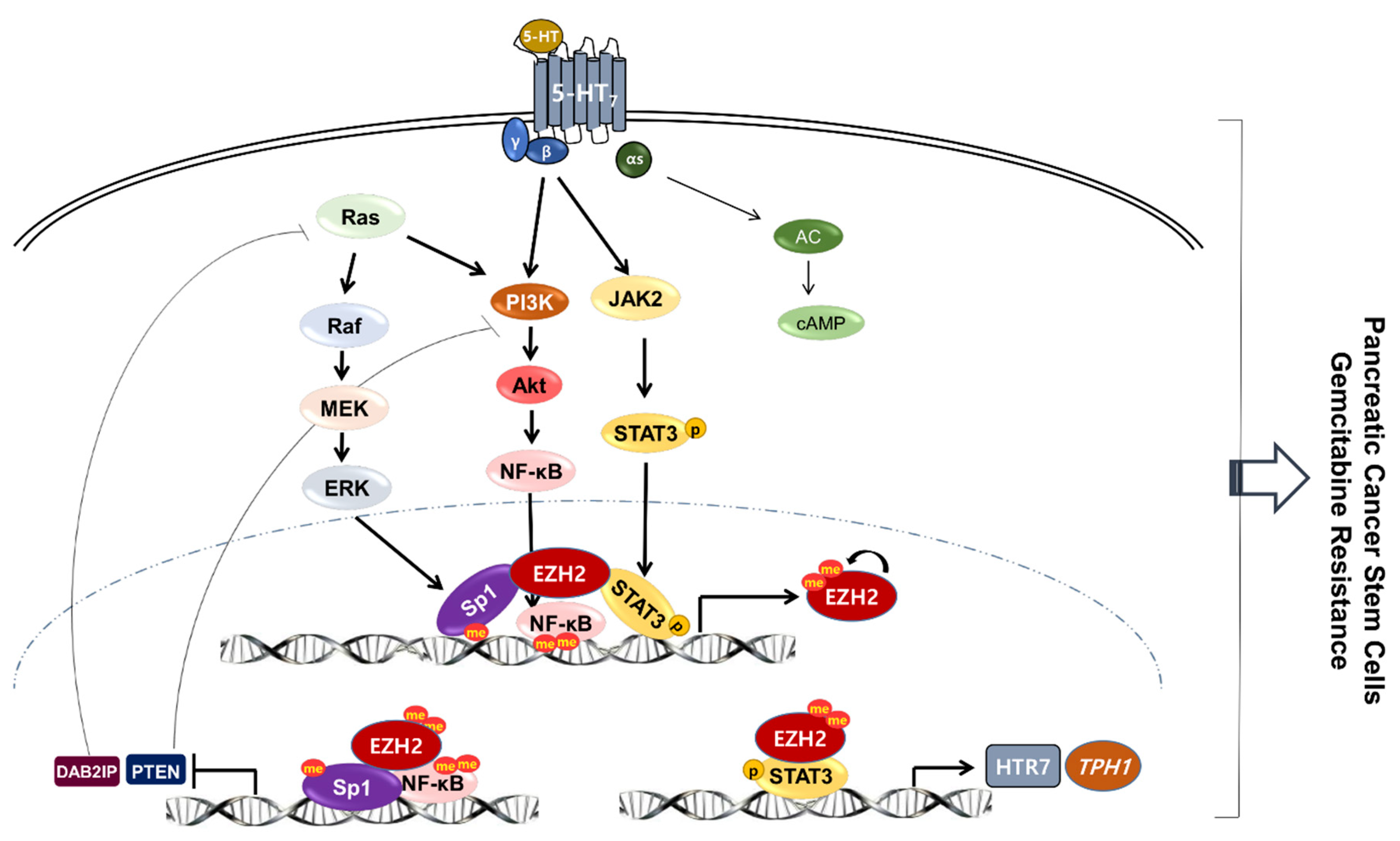
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Howlader, N.; Noone, A.; Krapcho, M.; Miller, D.; Bishop, K.; Kosary, C. American Cancer Society Cancer Facts & Figures 2018. ACS 2018. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2018/cancer-facts-and-figures-2018.pdf (accessed on 13 September 2021).
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. Am. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, R.; Bodoky, G.; Ruhstaller, T.; Glimelius, B.; Bajetta, E.; Schuller, J.; Saletti, P.; Bauer, J.; Figer, A.; Pestalozzi, B.; et al. Gemcitabine plus capecitabine compared with gemcitabine alone in advanced pancreatic cancer: A randomized, multicenter, phase III trial of the Swiss Group for Clinical Cancer Research and the Central European Cooperative Oncology Group. Am. J. Clin. Oncol. 2007, 25, 2212–2217. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Scholz, A.; Heinze, S.; Detjen, K.M.; Peters, M.; Welzel, M.; Hauff, P.; Schirner, M.; Wiedenmann, B.; Rosewicz, S. Activated signal transducer and activator of transcription 3 (STAT3) supports the malignant phenotype of human pancreatic cancer. Gastroenterology 2003, 125, 891–905. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Wang, L.M.; Jove, R.; Vande Woude, G.F. Requirement of Stat3 signaling for HGF/SF-Met mediated tumorigenesis. Oncogene 2002, 21, 217–226. [Google Scholar] [CrossRef]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef]
- Khan, A.A.; Liu, X.; Yan, X.; Tahir, M.; Ali, S.; Huang, H. An overview of genetic mutations and epigenetic signatures in the course of pancreatic cancer progression. Cancer Metastasis Rev. 2021, 40, 245–272. [Google Scholar] [CrossRef]
- Paradise, B.D.; Barham, W.; Fernandez-Zapico, M.E. Targeting Epigenetic Aberrations in Pancreatic Cancer, a New Path to Improve Patient Outcomes? Cancers 2018, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Neureiter, D.; Jager, T.; Ocker, M.; Kiesslich, T. Epigenetics and pancreatic cancer: Pathophysiology and novel treatment aspects. World J. Gastroenterol. 2014, 20, 7830–7848. [Google Scholar] [CrossRef]
- Pasini, D.; Di Croce, L. Emerging roles for Polycomb proteins in cancer. Curr. Opin. Genet. Dev. 2016, 36, 50–58. [Google Scholar] [CrossRef]
- Duan, R.; Du, W.; Guo, W. EZH2: A novel target for cancer treatment. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef] [Green Version]
- Ougolkov, A.V.; Bilim, V.N.; Billadeau, D.D. Regulation of pancreatic tumor cell proliferation and chemoresistance by the histone methyltransferase enhancer of zeste homologue 2. Clin. Cancer Res. 2008, 14, 6790–6796. [Google Scholar] [CrossRef] [Green Version]
- Kocher, H.M.; Mears, L.; Lea, N.C.; Raj, K.; Mufti, G.J. JAK V617F missense mutation is absent in pancreatic cancer. Gut 2007, 56, 1174–1175. [Google Scholar] [CrossRef] [Green Version]
- Richly, H.; Aloia, L.; Di Croce, L. Roles of the Polycomb group proteins in stem cells and cancer. Cell Death Dis. 2011, 2, e204. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Cai, J.; Hou, Y.; Huang, Z.; Wang, Z. Role of EZH2 in cancer stem cells: From biological insight to a therapeutic target. Oncotarget 2017, 8, 37974–37990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.F.; Luo, X.; Xiang, L.S.; Li, H.T.; Zha, L.; Li, N.; He, J.M.; Xie, G.F.; Xie, X.; Liang, H.J. EZH2 promotes colorectal cancer stem-like cell expansion by activating p21cip1-Wnt/beta-catenin signaling. Oncotarget 2016, 7, 41540–41558. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Zaslavsky, A.; Fedele, G.; McLaughlin, S.K.; Reczek, E.E.; De Raedt, T.; Guney, I.; Strochlic, D.E.; Macconaill, L.E.; Beroukhim, R.; et al. An oncogene-tumor suppressor cascade drives metastattic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat. Med. 2010, 16, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Li, Z.; Wu, Z.; Aau, M.; Guan, P.; Karuturi, R.K.; Liou, Y.C.; Yu, Q. Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Mol. Cell 2011, 43, 798–810. [Google Scholar] [CrossRef]
- Iannetti, A.; Ledoux, A.C.; Tudhope, S.J.; Sellier, H.; Zhao, B.; Mowla, S.; Moore, A.; Hummerich, H.; Gewurz, B.E.; Cockell, S.J.; et al. Regulation of p53 and Rb links the alternative NF-kappaB pathway to EZH2 expression and cell senescence. PLoS Genet. 2014, 10, e1004642. [Google Scholar] [CrossRef] [Green Version]
- Mawe, G.M.; Hoffman, J.M. Serotonin signalling in the gut--functions, dysfunctions and therapeutic targets. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 473–486. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Toyofuku, Y.; Lynn, F.C.; Chak, E.; Uchida, T.; Mizukami, H.; Fujitani, Y.; Kawamori, R.; Miyatsuka, T.; Kosaka, Y.; et al. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat. Med. 2010, 16, 804–808. [Google Scholar] [CrossRef]
- Sarrouilhe, D.; Clarhaut, J.; Defamie, N.; Mesnil, M. Serotonin and cancer: What is the link? Curr. Mol. Med. 2015, 15, 62–77. [Google Scholar] [CrossRef] [PubMed]
- Lesurtel, M.; Graf, R.; Aleil, B.; Walther, D.J.; Tian, Y.; Jochum, W.; Gachet, C.; Bader, M.; Clavien, P.A. Platelet-derived serotonin mediates liver regeneration. Science 2006, 312, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Mammadova-Bach, E.; Mauler, M.; Braun, A.; Duerschmied, D. Autocrine and paracrine regulatory functions of platelet serotonin. Platelets 2018, 29, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Gautam, J.; Banskota, S.; Regmi, S.C.; Ahn, S.; Jeon, Y.H.; Jeong, H.; Kim, S.J.; Nam, T.G.; Jeong, B.S.; Kim, J.A. Tryptophan hydroxylase 1 and 5-HT7 receptor preferentially expressed in triple-negative breast cancer promote cancer progression through autocrine serotonin signaling. Mol. Cancer 2016, 15, 75. [Google Scholar] [CrossRef] [Green Version]
- Pepin, M.E.; Bickerton, H.H.; Bethea, M.; Hunter, C.S.; Wende, A.R.; Banerjee, R.R. Prolactin Receptor Signaling Regulates a Pregnancy-Specific Transcriptional Program in Mouse Islets. Endocrinology 2019, 160, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Saponara, E.; Grabliauskaite, K.; Bombardo, M.; Buzzi, R.; Silva, A.B.; Malagola, E.; Tian, Y.; Hehl, A.B.; Schraner, E.M.; Seleznik, G.M.; et al. Serotonin promotes acinar dedifferentiation following pancreatitis-induced regeneration in the adult pancreas. J. Pathol. 2015, 237, 495–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurbuz, N.; Ashour, A.A.; Alpay, S.N.; Ozpolat, B. Down-regulation of 5-HT1B and 5-HT1D receptors inhibits proliferation, clonogenicity and invasion of human pancreatic cancer cells. PLoS ONE 2014, 9, e110067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Van Vlerken, L.E.; Kiefer, C.M.; Morehouse, C.; Li, Y.; Groves, C.; Wilson, S.D.; Yao, Y.; Hollingsworth, R.E.; Hurt, E.M. EZH2 is required for breast and pancreatic cancer stem cell maintenance and can be used as a functional cancer stem cell reporter. Stem Cells Transl. Med. 2013, 2, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Zong, X.; Wang, W.; Ozes, A.; Fang, F.; Sandusky, G.E.; Nephew, K.P. EZH2-Mediated Downregulation of the Tumor Suppressor DAB2IP Maintains Ovarian Cancer Stem Cells. Cancer Res. 2020, 80, 4371–4385. [Google Scholar] [CrossRef] [PubMed]
- Jarome, T.J.; Perez, G.A.; Hauser, R.M.; Hatch, K.M.; Lubin, F.D. EZH2 Methyltransferase Activity Controls Pten Expression and mTOR Signaling during Fear Memory Reconsolidation. J. Neurosci. Res. 2018, 38, 7635–7648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pappas, K.; Martin, T.C.; Wolfe, A.L.; Nguyen, C.B.; Su, T.; Jin, J.; Hibshoosh, H.; Parsons, R. NOTCH and EZH2 collaborate to repress PTEN expression in breast cancer. Commun. Biol. 2021, 4, 312. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Yin, X.; Lai, X.; Liu, C.; Nie, W.; Li, D.; Xie, Z.; Li, Z.; Meng, F. Upregulation of DAB2IP Inhibits Ras Activity and Tumorigenesis in Human Pancreatic Cancer Cells. Cancer Res. Treat. 2020, 19, 1533033819895494. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Holder, M.; Grau, D.; Saldana-Meyer, R.; Yu, J.R.; Ganai, R.A.; Zhang, J.; Wang, M.; LeRoy, G.; Dobenecker, M.W.; et al. Distinct Stimulatory Mechanisms Regulate the Catalytic Activity of Polycomb Repressive Complex 2. Mol. Cell 2018, 70, 435–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.H.; Li, J.; Dong, F.Y.; Yang, J.Y.; Liu, D.J.; Yang, X.M.; Wang, Y.H.; Yang, M.W.; Fu, X.L.; Zhang, X.X.; et al. Increased Serotonin Signaling Contributes to the Warburg Effect in Pancreatic Tumor Cells Under Metabolic Stress and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 2017, 153, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Alpini, G.; Invernizzi, P.; Gaudio, E.; Venter, J.; Kopriva, S.; Bernuzzi, F.; Onori, P.; Franchitto, A.; Coufal, M.; Frampton, G.; et al. Serotonin metabolism is dysregulated in cholangiocarcinoma, which has implications for tumor growth. Cancer Res. 2008, 68, 9184–9193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soll, C.; Jang, J.H.; Riener, M.O.; Moritz, W.; Wild, P.J.; Graf, R.; Clavien, P.A. Serotonin promotes tumor growth in human hepatocellular cancer. Hepatology 2010, 51, 1244–1254. [Google Scholar] [CrossRef]
- Karmakar, S.; Lal, G. Role of serotonin receptor signaling in cancer cells and anti-tumor immunity. Theranostics 2021, 11, 5296–5312. [Google Scholar] [CrossRef]
- Du, X.; Wang, T.; Wang, Z.; Wu, X.; Gu, Y.; Huang, Q.; Wang, J.; Xie, J. 5-HT7 Receptor Contributes to Proliferation, Migration and Invasion in NSCLC Cells. OncoTargets Ther. 2020, 13, 2139–2151. [Google Scholar] [CrossRef] [Green Version]
- Cinar, I.; Sirin, B.; Halici, Z.; Palabiyik-Yucelik, S.S.; Akpinar, E.; Cadirci, E. 5-HT7 receptors as a new target for prostate cancer physiopathology and treatment: An experimental study on PC-3 cells and FFPE tissues. Naunyn Schmiedebergs Arch. Pharmacol. 2021, 394, 1205–1213. [Google Scholar] [CrossRef]
- Margueron, R.; Li, G.; Sarma, K.; Blais, A.; Zavadil, J.; Woodcock, C.L.; Dynlacht, B.D.; Reinberg, D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 2008, 32, 503–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Liu, Y.; Hsu, Y.-J.; Fujiwara, Y.; Kim, J.; Mao, X.; Yuan, G.-C.; Orkin, S.H. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol. Cell 2008, 32, 491–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagishi, M.; Hori, M.; Fujikawa, D.; Ohsugi, T.; Honma, D.; Adachi, N.; Katano, H.; Hishima, T.; Kobayashi, S.; Nakano, K. Targeting excessive EZH1 and EZH2 activities for abnormal histone methylation and transcription network in malignant lymphomas. Cell Rep. 2019, 29, 2321–2337. [Google Scholar] [CrossRef] [PubMed]
- Iida, H.; Ogihara, T.; Min, M.K.; Hara, A.; Kim, Y.G.; Fujimaki, K.; Tamaki, M.; Fujitani, Y.; Kim, H.; Watada, H. Expression mechanism of tryptophan hydroxylase 1 in mouse islets during pregnancy. J. Mol. Endocrinol. 2015, 55, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anwar, T.; Gonzalez, M.E.; Kleer, C.G. Noncanonical Functions of the Polycomb Group Protein EZH2 in Breast Cancer. Am. J. Pathol. 2021, 191, 774–783. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaudhary, P.; Guragain, D.; Chang, J.-H.; Kim, J.-A. TPH1 and 5-HT7 Receptor Overexpression Leading to Gemcitabine-Resistance Requires Non-Canonical Permissive Action of EZH2 in Pancreatic Ductal Adenocarcinoma. Cancers 2021, 13, 5305. https://doi.org/10.3390/cancers13215305
Chaudhary P, Guragain D, Chang J-H, Kim J-A. TPH1 and 5-HT7 Receptor Overexpression Leading to Gemcitabine-Resistance Requires Non-Canonical Permissive Action of EZH2 in Pancreatic Ductal Adenocarcinoma. Cancers. 2021; 13(21):5305. https://doi.org/10.3390/cancers13215305
Chicago/Turabian StyleChaudhary, Prakash, Diwakar Guragain, Jae-Hoon Chang, and Jung-Ae Kim. 2021. "TPH1 and 5-HT7 Receptor Overexpression Leading to Gemcitabine-Resistance Requires Non-Canonical Permissive Action of EZH2 in Pancreatic Ductal Adenocarcinoma" Cancers 13, no. 21: 5305. https://doi.org/10.3390/cancers13215305