
Atlas of Lobular Breast Cancer Models: Challenges and Strategic Directions

 , , , , , and
, , , , , and
Abstract
:Simple Summary
Abstract
1. Search Strategy and Selection Criteria
2. Introduction
3. In Vitro Cell-Based ILC Models
3.1. E-cadherin-Deficient Well-Characterized ILC Cell Lines
3.2. ILC-like Cell Lines Derived from Clinical NSTs with ILC Molecular Features (Genetic and Expression Profile)
3.3. Epigenetic Inactivation of E-Cadherin
3.4. ILC Lines with Proficient E-Cadherin and Defective Adherens Junctions
3.5. ILC Cell Lines Developed from Patient-Derived Xenografts (PDXs)
3.6. Experimental CDH1 Downregulation/Deletion in E-Cadherin-Positive Non-ILC Cells
3.7. Ongoing Efforts for the Generation of Additional ILC Lines
4. ILC Cell Line-Based 3D Models and Ex Vivo Patient-Derived 3D Tumor Organoids (PDOs)
5. ILC Mouse Models
5.1. Genetically Engineered ILC Mouse Models
5.1.1. Orthotopic Transplantation of Mouse ILC Tumor Fragments into Syngeneic Mice
5.1.2. GEMM-Derived ILC Lines
5.1.3. Somatic ILC Models
6. Modeling ILC with Xenografts
6.1. Cell Line-Derived Xenografts
6.2. Patient-Derived Xenografts (PDXs)

{kind=link}
{kind=link}
{kind=link}
| Name | Tumor | Tissue | Biomarker (Model) | Laboratory/Institute | Implantation Site | Ref. |
|---|---|---|---|---|---|---|
| Xenografts (cell lines) | ||||||
| SUM-44 PE | ILC | PE | ER+/PR+/−/HER2− | Prof. C. Brisken/EPFL-ISREC | Milk ducts | [160] |
| MDA-MB-134-VI | ILC | PE | ER+/PR+/−/HER2− | Prof. C. Brisken/EPFL-ISREC | Milk ducts | [160] |
| IPH-926 | ILC | MA | ER−/PR−/HER2− | Prof. M. Christgen/MHH | Subcutaneous | [34] |
| PDXs | ||||||
| T69 | ILC | PrBC | ER+/PR+/HER2− | Prof. C. Brisken/EPFL-ISREC | Milk ducts | [176] |
| T73 | ILC | PrBC | ER+/PR−/HER2+ | Prof. C. Brisken/EPFL-ISREC | Milk ducts | [176] |
| T78 | ILC | PrBC | ER+/PR+/HER2− | Prof. C. Brisken/EPFL-ISREC | Milk ducts | [176] |
| T85 | ILC | PrBC | ER+/PR−/HER2− | Prof. C. Brisken/EPFL-ISREC | Milk ducts | [176] |
| T86 | ILC | PrBC | ER+/PR+/HER2− | Prof. C. Brisken/EPFL-ISREC | Milk ducts | [176] |
| LA-PDX1 | ILC | PrBC | ER+/PR+/HER2− | Prof. R. Iggo/BCI | Milk ducts | [177] |
| LA-PDX2 | ILC | PrBC | ER+/PR+/HER2− | Prof. R. Iggo/BCI | Milk ducts | [177] |
| LA-PDX3 | ILC | PrBC | ER+/PR+/HER2− | Prof. R. Iggo/BCI | Milk ducts | [177] |
| LA-PDX4 | ILC | PrBC | ER+/PR+/HER2− | Prof. R. Iggo/BCI | Milk ducts | [177] |
| BCM-3561 | ILC | un | ER−/PR−/HER2ENRICHED | Prof. M.T. Lewis/BCM | Fat pad (mammary) | [178] |
| BCM-4189 | LCIS | MA | ER−/PR−/HER2ENRICHED | Prof. M.T. Lewis/BCM | Fat pad (mammary) | [178] |
| HCI-005 | Mixed NST/ILC | PE | ER+/PR+/HER2+ | Prof. A.L. Welm/HCI | Fat pad (mammary) | [179] |
| HCI-006 | Mixed NST/ILC | PE | ER+/PR+/HER2 (n/a) | Prof. A.L. Welm/HCI | Fat pad (mammary) | [179] |
| HCI-011 | NST | PE | ER+/PR+/HER2− | Prof. A.L. Welm/HCI | Fat pad (mammary) | [179] |
| HCI-013 | ILC | PE | ER+/PR+/HER2− | Prof. A.L. Welm/HCI | Fat pad (mammary) | [179] |
| HCI-013-EI | ILC | PE | ER−/PR−/HER2− | Prof. A.L. Welm/HCI | Fat pad (mammary) | [168,179] |
| HCI-014 | ILC | PE | ER−/PR−/HER2− | Prof. A.L. Welm/HCI | Fat pad (mammary) | [179] |
| HCI-018 | n/a | BrM | ER−/PR−/HER2− | Prof. A.L. Welm/HCI | Fat pad (mammary) | [179] |
| HCI-031 | ILC/LCIS | PE | ER−/PR−/HER2− | Prof. A.L. Welm/HCI | Fat pad (mammary) | [168] |
| HCI-031OV | ILC | OvM * | ER−/PR−/HER2− | Prof. A.L. Welm/HCI | Fat pad (mammary) | [168] |
| WHIM2/5 | Mixed NST/ILC | BrM | ER−/PR−/HER2− | Prof. M. Ellis/WUSTL | Fat pad (mammary) | [180] |
| WHIM9 | Mixed NST/ILC | ns | ER+/PR+/HER2− | Prof. M. Ellis/WUSTL | Fat pad (mammary) | [180] |
| WHIM13 | NST (ILC features) | SkR | ER−/PR−/HER2− | Prof. M. Ellis/WUSTL | Fat pad (mammary) | [180] |
| WHIM20 | Mixed NST/ILC | SkC | ER−/PR−/HER2+ | Prof. M. Ellis/WUSTL | Fat pad (mammary) | [180] |
| WHIM23 | Mixed NST/ILC | SkC | ER−/PR+/HER2− | Prof. M. Ellis/WUSTL | Fat pad (mammary) | [180] |
| HBCx-7 | ILC | PrBC | ER−/PR−/HER2− | Prof. E. Marangoni, Prof. MF. Poupon/IC | Fat pad (Interscapular) | [172] |
| HBCx-19 | ILC | PrBC | ER+/PR−/HER2+ | Prof. E. Marangoni, Prof. MF. Poupon/IC | Fat pad (Interscapular) | [172] |
| HBCx-36 | ILC | PrBC | ER−/PR−/HER2+ | Prof. E. Marangoni, Prof. MF. Poupon/IC | Fat pad (Interscapular) | [172] |
| Met BC 5 | ILC | AF | ER+/PR+/HER2− | Prof. R. Clarke/MBRC | Subcutaneous | [174] |
| Met BC 9 | ILC | AF | ER+/PR+/HER2− | Prof. R. Clarke/MBRC | Subcutaneous | [174] |
| Met BC 11 | ILC | AF | ER+/PR+/HER2− | Prof. R. Clarke/MBRC | Subcutaneous | [174] |
| Met BC 11 | ILC | AF | ER+/PR+/HER2− | Prof. R. Clarke/MBRC | Subcutaneous | [174] |
7. Other Animals
8. ILC Challenges
8.1. Molecular Mechanisms of ILC Drug Resistance
8.2. ILC Tumor Microenvironment
8.3. Modeling ILC Dormancy and Metastasis
9. Strengths and Limitations of Different ILC Models
10. ILC Initiatives and Repositories of ILC Models
11. Discussion
12. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ER | Estrogen Receptor |
| PR | Progesterone Receptor |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| PDX | Patient-Derived Xenograft |
| PDO | Patient-Derived Organoid |
| GEMMs | Genetically Engineered Mouse Models |
| Xenograft | Human-derived cells and tissues transplanted into a recipient of another species (mice, rats, zebrafish, etc.) |
| CaMa | Cancer Mammary |
| NSG | NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ |
| ELBCC | The European Lobular Breast Cancer Consortium |
| COSMIC | Catalogue Omf Somatic Mutations In Cancer |
| ECM | Extracellular matrix |
| CTC | Circulating Tumor Cell |
| DTC | Disseminating Tumor Cell |
| HCMI | Human Cancer Models Initiative |
| KCLB | Korean Cell Line Bank |
| CCLE | Cancer Cell Line Encyclopedia |
| RIKEN | Rikagaku Kenkyūjo Cell Bank |
| ATCC | American Type Culture Collection |
Appendix A
Appendix A.1. Initiatives and Models Repositories
- Platform to bring together discovery scientists, translational researchers, and clinical experts, with the ultimate goal to improve the understanding, diagnosis, treatment, and prognosis of women who have Invasive Lobular Breast Cancer—http://www.elbcc.org/
- Lobular Breast Cancer: Discovery Science, Translational Goals, Clinical Impact—https://www.cost.eu/actions/CA19138/#tabs|Name:overview
- The Lobular Breast Cancer Alliance (LBCA) has compiled a reference list of research publications related to lobular breast cancer—https://lobularbreastcancer.org/
- Patient-Derived Xenograft and Advanced In Vivo Models (PDX-AIM) Core of Baylor College of Medicine. PDX Portal is designed to allow investigators to browse the various organ site-specific PDX collections—https://pdxportal.research.bcm.edu/pdxportal/?dswid=8822
- PDX Finder—a comprehensive open global catalogue of PDX models and their associated data across resources—https://www.pdxfinder.org/
- The PDXNet comprises six PDX Development and Trial Centers (PDTCs) and the PDX Data Commons and Coordinating Center (PDCCC), and is working with the National Cancer Institute to translate PDX studies to clinical trials.
- EurOPDX Consortium is an initiative of translational and clinical researchers counting 18 not-for-profit cancer centers and universities as members across Europe and the US, https://www.europdx.eu/
- The Human and Mouse Linked Evaluation of Tumors (HAMLET) Core developed by Prof. Matthew Ellis, M.D., Ph.D., and Shunqiang Li (Director: Shunqiang Li, Ph.D.—https://digitalcommons.wustl.edu/hamlet/
- Public Repository of Xenografts (ProXe) is an open-source website designed to disseminate information relevant to patient-derived xenografts (PDXs) and PDXs from patients with leukemia or lymphoma by the Weinstock Laboratory. Many models are being licensed to the Jackson Laboratories for industry-scale purposes, including distribution on a fee-for-service basis.
- Cell Model Passports—a hub for clinical, genetic, and functional datasets of preclinical cancer models, https://cellmodelpassports.sanger.ac.uk/ [222].
- NCI Patient-Derived Models Repository (PDMR)—https://pdmr.cancer.gov/
Appendix A.2. Companies
- Novartis Institutes for Biomedical Research PDX Encyclopedia (NIBR PDXE), an industry-led initiative that includes approximately 1000 models
- Horizon (The WHIM, Washington University Human-in-Mouse, collection developed by Shunqiang Li and Matthew Ellis at Washington University and published in Cell Reports). ILC models are reported in Table 5 and Supplementary File S1.4.
- XenOPAT (a spin-off of the Institute of Biomedical Research (IDIBELL), the Catalan Institute of Oncology (ICO) and then University Hospital of Bellvitge).
- Crown Bioscience Inc., http://hubase.crownbio.com/
- Champions TumorGraft® Database—https://database.championsoncology.com/models/filter. *Run PDX studies (in-house) to test experimental oncology compounds.
- Charles River has more than 400 fully characterized proprietary patient-derived xenografts (PDXs), representing major histotypes and tumors and providing extensive background and characterization. No ILC model was found in the database.
References
- Bairoch, A. The Cellosaurus, a Cell-Line Knowledge Resource. J. Biomol. Tech. JBT 2018, 29, 25–38. [Google Scholar] [CrossRef]
- Christgen, M.; Steinemann, D.; Kühnle, E.; Länger, F.; Gluz, O.; Harbeck, N.; Kreipe, H. Lobular Breast Cancer: Clinical, Molecular and Morphological Characteristics. Pathol. Res. Pract. 2016, 212, 583–597. [Google Scholar] [CrossRef]
- Desmedt, C.; Zoppoli, G.; Sotiriou, C.; Salgado, R. Transcriptomic and Genomic Features of Invasive Lobular Breast Cancer. Semin. Cancer Biol. 2017, 44, 98–105. [Google Scholar] [CrossRef]
- Vlug, E.; Ercan, C.; van der Wall, E.; van Diest, P.J.; Derksen, P.W.B. Lobular Breast Cancer: Pathology, Biology, and Options for Clinical Intervention. Arch. Immunol. Ther. Exp. 2014, 62, 7–21. [Google Scholar] [CrossRef]
- Wilson, N.; Ironside, A.; Diana, A.; Oikonomidou, O. Lobular Breast Cancer: A Review. Front. Oncol. 2020, 10, 591399. [Google Scholar] [CrossRef] [PubMed]
- Trapani, D.; Gandini, S.; Corti, C.; Crimini, E.; Bellerba, F.; Minchella, I.; Criscitiello, C.; Tarantino, P.; Curigliano, G. Benefit of Adjuvant Chemotherapy in Patients with Lobular Breast Cancer: A Systematic Review of the Literature and Metanalysis. Cancer Treat. Rev. 2021, 97, 102205. [Google Scholar] [CrossRef]
- Hovis, K.K.; Lee, J.M.; Hippe, D.S.; Linden, H.; Flanagan, M.R.; Kilgore, M.R.; Yee, J.; Partridge, S.C.; Rahbar, H. Accuracy of Preoperative Breast MRI versus Conventional Imaging in Measuring Pathologic Extent of Invasive Lobular Carcinoma. J. Breast Imaging 2021, 3, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Nunes, R.; Sella, T.; Treuner, K.; Atkinson, J.M.; Wong, J.; Zhang, Y.; Exman, P.; Dabbs, D.; Richardson, A.L.; Schnabel, C.A.; et al. Prognostic Utility of Breast Cancer Index to Stratify Distant Recurrence Risk in Invasive Lobular Carcinoma. Clin. Cancer Res. 2021, 27, 5688–5696. [Google Scholar] [CrossRef]
- McCart Reed, A.E.; Kalinowski, L.; Simpson, P.T.; Lakhani, S.R. Invasive Lobular Carcinoma of the Breast: The Increasing Importance of This Special Subtype. Breast Cancer Res. 2021, 23, 6. [Google Scholar] [CrossRef] [PubMed]
- Abouharb, S.; Ensor, J.; Loghin, M.E.; Katz, R.; Moulder, S.L.; Esteva, F.J.; Smith, B.; Valero, V.; Hortobagyi, G.N.; Melhem-Bertrandt, A. Leptomeningeal Disease and Breast Cancer: The Importance of Tumor Subtype. Breast Cancer Res. Treat. 2014, 146, 477–486. [Google Scholar] [CrossRef]
- Blohmer, M.; Zhu, L.; Atkinson, J.M.; Beriwal, S.; Rodríguez-López, J.L.; Rosenzweig, M.; Brufsky, A.M.; Tseng, G.; Lucas, P.C.; Lee, A.V.; et al. Patient Treatment and Outcome after Breast Cancer Orbital and Periorbital Metastases: A Comprehensive Case Series Including Analysis of Lobular versus Ductal Tumor Histology. Breast Cancer Res. 2020, 22, 70. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.; Howell, A.; Chrissohou, M.; Swindell, R.I.; Hudson, M.; Sellwood, R.A. A Comparison of the Metastatic Pattern of Infiltrating Lobular Carcinoma and Infiltrating Duct Carcinoma of the Breast. Br. J. Cancer 1984, 50, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, A.; Rajagopal, P.S.; Villgran, V.; Sandhu, G.S.; Jankowitz, R.C.; Jacob, M.; Rosenzweig, M.; Oesterreich, S.; Brufsky, A. Distinct Pattern of Metastases in Patients with Invasive Lobular Carcinoma of the Breast. Geburtshilfe Frauenheilkd. 2017, 77, 660–666. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, B.; Fong, C.; Luthra, A.; Smith, S.A.; DiNatale, R.G.; Nandakumar, S.; Walch, H.; Chatila, W.K.; Madupuri, R.; Kundra, R.; et al. Genomic Characterization of Metastatic Patterns from Prospective Clinical Sequencing of 25,000. Available online: https://www.biorxiv.org/content/10.1101/2021.06.28.450217v1 (accessed on 1 October 2021).
- Raap, M.; Antonopoulos, W.; Dämmrich, M.; Christgen, H.; Steinmann, D.; Länger, F.; Lehmann, U.; Kreipe, H.; Christgen, M. High Frequency of Lobular Breast Cancer in Distant Metastases to the Orbit. Cancer Med. 2015, 4, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Sledge, G.W.; Chagpar, A.; Perou, C. Collective Wisdom: Lobular Carcinoma of the Breast. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, 18–21. [Google Scholar] [CrossRef]
- Borst, M.J.; Ingold, J.A. Metastatic Patterns of Invasive Lobular versus Invasive Ductal Carcinoma of the Breast. Surgery 1993, 114, 637–641; discussion 641–642. [Google Scholar]
- Inoue, M.; Nakagomi, H.; Nakada, H.; Furuya, K.; Ikegame, K.; Watanabe, H.; Omata, M.; Oyama, T. Specific Sites of Metastases in Invasive Lobular Carcinoma: A Retrospective Cohort Study of Metastatic Breast Cancer. Breast Cancer 2017, 24, 667–672. [Google Scholar] [CrossRef]
- Kutasovic, J.R.; McCart Reed, A.E.; Males, R.; Sim, S.; Saunus, J.M.; Dalley, A.; McEvoy, C.R.; Dedina, L.; Miller, G.; Peyton, S.; et al. Breast Cancer Metastasis to Gynaecological Organs: A Clinico-Pathological and Molecular Profiling Study. J. Pathol. Clin. Res. 2019, 5, 25–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruner, H.C.; Derksen, P.W.B. Loss of E-Cadherin-Dependent Cell-Cell Adhesion and the Development and Progression of Cancer. Cold Spring Harb. Perspect. Biol. 2018, 10, a029330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sflomos, G.; Brisken, C. Breast Cancer Microenvironment and the Metastatic Process. In Breast Cancer; Veronesi, U., Goldhirsch, A., Veronesi, P., Gentilini, O.D., Leonardi, M.C., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 39–48. ISBN 978-3-319-48846-2. [Google Scholar]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [Green Version]
- Desmedt, C.; Zoppoli, G.; Gundem, G.; Pruneri, G.; Larsimont, D.; Fornili, M.; Fumagalli, D.; Brown, D.; Rothé, F.; Vincent, D.; et al. Genomic Characterization of Primary Invasive Lobular Breast Cancer. J. Clin. Oncol. 2016, 34, 1872–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaut, M.; Chin, S.-F.; Majewski, I.; Severson, T.M.; Bismeijer, T.; de Koning, L.; Peeters, J.K.; Schouten, P.C.; Rueda, O.M.; Bosma, A.J.; et al. Integration of Genomic, Transcriptomic and Proteomic Data Identifies Two Biologically Distinct Subtypes of Invasive Lobular Breast Cancer. Sci. Rep. 2016, 6, 18517. [Google Scholar] [CrossRef] [PubMed]
- Derksen, P.W.B.; Liu, X.; Saridin, F.; van der Gulden, H.; Zevenhoven, J.; Evers, B.; van Beijnum, J.R.; Griffioen, A.W.; Vink, J.; Krimpenfort, P.; et al. Somatic Inactivation of E-Cadherin and P53 in Mice Leads to Metastatic Lobular Mammary Carcinoma through Induction of Anoikis Resistance and Angiogenesis. Cancer Cell 2006, 10, 437–449. [Google Scholar] [CrossRef] [Green Version]
- Nagle, A.M.; Levine, K.M.; Tasdemir, N.; Scott, J.A.; Burlbaugh, K.; Kehm, J.; Katz, T.A.; Boone, D.N.; Jacobsen, B.M.; Atkinson, J.M.; et al. Loss of E-Cadherin Enhances IGF1-IGF1R Pathway Activation and Sensitizes Breast Cancers to Anti-IGF1R/InsR Inhibitors. Clin. Cancer Res. 2018, 24, 5165–5177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teo, K.; Gómez-Cuadrado, L.; Tenhagen, M.; Byron, A.; Rätze, M.; van Amersfoort, M.; Renes, J.; Strengman, E.; Mandoli, A.; Singh, A.A.; et al. E-Cadherin Loss Induces Targetable Autocrine Activation of Growth Factor Signalling in Lobular Breast Cancer. Sci. Rep. 2018, 8, 15454. [Google Scholar] [CrossRef] [Green Version]
- Christgen, M.; Cserni, G.; Floris, G.; Marchio, C.; Djerroudi, L.; Kreipe, H.; Derksen, P.W.B.; Vincent-Salomon, A. Lobular Breast Cancer: Histomorphology and Different Concepts of a Special Spectrum of Tumors. Cancers 2021, 13, 3695. [Google Scholar] [CrossRef] [PubMed]
- Desmedt, C.; Salgado, R.; Fornili, M.; Pruneri, G.; Van den Eynden, G.; Zoppoli, G.; Rothé, F.; Buisseret, L.; Garaud, S.; Willard-Gallo, K.; et al. Immune Infiltration in Invasive Lobular Breast Cancer. J. Natl. Cancer Inst. 2018, 110, 768–776. [Google Scholar] [CrossRef]
- Ben-David, U.; Beroukhim, R.; Golub, T.R. Genomic Evolution of Cancer Models: Perils and Opportunities. Nat. Rev. Cancer 2019, 19, 97–109. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Sflomos, G.; Brisken, C. The Challenges of Modeling Hormone Receptor-Positive Breast Cancer in Mice. Endocr. Relat. Cancer 2018, 25, R319–R330. [Google Scholar] [CrossRef] [Green Version]
- Brueffer, C.; Vallon-Christersson, J.; Grabau, D.; Ehinger, A.; Häkkinen, J.; Hegardt, C.; Malina, J.; Chen, Y.; Bendahl, P.-O.; Manjer, J.; et al. Clinical Value of RNA Sequencing-Based Classifiers for Prediction of the Five Conventional Breast Cancer Biomarkers: A Report from the Population-Based Multicenter Sweden Cancerome Analysis Network—Breast Initiative. JCO Precis. Oncol. 2018, 2, 1–18. [Google Scholar] [CrossRef]
- Ethier, S.P.; Mahacek, M.L.; Gullick, W.J.; Frank, T.S.; Weber, B.L. Differential Isolation of Normal Luminal Mammary Epithelial Cells and Breast Cancer Cells from Primary and Metastatic Sites Using Selective Media. Cancer Res. 1993, 53, 627–635. [Google Scholar] [PubMed]
- Christgen, M.; Bruchhardt, H.; Hadamitzky, C.; Rudolph, C.; Steinemann, D.; Gadzicki, D.; Hasemeier, B.; Römermann, D.; Focken, T.; Krech, T.; et al. Comprehensive Genetic and Functional Characterization of IPH-926: A Novel CDH1-Null Tumour Cell Line from Human Lobular Breast Cancer. J. Pathol. 2009, 217, 620–632. [Google Scholar] [CrossRef]
- Fogh, J.; Fogh, J.M.; Orfeo, T. One Hundred and Twenty-Seven Cultured Human Tumor Cell Lines Producing Tumors in Nude Mice. J. Natl. Cancer Inst. 1977, 59, 221–226. [Google Scholar] [CrossRef]
- Domann, F.E.; Rice, J.C.; Hendrix, M.J.; Futscher, B.W. Epigenetic Silencing of Maspin Gene Expression in Human Breast Cancers. Int. J. Cancer 2000, 85, 805–810. [Google Scholar] [CrossRef]
- Rye, P.D.; Norum, L.; Olsen, D.R.; Garman-Vik, S.; Kaul, S.; Fodstad, O. Brain Metastasis Model in Athymic Nude Mice Using a Novel MUC1-Secreting Human Breast-Cancer Cell Line, MA11. Int. J. Cancer 1996, 68, 682–687. [Google Scholar] [CrossRef]
- Jambal, P.; Badtke, M.M.; Harrell, J.C.; Borges, V.F.; Post, M.D.; Sollender, G.E.; Spillman, M.A.; Horwitz, K.B.; Jacobsen, B.M. Estrogen Switches Pure Mucinous Breast Cancer to Invasive Lobular Carcinoma with Mucinous Features. Breast Cancer Res. Treat. 2013, 137, 431–448. [Google Scholar] [CrossRef] [Green Version]
- Cailleau, R.; Olivé, M.; Cruciger, Q.V. Long-Term Human Breast Carcinoma Cell Lines of Metastatic Origin: Preliminary Characterization. In Vitro 1978, 14, 911–915. [Google Scholar] [CrossRef] [PubMed]
- Mattes, M.J.; Cordon-Cardo, C.; Lewis, J.L.; Old, L.J.; Lloyd, K.O. Cell Surface Antigens of Human Ovarian and Endometrial Carcinoma Defined by Mouse Monoclonal Antibodies. Proc. Natl. Acad. Sci. USA 1984, 81, 568–572. [Google Scholar] [CrossRef] [Green Version]
- Lippman, M.; Bolan, G.; Huff, K. The Effects of Estrogens and Antiestrogens on Hormone-Responsive Human Breast Cancer in Long-Term Tissue Culture. Cancer Res. 1976, 36, 4595–4601. [Google Scholar]
- Rostagno, P.; Moll, J.L.; Birtwisle-Peyrottes, I.; Ettore, F.; Lagrange, J.L.; Gioanni, J.; Caldani, C. Effects of Tamoxifen on Potential Doubling Time of Human Breast Cancer Cell Line Determined by Image Cytometry of Double Fluorescent BrdU and DNA Labeling. Anticancer Res. 1994, 14, 2025–2032. [Google Scholar]
- Engel, L.W.; Young, N.A.; Tralka, T.S.; Lippman, M.E.; O’Brien, S.J.; Joyce, M.J. Establishment and Characterization of Three New Continuous Cell Lines Derived from Human Breast Carcinomas. Cancer Res. 1978, 38, 3352–3364. [Google Scholar]
- Gazdar, A.F.; Kurvari, V.; Virmani, A.; Gollahon, L.; Sakaguchi, M.; Westerfield, M.; Kodagoda, D.; Stasny, V.; Cunningham, H.T.; Wistuba, I.I.; et al. Characterization of Paired Tumor and Non-Tumor Cell Lines Established from Patients with Breast Cancer. Int. J. Cancer 1998, 78, 766–774. [Google Scholar] [CrossRef]
- Smith, H.S.; Wolman, S.R.; Dairkee, S.H.; Hancock, M.C.; Lippman, M.; Leff, A.; Hackett, A.J. Immortalization in Culture: Occurrence at a Late Stage in the Progression of Breast Cancer. J. Natl. Cancer Inst. 1987, 78, 611–615. [Google Scholar]
- Lasfargues, E.Y.; Coutinho, W.G. Human Breast Tumor Cells in Culture; New Concepts in Mammary Carcinogenesis. In New Frontiers in Mammary Pathology; Hollmann, K.H., de Brux, J., Verley, J.M., Eds.; Springer US: Boston, MA, USA, 1981; pp. 117–143. ISBN 978-1-4757-0021-3. [Google Scholar]
- Sawada, T.; Chung, Y.S.; Nakata, B.; Kubo, T.; Kondo, Y.; Sogabe, T.; Onoda, N.; Ogawa, Y.; Yamada, N.; Sowa, M. Establishment and characterization of a human breast cancer cell line, OCUB-1. Hum. Cell 1994, 7, 138–144. [Google Scholar]
- Wasielewski, M.; Elstrodt, F.; Klijn, J.G.M.; Berns, E.M.J.J.; Schutte, M. Thirteen New P53 Gene Mutants Identified among 41 Human Breast Cancer Cell Lines. Breast Cancer Res. Treat. 2006, 99, 97–101. [Google Scholar] [CrossRef]
- Barnabas, N.; Cohen, D. Phenotypic and Molecular Characterization of MCF10DCIS and SUM Breast Cancer Cell Lines. Int. J. Breast Cancer 2013, 2013, 872743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmedt, C.; Pingitore, J.; Rothé, F.; Marchio, C.; Clatot, F.; Rouas, G.; Richard, F.; Bertucci, F.; Mariani, O.; Galant, C.; et al. ESR1 Mutations in Metastatic Lobular Breast Cancer Patients. NPJ Breast Cancer 2019, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.-A.; Ribas, R.; Simigdala, N.; Schuster, E.; Pancholi, S.; Tenev, T.; Gellert, P.; Buluwela, L.; Harrod, A.; Thornhill, A.; et al. Discovery of Naturally Occurring ESR1 Mutations in Breast Cancer Cell Lines Modelling Endocrine Resistance. Nat. Commun. 2017, 8, 1865. [Google Scholar] [CrossRef]
- Ethier, S.P.; Guest, S.T.; Garrett-Mayer, E.; Armeson, K.; Wilson, R.C.; Duchinski, K.; Couch, D.; Gray, J.W.; Kappler, C. Development and Implementation of the SUM Breast Cancer Cell Line Functional Genomics Knowledge Base. NPJ Breast Cancer 2020, 6, 30. [Google Scholar] [CrossRef]
- Christgen, M.; Geffers, R.; Kreipe, H.; Lehmann, U. IPH-926 Lobular Breast Cancer Cells Are Triple-Negative but Their Microarray Profile Uncovers a Luminal Subtype. Cancer Sci. 2013, 104, 1726–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christgen, M.; Noskowicz, M.; Heil, C.; Schipper, E.; Christgen, H.; Geffers, R.; Kreipe, H.; Lehmann, U. IPH-926 Lobular Breast Cancer Cells Harbor a P53 Mutant with Temperature-Sensitive Functional Activity and Allow for Profiling of P53-Responsive Genes. Lab. Investig. 2012, 92, 1635–1647. [Google Scholar] [CrossRef] [Green Version]
- Van Agthoven, T.; Dorssers, L.C.J.; Lehmann, U.; Kreipe, H.; Looijenga, L.H.J.; Christgen, M. Breast Cancer Anti-Estrogen Resistance 4 (BCAR4) Drives Proliferation of IPH-926 Lobular Carcinoma Cells. PLoS ONE 2015, 10, e0136845. [Google Scholar] [CrossRef] [Green Version]
- Krech, T.; Scheuerer, E.; Geffers, R.; Kreipe, H.; Lehmann, U.; Christgen, M. ABCB1/MDR1 Contributes to the Anticancer Drug-Resistant Phenotype of IPH-926 Human Lobular Breast Cancer Cells. Cancer Lett. 2012, 315, 153–160. [Google Scholar] [CrossRef]
- Meijer, D.; van Agthoven, T.; Bosma, P.T.; Nooter, K.; Dorssers, L.C.J. Functional Screen for Genes Responsible for Tamoxifen Resistance in Human Breast Cancer Cells. Mol. Cancer Res. MCR 2006, 4, 379–386. [Google Scholar] [CrossRef] [Green Version]
- Reis-Filho, J.S.; Simpson, P.T.; Turner, N.C.; Lambros, M.B.; Jones, C.; Mackay, A.; Grigoriadis, A.; Sarrio, D.; Savage, K.; Dexter, T.; et al. FGFR1 Emerges as a Potential Therapeutic Target for Lobular Breast Carcinomas. Clin. Cancer Res. 2006, 12, 6652–6662. [Google Scholar] [CrossRef] [Green Version]
- Sikora, M.J.; Cooper, K.L.; Bahreini, A.; Luthra, S.; Wang, G.; Chandran, U.R.; Davidson, N.E.; Dabbs, D.J.; Welm, A.L.; Oesterreich, S. Invasive Lobular Carcinoma Cell Lines Are Characterized by Unique Estrogen-Mediated Gene Expression Patterns and Altered Tamoxifen Response. Cancer Res. 2014, 74, 1463–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikora, M.J.; Jankowitz, R.C.; Dabbs, D.J.; Oesterreich, S. Invasive Lobular Carcinoma of the Breast: Patient Response to Systemic Endocrine Therapy and Hormone Response in Model Systems. Steroids 2013, 78, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Pearson, A.; Sharpe, R.; Lambros, M.; Geyer, F.; Lopez-Garcia, M.A.; Natrajan, R.; Marchio, C.; Iorns, E.; Mackay, A.; et al. FGFR1 Amplification Drives Endocrine Therapy Resistance and Is a Therapeutic Target in Breast Cancer. Cancer Res. 2010, 70, 2085–2094. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Basudan, A.; Sikora, M.J.; Bahreini, A.; Tasdemir, N.; Levine, K.M.; Jankowitz, R.C.; McAuliffe, P.F.; Dabbs, D.; Haupt, S.; et al. Frequent Amplifications of ESR1, ERBB2 and MDM4 in Primary Invasive Lobular Breast Carcinoma. Cancer Lett. 2019, 461, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, N.; Bossart, E.A.; Li, Z.; Zhu, L.; Sikora, M.J.; Levine, K.M.; Jacobsen, B.M.; Tseng, G.C.; Davidson, N.E.; Oesterreich, S. Comprehensive Phenotypic Characterization of Human Invasive Lobular Carcinoma Cell Lines in 2D and 3D Cultures. Cancer Res. 2018, 78, 6209–6222. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Pal, A.; Bornmann, W.G.; Yamasaki, F.; Esteva, F.J.; Hortobagyi, G.N.; Bartholomeusz, C.; Ueno, N.T. Activity of Lapatinib Is Independent of EGFR Expression Level in HER2-Overexpressing Breast Cancer Cells. Mol. Cancer Ther. 2008, 7, 1846–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacroix, M.; Leclercq, G. Relevance of Breast Cancer Cell Lines as Models for Breast Tumours: An Update. Breast Cancer Res. Treat. 2004, 83, 249–289. [Google Scholar] [CrossRef] [PubMed]
- Froehlich, K.; Haeger, J.-D.; Heger, J.; Pastuschek, J.; Photini, S.M.; Yan, Y.; Lupp, A.; Pfarrer, C.; Mrowka, R.; Schleußner, E.; et al. Generation of Multicellular Breast Cancer Tumor Spheroids: Comparison of Different Protocols. J. Mammary Gland Biol. Neoplasia 2016, 21, 89–98. [Google Scholar] [CrossRef]
- Pierceall, W.E.; Woodard, A.S.; Morrow, J.S.; Rimm, D.; Fearon, E.R. Frequent Alterations in E-Cadherin and Alpha- and Beta-Catenin Expression in Human Breast Cancer Cell Lines. Oncogene 1995, 11, 1319–1326. [Google Scholar]
- Du Manoir, J.M.; Francia, G.; Man, S.; Mossoba, M.; Medin, J.A.; Viloria-Petit, A.; Hicklin, D.J.; Emmenegger, U.; Kerbel, R.S. Strategies for Delaying or Treating in Vivo Acquired Resistance to Trastuzumab in Human Breast Cancer Xenografts. Clin. Cancer Res. 2006, 12, 904–916. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Pu, X.; Shi, M.; Chen, L.; Qian, L.; Song, Y.; Yuan, G.; Zhang, H.; Yu, M.; Hu, M.; et al. C-Jun, a Crucial Molecule in Metastasis of Breast Cancer and Potential Target for Biotherapy. Oncol. Rep. 2007, 18, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- Vranic, S.; Gatalica, Z.; Wang, Z.-Y. Update on the Molecular Profile of the MDA-MB-453 Cell Line as a Model for Apocrine Breast Carcinoma Studies. Oncol. Lett. 2011, 2, 1131–1137. [Google Scholar] [CrossRef] [Green Version]
- Eusebi, V.; Magalhaes, F.; Azzopardi, J.G. Pleomorphic Lobular Carcinoma of the Breast: An Aggressive Tumor Showing Apocrine Differentiation. Hum. Pathol. 1992, 23, 655–662. [Google Scholar] [CrossRef]
- Singh, G.; Odriozola, L.; Guan, H.; Kennedy, C.R.; Chan, A.M. Characterization of a Novel PTEN Mutation in MDA-MB-453 Breast Carcinoma Cell Line. BMC Cancer 2011, 11, 490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenny, P.A.; Lee, G.Y.; Myers, C.A.; Neve, R.M.; Semeiks, J.R.; Spellman, P.T.; Lorenz, K.; Lee, E.H.; Barcellos-Hoff, M.H.; Petersen, O.W.; et al. The Morphologies of Breast Cancer Cell Lines in Three-Dimensional Assays Correlate with Their Profiles of Gene Expression. Mol. Oncol. 2007, 1, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Koyama, K.; Kamai, Y.; Hirotani, K.; Ogitani, Y.; Zembutsu, A.; Abe, M.; Kaneda, Y.; Maeda, N.; Shiose, Y.; et al. A Novel HER3-Targeting Antibody-Drug Conjugate, U3-1402, Exhibits Potent Therapeutic Efficacy through the Delivery of Cytotoxic Payload by Efficient Internalization. Clin. Cancer Res. 2019, 25, 7151–7161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollestelle, A.; Nagel, J.H.A.; Smid, M.; Lam, S.; Elstrodt, F.; Wasielewski, M.; Ng, S.S.; French, P.J.; Peeters, J.K.; Rozendaal, M.J.; et al. Distinct Gene Mutation Profiles among Luminal-Type and Basal-Type Breast Cancer Cell Lines. Breast Cancer Res. Treat. 2010, 121, 53–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charafe-Jauffret, E.; Ginestier, C.; Monville, F.; Finetti, P.; Adélaïde, J.; Cervera, N.; Fekairi, S.; Xerri, L.; Jacquemier, J.; Birnbaum, D.; et al. Gene Expression Profiling of Breast Cell Lines Identifies Potential New Basal Markers. Oncogene 2006, 25, 2273–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, I.; Batty, E.M.; Pole, J.C.; Blood, K.A.; Mo, S.; Cooke, S.L.; Ng, C.; Howe, K.L.; Chin, S.-F.; Brenton, J.D.; et al. Structural Analysis of the Genome of Breast Cancer Cell Line ZR-75-30 Identifies Twelve Expressed Fusion Genes. BMC Genom. 2012, 13, 719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Wetering, M.; Barker, N.; Harkes, I.C.; van der Heyden, M.; Dijk, N.J.; Hollestelle, A.; Klijn, J.G.; Clevers, H.; Schutte, M. Mutant E-Cadherin Breast Cancer Cells Do Not Display Constitutive Wnt Signaling. Cancer Res. 2001, 61, 278–284. [Google Scholar] [PubMed]
- Dobrynin, Y.V. Establishment and Characteristics of Cell Strains from Some Epithelial Tumors of Human Origin. J. Natl. Cancer Inst. 1963, 31, 1173–1195. [Google Scholar]
- Koch, C.; Kuske, A.; Joosse, S.A.; Yigit, G.; Sflomos, G.; Thaler, S.; Smit, D.J.; Werner, S.; Borgmann, K.; Gärtner, S.; et al. Characterization of Circulating Breast Cancer Cells with Tumorigenic and Metastatic Capacity. EMBO Mol. Med. 2020, 12, e11908. [Google Scholar] [CrossRef]
- Bergeron, A.; MacGrogan, G.; Bertaut, A.; Ladoire, S.; Arveux, P.; Desmoulins, I.; Bonnefoi, H.; Loustalot, C.; Auriol, S.; Beltjens, F.; et al. Triple-Negative Breast Lobular Carcinoma: A Luminal Androgen Receptor Carcinoma with Specific ESRRA Mutations. Mod. Pathol. 2021, 34, 1282–1296. [Google Scholar] [CrossRef]
- Christgen, M.; Bartels, S.; Luft, A.; Persing, S.; Henkel, D.; Lehmann, U.; Kreipe, H. Activating Human Epidermal Growth Factor Receptor 2 (HER2) Gene Mutation in Bone Metastases from Breast Cancer. Virchows Arch. 2018, 473, 577–582. [Google Scholar] [CrossRef]
- Ding, Q.; Chen, H.; Lim, B.; Damodaran, S.; Chen, W.; Tripathy, D.; Piha-Paul, S.; Luthra, R.; Meric-Bernstam, F.; Sahin, A.A. HER2 Somatic Mutation Analysis in Breast Cancer: Correlation with Clinicopathological Features. Hum. Pathol. 2019, 92, 32–38. [Google Scholar] [CrossRef]
- Lien, H.-C.; Chen, Y.-L.; Juang, Y.-L.; Jeng, Y.-M. Frequent Alterations of HER2 through Mutation, Amplification, or Overexpression in Pleomorphic Lobular Carcinoma of the Breast. Breast Cancer Res. Treat. 2015, 150, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Riedlinger, G.M.; Joshi, S.; Hirshfield, K.M.; Barnard, N.; Ganesan, S. Targetable Alterations in Invasive Pleomorphic Lobular Carcinoma of the Breast. Breast Cancer Res. 2021, 23, 7. [Google Scholar] [CrossRef]
- Ross, J.S.; Gay, L.M.; Wang, K.; Ali, S.M.; Chumsri, S.; Elvin, J.A.; Bose, R.; Vergilio, J.-A.; Suh, J.; Yelensky, R.; et al. Nonamplification ERBB2 Genomic Alterations in 5605 Cases of Recurrent and Metastatic Breast Cancer: An Emerging Opportunity for Anti-HER2 Targeted Therapies. Cancer 2016, 122, 2654–2662. [Google Scholar] [CrossRef]
- Chan, A.O.-O.; Lam, S.-K.; Wong, B.C.-Y.; Wong, W.-M.; Yuen, M.-F.; Yeung, Y.-H.; Hui, W.-M.; Rashid, A.; Kwong, Y.-L. Promoter Methylation of E-Cadherin Gene in Gastric Mucosa Associated with Helicobacter Pylori Infection and in Gastric Cancer. Gut 2003, 52, 502–506. [Google Scholar] [CrossRef] [Green Version]
- Chiles, M.C.; Ai, L.; Zuo, C.; Fan, C.-Y.; Smoller, B.R. E-Cadherin Promoter Hypermethylation in Preneoplastic and Neoplastic Skin Lesions. Mod. Pathol. 2003, 16, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Droufakou, S.; Deshmane, V.; Roylance, R.; Hanby, A.; Tomlinson, I.; Hart, I.R. Multiple Ways of Silencing E-Cadherin Gene Expression in Lobular Carcinoma of the Breast. Int. J. Cancer 2001, 92, 404–408. [Google Scholar] [CrossRef] [Green Version]
- Bajrami, I.; Marlow, R.; van de Ven, M.; Brough, R.; Pemberton, H.N.; Frankum, J.; Song, F.; Rafiq, R.; Konde, A.; Krastev, D.B.; et al. E-Cadherin/ROS1 Inhibitor Synthetic Lethality in Breast Cancer. Cancer Discov. 2018, 8, 498–515. [Google Scholar] [CrossRef] [Green Version]
- Bignell, G.R.; Greenman, C.D.; Davies, H.; Butler, A.P.; Edkins, S.; Andrews, J.M.; Buck, G.; Chen, L.; Beare, D.; Latimer, C.; et al. Signatures of Mutation and Selection in the Cancer Genome. Nature 2010, 463, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Cailleau, R.; Young, R.; Olivé, M.; Reeves, W.J. Breast Tumor Cell Lines from Pleural Effusions. J. Natl. Cancer Inst. 1974, 53, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Young, R.K.; Cailleau, R.M.; Mackay, B.; Reeves, W.J. Establishment of Epithelial Cell Line MDA-MB-157 from Metastatic Pleural Effusion of Human Breast Carcinoma. In Vitro 1974, 9, 239–245. [Google Scholar] [CrossRef]
- Hackett, A.J.; Smith, H.S.; Springer, E.L.; Owens, R.B.; Nelson-Rees, W.A.; Riggs, J.L.; Gardner, M.B. Two Syngeneic Cell Lines from Human Breast Tissue: The Aneuploid Mammary Epithelial (Hs578T) and the Diploid Myoepithelial (Hs578Bst) Cell Lines. J. Natl. Cancer Inst. 1977, 58, 1795–1806. [Google Scholar] [CrossRef]
- Morton, R.A.; Ewing, C.M.; Nagafuchi, A.; Tsukita, S.; Isaacs, W.B. Reduction of E-Cadherin Levels and Deletion of the Alpha-Catenin Gene in Human Prostate Cancer Cells. Cancer Res. 1993, 53, 3585–3590. [Google Scholar]
- Zhao, C.; Thompson, B.J.; Chen, K.; Gao, F.; Blouw, B.; Marella, M.; Zimmerman, S.; Kimbler, T.; Garrovillo, S.; Bahn, J.; et al. The Growth of a Xenograft Breast Cancer Tumor Model with Engineered Hyaluronan-Accumulating Stroma Is Dependent on Hyaluronan and Independent of CD44. Oncotarget 2019, 10, 6561–6576. [Google Scholar] [CrossRef]
- Saunus, J.M.; Smart, C.E.; Kutasovic, J.R.; Johnston, R.L.; Kalita-de Croft, P.; Miranda, M.; Rozali, E.N.; Vargas, A.C.; Reid, L.E.; Lorsy, E.; et al. Multidimensional Phenotyping of Breast Cancer Cell Lines to Guide Preclinical Research. Breast Cancer Res. Treat. 2018, 167, 289–301. [Google Scholar] [CrossRef] [Green Version]
- Finlay-Schultz, J.; Jacobsen, B.M.; Riley, D.; Paul, K.V.; Turner, S.; Ferreira-Gonzalez, A.; Harrell, J.C.; Kabos, P.; Sartorius, C.A. New Generation Breast Cancer Cell Lines Developed from Patient-Derived Xenografts. Breast Cancer Res. 2020, 22, 68. [Google Scholar] [CrossRef] [PubMed]
- Ilina, O.; Gritsenko, P.G.; Syga, S.; Lippoldt, J.; La Porta, C.A.M.; Chepizhko, O.; Grosser, S.; Vullings, M.; Bakker, G.-J.; Starruß, J.; et al. Cell-Cell Adhesion and 3D Matrix Confinement Determine Jamming Transitions in Breast Cancer Invasion. Nat. Cell Biol. 2020, 22, 1103–1115. [Google Scholar] [CrossRef]
- Chen, A.; Beetham, H.; Black, M.A.; Priya, R.; Telford, B.J.; Guest, J.; Wiggins, G.A.R.; Godwin, T.D.; Yap, A.S.; Guilford, P.J. E-Cadherin Loss Alters Cytoskeletal Organization and Adhesion in Non-Malignant Breast Cells but Is Insufficient to Induce an Epithelial-Mesenchymal Transition. BMC Cancer 2014, 14, 552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Ding, K.; Priedigkeit, N.; Elangovan, A.; Levine, K.M.; Carleton, N.; Savariau, L.; Atkinson, J.M.; Oesterreich, S.; Lee, A.V. Single-Cell Transcriptomic Heterogeneity in Invasive Ductal and Lobular Breast Cancer Cells. Cancer Res. 2021, 81, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Ganz, H.M.; Buchmann, B.; Engelbrecht, L.K.; Jesinghaus, M.; Eichelberger, L.; Gabka, C.J.; Schmidt, G.P.; Muckenhuber, A.; Weichert, W.; Bausch, A.R.; et al. Generation of Ductal Organoids from Normal Mammary Luminal Cells Reveals Invasive Potential. J. Pathol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hugo, H.J.; Gunasinghe, N.P.a.D.; Hollier, B.G.; Tanaka, T.; Blick, T.; Toh, A.; Hill, P.; Gilles, C.; Waltham, M.; Thompson, E.W. Epithelial Requirement for in vitro Proliferation and Xenograft Growth and Metastasis of MDA-MB-468 Human Breast Cancer Cells: Oncogenic Rather than Tumor-Suppressive Role of E-Cadherin. Breast Cancer Res. 2017, 19, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol, E.S.; Feng, Y.X.; Jin, D.X.; Basudan, A.; Lee, A.V.; Atkinson, J.M.; Chen, J.; Stephens, P.J.; Frampton, G.M.; Gupta, P.B.; et al. Loss of Function of NF1 Is a Mechanism of Acquired Resistance to Endocrine Therapy in Lobular Breast Cancer. Ann. Oncol. 2019, 30, 115–123. [Google Scholar] [CrossRef]
- Telford, B.J.; Chen, A.; Beetham, H.; Frick, J.; Brew, T.P.; Gould, C.M.; Single, A.; Godwin, T.; Simpson, K.J.; Guilford, P. Synthetic Lethal Screens Identify Vulnerabilities in GPCR Signaling and Cytoskeletal Organization in E-Cadherin-Deficient Cells. Mol. Cancer Ther. 2015, 14, 1213–1223. [Google Scholar] [CrossRef] [Green Version]
- Aouad, P.; Zhang, Y.; Stibolt, C.; Mani, S.A.; Sflomos, G.; Brisken, C. Epithelial-Mesenchymal Plasticity Determines Estrogen Receptor Positive (ER+) Breast Cancer Dormancy and Reacquisition of an Epithelial State Drives Awakening. Available online: https://www.biorxiv.org/content/10.1101/2021.07.22.453458v1 (accessed on 1 October 2021).
- Padmanaban, V.; Krol, I.; Suhail, Y.; Szczerba, B.M.; Aceto, N.; Bader, J.S.; Ewald, A.J. E-Cadherin Is Required for Metastasis in Multiple Models of Breast Cancer. Nature 2019, 573, 439–444. [Google Scholar] [CrossRef]
- Hornsveld, M.; Tenhagen, M.; van de Ven, R.A.; Smits, A.M.M.; van Triest, M.H.; van Amersfoort, M.; Kloet, D.E.A.; Dansen, T.B.; Burgering, B.M.; Derksen, P.W.B. Restraining FOXO3-Dependent Transcriptional BMF Activation Underpins Tumour Growth and Metastasis of E-Cadherin-Negative Breast Cancer. Cell Death Differ. 2016, 23, 1483–1492. [Google Scholar] [CrossRef] [Green Version]
- Settleman, J.; Neto, J.M.F.; Bernards, R. Thinking Differently about Cancer Treatment Regimens. Cancer Discov. 2021, 11, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Filipe, E.C.; Parker, A.L.; Cadell, A.L.; Major, G.; Croucher, D.R.; Cox, T.R. In vitro 3D Models of Tunable Stiffness. Methods Mol. Biol. Clifton NJ 2021, 2294, 27–42. [Google Scholar] [CrossRef]
- Kim, J.; Koo, B.-K.; Knoblich, J.A. Human Organoids: Model Systems for Human Biology and Medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Kutys, M.L.; Polacheck, W.J.; Welch, M.K.; Gagnon, K.A.; Koorman, T.; Kim, S.; Li, L.; McClatchey, A.I.; Chen, C.S. Uncovering Mutation-Specific Morphogenic Phenotypes and Paracrine-Mediated Vessel Dysfunction in a Biomimetic Vascularized Mammary Duct Platform. Nat. Commun. 2020, 11, 3377. [Google Scholar] [CrossRef] [PubMed]
- Santo, V.E.; Rebelo, S.P.; Estrada, M.F.; Alves, P.M.; Boghaert, E.; Brito, C. Drug Screening in 3D in vitro Tumor Models: Overcoming Current Pitfalls of Efficacy Read-Outs. Biotechnol. J. 2017, 12, 1600505. [Google Scholar] [CrossRef]
- Stock, K.; Estrada, M.F.; Vidic, S.; Gjerde, K.; Rudisch, A.; Santo, V.E.; Barbier, M.; Blom, S.; Arundkar, S.C.; Selvam, I.; et al. Capturing Tumor Complexity in vitro: Comparative Analysis of 2D and 3D Tumor Models for Drug Discovery. Sci. Rep. 2016, 6, 28951. [Google Scholar] [CrossRef] [Green Version]
- Franchi-Mendes, T.; Eduardo, R.; Domenici, G.; Brito, C. 3D Cancer Models: Depicting Cellular Crosstalk within the Tumour Microenvironment. Cancers 2021, 13, 4610. [Google Scholar] [CrossRef]
- Manuel Iglesias, J.; Beloqui, I.; Garcia-Garcia, F.; Leis, O.; Vazquez-Martin, A.; Eguiara, A.; Cufi, S.; Pavon, A.; Menendez, J.A.; Dopazo, J.; et al. Mammosphere Formation in Breast Carcinoma Cell Lines Depends upon Expression of E-Cadherin. PLoS ONE 2013, 8, e77281. [Google Scholar] [CrossRef] [Green Version]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386. [Google Scholar] [CrossRef] [Green Version]
- Campaner, E.; Zannini, A.; Santorsola, M.; Bonazza, D.; Bottin, C.; Cancila, V.; Tripodo, C.; Bortul, M.; Zanconati, F.; Schoeftner, S.; et al. Breast Cancer Organoids Model Patient-Specific Response to Drug Treatment. Cancers 2020, 12, 3869. [Google Scholar] [CrossRef]
- Rosenbluth, J.M.; Schackmann, R.C.J.; Gray, G.K.; Selfors, L.M.; Li, C.M.-C.; Boedicker, M.; Kuiken, H.J.; Richardson, A.; Brock, J.; Garber, J.; et al. Organoid Cultures from Normal and Cancer-Prone Human Breast Tissues Preserve Complex Epithelial Lineages. Nat. Commun. 2020, 11, 1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livingston, M.K.; Morgan, M.M.; Daly, W.T.; Murphy, W.L.; Johnson, B.P.; Beebe, D.J.; Virumbrales-Muñoz, M. Evaluation of PEG-Based Hydrogel Influence on Estrogen Receptor Driven Responses in MCF7 Breast Cancer Cells. ACS Biomater. Sci. Eng. 2019, 5, 6089–6098. [Google Scholar] [CrossRef] [PubMed]
- Zimoch, J.; Padial, J.S.; Klar, A.S.; Vallmajo-Martin, Q.; Meuli, M.; Biedermann, T.; Wilson, C.J.; Rowan, A.; Reichmann, E. Polyisocyanopeptide Hydrogels: A Novel Thermo-Responsive Hydrogel Supporting Pre-Vascularization and the Development of Organotypic Structures. Acta Biomater. 2018, 70, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; van Vliet, E.J.; Sachs, N.; Rosenbluth, J.M.; Kopper, O.; Rebel, H.G.; Wehrens, E.J.; Piani, C.; Visvader, J.E.; Verissimo, C.S.; et al. Long-Term Culture, Genetic Manipulation and Xenotransplantation of Human Normal and Breast Cancer Organoids. Nat. Protoc. 2021, 16, 1936–1965. [Google Scholar] [CrossRef] [PubMed]
- Cartaxo, A.L.; Estrada, M.F.; Domenici, G.; Roque, R.; Silva, F.; Gualda, E.J.; Loza-Alvarez, P.; Sflomos, G.; Brisken, C.; Alves, P.M.; et al. A Novel Culture Method That Sustains ERα Signaling in Human Breast Cancer Tissue Microstructures. J. Exp. Clin. Cancer Res. 2020, 39, 161. [Google Scholar] [CrossRef] [PubMed]
- Boussadia, O.; Kutsch, S.; Hierholzer, A.; Delmas, V.; Kemler, R. E-Cadherin Is a Survival Factor for the Lactating Mouse Mammary Gland. Mech. Dev. 2002, 115, 53–62. [Google Scholar] [CrossRef]
- Boelens, M.C.; Nethe, M.; Klarenbeek, S.; de Ruiter, J.R.; Schut, E.; Bonzanni, N.; Zeeman, A.L.; Wientjens, E.; van der Burg, E.; Wessels, L.; et al. PTEN Loss in E-Cadherin-Deficient Mouse Mammary Epithelial Cells Rescues Apoptosis and Results in Development of Classical Invasive Lobular Carcinoma. Cell Rep. 2016, 16, 2087–2101. [Google Scholar] [CrossRef] [Green Version]
- An, Y.; Adams, J.R.; Hollern, D.P.; Zhao, A.; Chang, S.G.; Gams, M.S.; Chung, P.E.D.; He, X.; Jangra, R.; Shah, J.S.; et al. Cdh1 and Pik3ca Mutations Cooperate to Induce Immune-Related Invasive Lobular Carcinoma of the Breast. Cell Rep. 2018, 25, 702–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annunziato, S.; Kas, S.M.; Nethe, M.; Yücel, H.; Del Bravo, J.; Pritchard, C.; Bin Ali, R.; van Gerwen, B.; Siteur, B.; Drenth, A.P.; et al. Modeling Invasive Lobular Breast Carcinoma by CRISPR/Cas9-Mediated Somatic Genome Editing of the Mammary Gland. Genes Dev. 2016, 30, 1470–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kas, S.M.; de Ruiter, J.R.; Schipper, K.; Annunziato, S.; Schut, E.; Klarenbeek, S.; Drenth, A.P.; van der Burg, E.; Klijn, C.; Ten Hoeve, J.J.; et al. Insertional Mutagenesis Identifies Drivers of a Novel Oncogenic Pathway in Invasive Lobular Breast Carcinoma. Nat. Genet. 2017, 49, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Schipper, K.; Drenth, A.P.; van der Burg, E.; Cornelissen, S.; Klarenbeek, S.; Nethe, M.; Jonkers, J. Truncated ASPP2 Drives Initiation and Progression of Invasive Lobular Carcinoma via Distinct Mechanisms. Cancer Res. 2020, 80, 1486–1497. [Google Scholar] [CrossRef] [Green Version]
- Cornelissen, L.M.; Drenth, A.P.; van der Burg, E.; de Bruijn, R.; Pritchard, C.E.J.; Huijbers, I.J.; Zwart, W.; Jonkers, J. TRPS1 Acts as a Context-Dependent Regulator of Mammary Epithelial Cell Growth/Differentiation and Breast Cancer Development. Genes Dev. 2020, 34, 179–193. [Google Scholar] [CrossRef]
- Derksen, P.W.B.; Braumuller, T.M.; van der Burg, E.; Hornsveld, M.; Mesman, E.; Wesseling, J.; Krimpenfort, P.; Jonkers, J. Mammary-Specific Inactivation of E-Cadherin and P53 Impairs Functional Gland Development and Leads to Pleomorphic Invasive Lobular Carcinoma in Mice. Dis. Model. Mech. 2011, 4, 347–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schackmann, R.C.J.; van Amersfoort, M.; Haarhuis, J.H.I.; Vlug, E.J.; Halim, V.A.; Roodhart, J.M.L.; Vermaat, J.S.; Voest, E.E.; van der Groep, P.; van Diest, P.J.; et al. Cytosolic P120-Catenin Regulates Growth of Metastatic Lobular Carcinoma through Rock1-Mediated Anoikis Resistance. J. Clin. Investig. 2011, 121, 3176–3188. [Google Scholar] [CrossRef]
- Tenhagen, M.; Klarenbeek, S.; Braumuller, T.M.; Hofmann, I.; van der Groep, P.; Ter Hoeve, N.; van der Wall, E.; Jonkers, J.; Derksen, P.W.B. P120-Catenin Is Critical for the Development of Invasive Lobular Carcinoma in Mice. J. Mammary Gland Biol. Neoplasia 2016, 21, 81–88. [Google Scholar] [CrossRef] [Green Version]
- De Groot, J.S.; Ratze, M.A.; van Amersfoort, M.; Eisemann, T.; Vlug, E.J.; Niklaas, M.T.; Chin, S.-F.; Caldas, C.; van Diest, P.J.; Jonkers, J.; et al. AE-Catenin Is a Candidate Tumor Suppressor for the Development of E-Cadherin-Expressing Lobular-Type Breast Cancer. J. Pathol. 2018, 245, 456–467. [Google Scholar] [CrossRef]
- Kas, S.M.; de Ruiter, J.R.; Schipper, K.; Schut, E.; Bombardelli, L.; Wientjens, E.; Drenth, A.P.; de Korte-Grimmerink, R.; Mahakena, S.; Phillips, C.; et al. Transcriptomics and Transposon Mutagenesis Identify Multiple Mechanisms of Resistance to the FGFR Inhibitor AZD4547. Cancer Res. 2018, 78, 5668–5679. [Google Scholar] [CrossRef] [Green Version]
- Klarenbeek, S.; Doornebal, C.W.; Kas, S.M.; Bonzanni, N.; Bhin, J.; Braumuller, T.M.; van der Heijden, I.; Opdam, M.; Schouten, P.C.; Kersten, K.; et al. Response of Metastatic Mouse Invasive Lobular Carcinoma to MTOR Inhibition Is Partly Mediated by the Adaptive Immune System. Oncoimmunology 2020, 9, 1724049. [Google Scholar] [CrossRef] [Green Version]
- Doornebal, C.W.; Klarenbeek, S.; Braumuller, T.M.; Klijn, C.N.; Ciampricotti, M.; Hau, C.-S.; Hollmann, M.W.; Jonkers, J.; de Visser, K.E. A Preclinical Mouse Model of Invasive Lobular Breast Cancer Metastasis. Cancer Res. 2013, 73, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffelt, S.B.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.-S.; Verstegen, N.J.M.; Ciampricotti, M.; Hawinkels, L.J.A.C.; Jonkers, J.; et al. IL-17-Producing Γδ T Cells and Neutrophils Conspire to Promote Breast Cancer Metastasis. Nature 2015, 522, 345–348. [Google Scholar] [CrossRef]
- Kersten, K.; Coffelt, S.B.; Hoogstraat, M.; Verstegen, N.J.M.; Vrijland, K.; Ciampricotti, M.; Doornebal, C.W.; Hau, C.-S.; Wellenstein, M.D.; Salvagno, C.; et al. Mammary Tumor-Derived CCL2 Enhances pro-Metastatic Systemic Inflammation through Upregulation of IL1β in Tumor-Associated Macrophages. OncoImmunology 2017, 6, e1334744. [Google Scholar] [CrossRef] [PubMed]
- Richard, F.; Majjaj, S.; Venet, D.; Rothé, F.; Pingitore, J.; Boeckx, B.; Marchio, C.; Clatot, F.; Bertucci, F.; Mariani, O.; et al. Characterization of Stromal Tumor-Infiltrating Lymphocytes and Genomic Alterations in Metastatic Lobular Breast Cancer. Clin. Cancer Res. 2020, 26, 6254–6265. [Google Scholar] [CrossRef] [PubMed]
- Hawinkels, L.J.A.C.; de Vinuesa, A.G.; Paauwe, M.; Kruithof-de Julio, M.; Wiercinska, E.; Pardali, E.; Mezzanotte, L.; Keereweer, S.; Braumuller, T.M.; Heijkants, R.C.; et al. Activin Receptor-like Kinase 1 Ligand Trap Reduces Microvascular Density and Improves Chemotherapy Efficiency to Various Solid Tumors. Clin. Cancer Res. 2016, 22, 96–106. [Google Scholar] [CrossRef] [Green Version]
- Xian, W.; Pappas, L.; Pandya, D.; Selfors, L.M.; Derksen, P.W.; de Bruin, M.; Gray, N.S.; Jonkers, J.; Rosen, J.M.; Brugge, J.S. Fibroblast Growth Factor Receptor 1-Transformed Mammary Epithelial Cells Are Dependent on RSK Activity for Growth and Survival. Cancer Res. 2009, 69, 2244–2251. [Google Scholar] [CrossRef] [Green Version]
- Huijbers, I.J.; Del Bravo, J.; Bin Ali, R.; Pritchard, C.; Braumuller, T.M.; van Miltenburg, M.H.; Henneman, L.; Michalak, E.M.; Berns, A.; Jonkers, J. Using the GEMM-ESC Strategy to Study Gene Function in Mouse Models. Nat. Protoc. 2015, 10, 1755–1785. [Google Scholar] [CrossRef]
- Linzell, J.L.; Peaker, M. Permeability of Mammary Ducts in the Lactating Goat. J. Physiol. 1971, 213, 48P–49P. [Google Scholar]
- Schipper, K.; Seinstra, D.; Paulien Drenth, A.; van der Burg, E.; Ramovs, V.; Sonnenberg, A.; van Rheenen, J.; Nethe, M.; Jonkers, J. Rebalancing of Actomyosin Contractility Enables Mammary Tumor Formation upon Loss of E-Cadherin. Nat. Commun. 2019, 10, 3800. [Google Scholar] [CrossRef] [Green Version]
- Annunziato, S.; Lutz, C.; Henneman, L.; Bhin, J.; Wong, K.; Siteur, B.; van Gerwen, B.; de Korte-Grimmerink, R.; Zafra, M.P.; Schatoff, E.M.; et al. In Situ CRISPR-Cas9 Base Editing for the Development of Genetically Engineered Mouse Models of Breast Cancer. EMBO J. 2020, 39, e102169. [Google Scholar] [CrossRef]
- Song, C.-Q.; Jiang, T.; Richter, M.; Rhym, L.H.; Koblan, L.W.; Zafra, M.P.; Schatoff, E.M.; Doman, J.L.; Cao, Y.; Dow, L.E.; et al. Adenine Base Editing in an Adult Mouse Model of Tyrosinaemia. Nat. Biomed. Eng. 2020, 4, 125–130. [Google Scholar] [CrossRef] [Green Version]
- Dobrolecki, L.E.; Airhart, S.D.; Alferez, D.G.; Aparicio, S.; Behbod, F.; Bentires-Alj, M.; Brisken, C.; Bult, C.J.; Cai, S.; Clarke, R.B.; et al. Patient-Derived Xenograft (PDX) Models in Basic and Translational Breast Cancer Research. Cancer Metastasis Rev. 2016, 35, 547–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrell, J.C.; Shroka, T.M.; Jacobsen, B.M. Estrogen Induces C-Kit and an Aggressive Phenotype in a Model of Invasive Lobular Breast Cancer. Oncogenesis 2017, 6, 396. [Google Scholar] [CrossRef]
- Gustin, J.P.; Miller, J.; Farag, M.; Rosen, D.M.; Thomas, M.; Scharpf, R.B.; Lauring, J. GATA3 Frameshift Mutation Promotes Tumor Growth in Human Luminal Breast Cancer Cells and Induces Transcriptional Changes Seen in Primary GATA3 Mutant Breast Cancers. Oncotarget 2017, 8, 103415–103427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinchy, B.; Gazdar, A.; Rabinovsky, R.; Yefenof, E.; Gordon, B.; Vitetta, E.S. The Growth and Metastasis of Human, HER-2/Neu-Overexpressing Tumor Cell Lines in Male SCID Mice. Breast Cancer Res. Treat. 2000, 61, 217–228. [Google Scholar] [CrossRef]
- Christenson, J.L.; O’Neill, K.I.; Williams, M.M.; Spoelstra, N.S.; Jones, K.L.; Trahan, G.D.; Reese, J.; Van Patten, E.T.; Elias, A.; Eisner, J.R.; et al. Activity of Combined Androgen Receptor Antagonism and Cell Cycle Inhibition in Androgen Receptor-Positive Triple-Negative Breast Cancer. Mol. Cancer Ther. 2021, 20, 1062–1071. [Google Scholar] [CrossRef]
- Haricharan, S.; Lei, J.; Ellis, M. Mammary Ductal Environment Is Necessary for Faithful Maintenance of Estrogen Signaling in ER+ Breast Cancer. Cancer Cell 2016, 29, 249–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sflomos, G.; Dormoy, V.; Metsalu, T.; Jeitziner, R.; Battista, L.; Scabia, V.; Raffoul, W.; Delaloye, J.-F.; Treboux, A.; Fiche, M.; et al. A Preclinical Model for ERα-Positive Breast Cancer Points to the Epithelial Microenvironment as Determinant of Luminal Phenotype and Hormone Response. Cancer Cell 2016, 29, 407–422. [Google Scholar] [CrossRef] [Green Version]
- Shamseddin, M.; De Martino, F.; Constantin, C.; Scabia, V.; Lancelot, A.-S.; Laszlo, C.; Ayyannan, A.; Battista, L.; Raffoul, W.; Gailloud-Matthieu, M.-C.; et al. Contraceptive Progestins with Androgenic Properties Stimulate Breast Epithelial Cell Proliferation. EMBO Mol. Med. 2021, 13, e14314. [Google Scholar] [CrossRef]
- Behbod, F.; Kittrell, F.S.; LaMarca, H.; Edwards, D.; Kerbawy, S.; Heestand, J.C.; Young, E.; Mukhopadhyay, P.; Yeh, H.-W.; Allred, D.C.; et al. An Intraductal Human-in-Mouse Transplantation Model Mimics the Subtypes of Ductal Carcinoma in Situ. Breast Cancer Res. 2009, 11, R66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdez, K.E.; Fan, F.; Smith, W.; Allred, D.C.; Medina, D.; Behbod, F. Human Primary Ductal Carcinoma in Situ (DCIS) Subtype-Specific Pathology Is Preserved in a Mouse Intraductal (MIND) Xenograft Model. J. Pathol. 2011, 225, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Clarke, R.; Jones, B.C.; Sevigny, C.M.; Hilakivi-Clarke, L.A.; Sengupta, S. Experimental Models of Endocrine Responsive Breast Cancer: Strengths, Limitations, and Use. Cancer Drug Resist. 2021, 4, 762–783. [Google Scholar] [CrossRef]
- Matthews, S.B.; Sartorius, C.A. Steroid Hormone Receptor Positive Breast Cancer Patient-Derived Xenografts. Horm. Cancer 2017, 8, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Sflomos, G.; Battista, L.; Aouad, P.; De Martino, F.; Scabia, V.; Stravodimou, A.; Ayyanan, A.; Ifticene-Treboux, A.; RLS; Bucher, P.; et al. Intraductal Xenografts Show Lobular Carcinoma Cells Rely on Their Own Extracellular Matrix and LOXL1. EMBO Mol. Med. 2021, 13, e13180. [Google Scholar] [CrossRef]
- Kozma, K.J.; Done, S.J.; Egan, S.E. The Tumor Cell-Derived Matrix of Lobular Breast Cancer: A New Vulnerability. EMBO Mol. Med. 2021, 13, e13807. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, S.; Hidalgo, M.; Kung, A.L. Examining the Utility of Patient-Derived Xenograft Mouse Models. Nat. Rev. Cancer 2015, 15, 311–316. [Google Scholar] [CrossRef]
- Guillen, K.P.; Fujita, M.; Butterfield, A.J.; Scherer, S.D.; Bailey, M.H.; Chu, Z.; DeRose, Y.S.; Zhao, L.; Cortes-Sanchez, E.; Yang, C.-H.; et al. A Breast Cancer Patient-Derived Xenograft and Organoid Platform for Drug Discovery and Precision Oncology. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ledford, H. US Cancer Institute to Overhaul Tumour Cell Lines. Nature 2016, 530, 391. [Google Scholar] [CrossRef] [Green Version]
- Eirew, P.; Steif, A.; Khattra, J.; Ha, G.; Yap, D.; Farahani, H.; Gelmon, K.; Chia, S.; Mar, C.; Wan, A.; et al. Dynamics of Genomic Clones in Breast Cancer Patient Xenografts at Single-Cell Resolution. Nature 2015, 518, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient-Derived Xenograft Models: An Emerging Platform for Translational Cancer Research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoge, A.C.H.; Getz, M.; Beroukhim, R.; Golub, T.R.; Ha, G.; Ben-David, U. DNA-Based Copy Number Analysis Confirms Genomic Evolution of PDX Models. Available online: https://www.biorxiv.org/content/10.1101/2021.01.15.426865v1 (accessed on 1 October 2021).
- Woo, X.Y.; Giordano, J.; Srivastava, A.; Zhao, Z.-M.; Lloyd, M.W.; de Bruijn, R.; Suh, Y.-S.; Patidar, R.; Chen, L.; Scherer, S.; et al. Conservation of Copy Number Profiles during Engraftment and Passaging of Patient-Derived Cancer Xenografts. Nat. Genet. 2021, 53, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-Throughput Screening Using Patient-Derived Tumor Xenografts to Predict Clinical Trial Drug Response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Meehan, T.F.; Conte, N.; Goldstein, T.; Inghirami, G.; Murakami, M.A.; Brabetz, S.; Gu, Z.; Wiser, J.A.; Dunn, P.; Begley, D.A.; et al. PDX-MI: Minimal Information for Patient-Derived Tumor Xenograft Models. Cancer Res. 2017, 77, e62–e66. [Google Scholar] [CrossRef] [Green Version]
- Cottu, P.; Marangoni, E.; Assayag, F.; de Cremoux, P.; Vincent-Salomon, A.; Guyader, C.; de Plater, L.; Elbaz, C.; Karboul, N.; Fontaine, J.J.; et al. Modeling of Response to Endocrine Therapy in a Panel of Human Luminal Breast Cancer Xenografts. Breast Cancer Res. Treat. 2012, 133, 595–606. [Google Scholar] [CrossRef]
- Marangoni, E.; Vincent-Salomon, A.; Auger, N.; Degeorges, A.; Assayag, F.; de Cremoux, P.; de Plater, L.; Guyader, C.; De Pinieux, G.; Judde, J.-G.; et al. A New Model of Patient Tumor-Derived Breast Cancer Xenografts for Preclinical Assays. Clin. Cancer Res. 2007, 13, 3989–3998. [Google Scholar] [CrossRef] [Green Version]
- Reyal, F.; Guyader, C.; Decraene, C.; Lucchesi, C.; Auger, N.; Assayag, F.; De Plater, L.; Gentien, D.; Poupon, M.-F.; Cottu, P.; et al. Molecular Profiling of Patient-Derived Breast Cancer Xenografts. Breast Cancer Res. 2012, 14, R11. [Google Scholar] [CrossRef] [PubMed]
- Simões, B.M.; O’Brien, C.S.; Eyre, R.; Silva, A.; Yu, L.; Sarmiento-Castro, A.; Alférez, D.G.; Spence, K.; Santiago-Gómez, A.; Chemi, F.; et al. Anti-Estrogen Resistance in Human Breast Tumors Is Driven by JAG1-NOTCH4-Dependent Cancer Stem Cell Activity. Cell Rep. 2015, 12, 1968–1977. [Google Scholar] [CrossRef] [Green Version]
- Bruna, A.; Rueda, O.M.; Greenwood, W.; Batra, A.S.; Callari, M.; Batra, R.N.; Pogrebniak, K.; Sandoval, J.; Cassidy, J.W.; Tufegdzic-Vidakovic, A.; et al. A Biobank of Breast Cancer Explants with Preserved Intra-Tumor Heterogeneity to Screen Anticancer Compounds. Cell 2016, 167, 260–274. [Google Scholar] [CrossRef] [Green Version]
- Fiche, M.; Scabia, V.; Aouad, P.; Battista, L.; Treboux, A.; Stravodimou, A.; Zaman, K.; Dormoy, V.; Ayyanan, A.; Sflomos, G.; et al. Intraductal Patient-Derived Xenografts of Estrogen Receptor α-Positive Breast Cancer Recapitulate the Histopathological Spectrum and Metastatic Potential of Human Lesions. J. Pathol. 2019, 247, 287–292. [Google Scholar] [CrossRef] [Green Version]
- Richard, E.; Grellety, T.; Velasco, V.; MacGrogan, G.; Bonnefoi, H.; Iggo, R. The Mammary Ducts Create a Favourable Microenvironment for Xenografting of Luminal and Molecular Apocrine Breast Tumours: Intraductal Xenografts of Patient-Derived Breast Cancer. J. Pathol. 2016, 240, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Claerhout, S.; Prat, A.; Dobrolecki, L.E.; Petrovic, I.; Lai, Q.; Landis, M.D.; Wiechmann, L.; Schiff, R.; Giuliano, M.; et al. A Renewable Tissue Resource of Phenotypically Stable, Biologically and Ethnically Diverse, Patient-Derived Human Breast Cancer Xenograft Models. Cancer Res. 2013, 73, 4885–4897. [Google Scholar] [CrossRef] [Green Version]
- DeRose, Y.S.; Wang, G.; Lin, Y.-C.; Bernard, P.S.; Buys, S.S.; Ebbert, M.T.W.; Factor, R.; Matsen, C.; Milash, B.A.; Nelson, E.; et al. Tumor Grafts Derived from Women with Breast Cancer Authentically Reflect Tumor Pathology, Growth, Metastasis and Disease Outcomes. Nat. Med. 2011, 17, 1514–1520. [Google Scholar] [CrossRef]
- Li, S.; Shen, D.; Shao, J.; Crowder, R.; Liu, W.; Prat, A.; He, X.; Liu, S.; Hoog, J.; Lu, C.; et al. Endocrine-Therapy-Resistant ESR1 Variants Revealed by Genomic Characterization of Breast-Cancer-Derived Xenografts. Cell Rep. 2013, 4, 1116–1130. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lewis, M.T. Establishment of Patient-Derived Xenograft (PDX) Models of Human Breast Cancer. Curr. Protoc. Mouse Biol. 2013, 3, 21–29. [Google Scholar] [CrossRef]
- DeRose, Y.S.; Gligorich, K.M.; Wang, G.; Georgelas, A.; Bowman, P.; Courdy, S.J.; Welm, A.L.; Welm, B.E. Patient-Derived Models of Human Breast Cancer: Protocols for in vitro and in Vivo Applications in Tumor Biology and Translational Medicine. Curr. Protoc. Pharmacol. 2013, 60, 14–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oesterreich, S.; Davidson, N.E. The Search for ESR1 Mutations in Breast Cancer. Nat. Genet. 2013, 45, 1415–1416. [Google Scholar] [CrossRef] [Green Version]
- Proia, D.A.; Kuperwasser, C. Reconstruction of Human Mammary Tissues in a Mouse Model. Nat. Protoc. 2006, 1, 206–214. [Google Scholar] [CrossRef]
- Guiu, S.; Wolfer, A.; Jacot, W.; Fumoleau, P.; Romieu, G.; Bonnetain, F.; Fiche, M. Invasive Lobular Breast Cancer and Its Variants: How Special Are They for Systemic Therapy Decisions? Crit. Rev. Oncol. Hematol. 2014, 92, 235–257. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, S.M.; Mohammed, S. Canine Mammary Tumors as a Model for Human Disease. Oncol. Lett. 2018, 15, 8195–8205. [Google Scholar] [CrossRef] [Green Version]
- Ressel, L.; Millanta, F.; Poli, A. Canine Invasive Lobular Carcinoma of the Mammary Gland: Morphological and Immunohistochemical Characterizations of Three Cases. J. Comp. Pathol. 2011, 144, 303–307. [Google Scholar] [CrossRef]
- Wood, C.E.; Usborne, A.L.; Starost, M.F.; Tarara, R.P.; Hill, L.R.; Wilkinson, L.M.; Geisinger, K.R.; Feiste, E.A.; Cline, J.M. Hyperplastic and Neoplastic Lesions of the Mammary Gland in Macaques. Vet. Pathol. 2006, 43, 471–483. [Google Scholar] [CrossRef]
- Costa, B.; Estrada, M.F.; Mendes, R.V.; Fior, R. Zebrafish Avatars towards Personalized Medicine—A Comparative Review between Avatar Models. Cells 2020, 9, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Brunson, D.C.; Tang, Q.; Do, D.; Iftimia, N.A.; Moore, J.C.; Hayes, M.N.; Welker, A.M.; Garcia, E.G.; Dubash, T.D.; et al. Visualizing Engrafted Human Cancer and Therapy Responses in Immunodeficient Zebrafish. Cell 2019, 177, 1903–1914. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Aceto, N.; Bersani, F.; Madden, M.W.; Donaldson, M.C.; Desai, R.; Zhu, H.; Comaills, V.; Zheng, Z.; et al. Ex Vivo Culture of Circulating Breast Tumor Cells for Individualized Testing of Drug Susceptibility. Science 2014, 345, 216–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riggins, R.B.; Lan, J.P.-J.; Zhu, Y.; Klimach, U.; Zwart, A.; Cavalli, L.R.; Haddad, B.R.; Chen, L.; Gong, T.; Xuan, J.; et al. ERRγ Mediates Tamoxifen Resistance in Novel Models of Invasive Lobular Breast Cancer. Cancer Res. 2008, 68, 8908–8917. [Google Scholar] [CrossRef] [Green Version]
- Sikora, M.J.; Jacobsen, B.M.; Levine, K.; Chen, J.; Davidson, N.E.; Lee, A.V.; Alexander, C.M.; Oesterreich, S. WNT4 Mediates Estrogen Receptor Signaling and Endocrine Resistance in Invasive Lobular Carcinoma Cell Lines. Breast Cancer Res. 2016, 18, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, T.; Sikora, M.J.; Levine, K.M.; Tasdemir, N.; Riggins, R.B.; Wendell, S.G.; Van Houten, B.; Oesterreich, S. Key Regulators of Lipid Metabolism Drive Endocrine Resistance in Invasive Lobular Breast Cancer. Breast Cancer Res. 2018, 20, 106. [Google Scholar] [CrossRef] [PubMed]
- Simigdala, N.; Gao, Q.; Pancholi, S.; Roberg-Larsen, H.; Zvelebil, M.; Ribas, R.; Folkerd, E.; Thompson, A.; Bhamra, A.; Dowsett, M.; et al. Cholesterol Biosynthesis Pathway as a Novel Mechanism of Resistance to Estrogen Deprivation in Estrogen Receptor-Positive Breast Cancer. Breast Cancer Res. 2016, 18, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, K.M.; Priedigkeit, N.; Basudan, A.; Tasdemir, N.; Sikora, M.J.; Sokol, E.S.; Hartmaier, R.J.; Ding, K.; Ahmad, N.Z.; Watters, R.J.; et al. FGFR4 Overexpression and Hotspot Mutations in Metastatic ER+ Breast Cancer Are Enriched in the Lobular Subtype. NPJ Breast Cancer 2019, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- De Andrade Natal, R.; Paiva, G.R.; Pelegati, V.B.; Marenco, L.; Alvarenga, C.A.; Vargas, R.F.; Derchain, S.F.; Sarian, L.O.; Franchet, C.; Cesar, C.L.; et al. Exploring Collagen Parameters in Pure Special Types of Invasive Breast Cancer. Sci. Rep. 2019, 9, 7715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pestalozzi, B.C.; Zahrieh, D.; Mallon, E.; Gusterson, B.A.; Price, K.N.; Gelber, R.D.; Holmberg, S.B.; Lindtner, J.; Snyder, R.; Thürlimann, B.; et al. Distinct Clinical and Prognostic Features of Infiltrating Lobular Carcinoma of the Breast: Combined Results of 15 International Breast Cancer Study Group Clinical Trials. J. Clin. Oncol. 2008, 26, 3006–3014. [Google Scholar] [CrossRef]
- Gómez-Cuadrado, L.; Tracey, N.; Ma, R.; Qian, B.; Brunton, V.G. Mouse Models of Metastasis: Progress and Prospects. Dis. Model. Mech. 2017, 10, 1061–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonkers, J.; Derksen, P.W.B. Modeling Metastatic Breast Cancer in Mice. J. Mammary Gland Biol. Neoplasia 2007, 12, 191–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kersten, K.; de Visser, K.E.; van Miltenburg, M.H.; Jonkers, J. Genetically Engineered Mouse Models in Oncology Research and Cancer Medicine. EMBO Mol. Med. 2017, 9, 137–153. [Google Scholar] [CrossRef]
- Buijs, J.T.; Matula, K.M.; Cheung, H.; Kruithof-de Julio, M.; van der Mark, M.H.; Snoeks, T.J.; Cohen, R.; Corver, W.E.; Mohammad, K.S.; Jonkers, J.; et al. Spontaneous Bone Metastases in a Preclinical Orthotopic Model of Invasive Lobular Carcinoma; the Effect of Pharmacological Targeting TGFβ Receptor I Kinase. J. Pathol. 2015, 235, 745–759. [Google Scholar] [CrossRef]
- Paauwe, M.; Heijkants, R.C.; Oudt, C.H.; van Pelt, G.W.; Cui, C.; Theuer, C.P.; Hardwick, J.C.H.; Sier, C.F.M.; Hawinkels, L.J.a.C. Endoglin Targeting Inhibits Tumor Angiogenesis and Metastatic Spread in Breast Cancer. Oncogene 2016, 35, 4069–4079. [Google Scholar] [CrossRef]
- Holen, I.; Speirs, V.; Morrissey, B.; Blyth, K. In vivo models in breast cancer research: Progress, challenges and future directions. Dis. Model. Mech. 2017, 10, 359–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyre, R.; Alférez, D.G.; Spence, K.; Kamal, M.; Shaw, F.L.; Simões, B.M.; Santiago-Gómez, A.; Sarmiento-Castro, A.; Bramley, M.; Absar, M.; et al. Patient-Derived Mammosphere and Xenograft Tumour Initiation Correlates with Progression to Metastasis. J. Mammary Gland Biol. Neoplasia 2016, 21, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Powley, I.R.; Patel, M.; Miles, G.; Pringle, H.; Howells, L.; Thomas, A.; Kettleborough, C.; Bryans, J.; Hammonds, T.; MacFarlane, M.; et al. Patient-Derived Explants (PDEs) as a Powerful Preclinical Platform for Anti-Cancer Drug and Biomarker Discovery. Br. J. Cancer 2020, 122, 735–744. [Google Scholar] [CrossRef] [Green Version]
- Tasdemir, N.; Scott, J.; Laotche, J.; Hou, W.; Bossart, E.; Atkinson, J.; Sflomos, G.; Sreekumar, S.; Castro, C.; Anderson, C.; et al. Abstract PD7-02: Novel Human Cell Line Xenograft Models of ERα-Positive Metastatic Invasive Lobular Breast Carcinoma as Pre-Clinical Platforms for Validating Candidate Genetic Drivers. In Proceedings of the 2018 San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 4–8 December 2018; American Association for Cancer Research: Philadelphia, PA, USA, 15 February 2019. [Google Scholar]
- Giuliano, M.; Herrera, S.; Christiny, P.; Shaw, C.; Creighton, C.J.; Mitchell, T.; Bhat, R.; Zhang, X.; Mao, S.; Dobrolecki, L.E.; et al. Circulating and Disseminated Tumor Cells from Breast Cancer Patient-Derived Xenograft-Bearing Mice as a Novel Model to Study Metastasis. Breast Cancer Res. 2015, 17, 3. [Google Scholar] [CrossRef] [Green Version]
- Pate, L.; Desmedt, C.; Metzger, O.; Burgess Hutcheson, L.; Turner, C.; Freeney, S.; Oesterreich, S. How Researchers, Clinicians and Patient Advocates Can Accelerate Lobular Breast Cancer Research. Cancers 2021, 13, 3094. [Google Scholar] [CrossRef]
- Christgen, M.; Derksen, P.W.B. Lobular Breast Cancer: Molecular Basis, Mouse and Cellular Models. Breast Cancer Res. 2015, 17, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Ruiter, J.R.; Wessels, L.F.A.; Jonkers, J. Mouse Models in the Era of Large Human Tumour Sequencing Studies. Open Biol. 2018, 8, 180080. [Google Scholar] [CrossRef] [Green Version]
- Blomberg, O.S.; Spagnuolo, L.; de Visser, K.E. Immune Regulation of Metastasis: Mechanistic Insights and Therapeutic Opportunities. Dis. Models Mech. 2018, 11, dmm036236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulter, L.; Bullock, E.; Mabruk, Z.; Brunton, V.G. The Fibrotic and Immune Microenvironments as Targetable Drivers of Metastasis. Br. J. Cancer 2021, 124, 27–36. [Google Scholar] [CrossRef]
- Chen, X.; Song, E. Turning Foes to Friends: Targeting Cancer-Associated Fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- Gómez-Cuadrado, L.; Zhao, H.; Souleimanova, M.; Noer, P.R.; Turnbull, A.K.; Oxvig, C.; Bertos, N.; Dixon, J.M.; Park, M.; Sims, A.H.; et al. Identification of Stromal Genes Differentially Expressed in Lobular Breast Cancer Highlights Role for Pregnancy-Associated-Plasma Protein-A. Available online: https://www.biorxiv.org/content/10.1101/2020.04.24.059386v1 (accessed on 1 October 2021).
- Iacobuzio-Donahue, C.A.; Michael, C.; Baez, P.; Kappagantula, R.; Hooper, J.E.; Hollman, T.J. Cancer Biology as Revealed by the Research Autopsy. Nat. Rev. Cancer 2019, 19, 686–697. [Google Scholar] [CrossRef] [Green Version]
- Reeves, G.K.; Beral, V.; Green, J.; Gathani, T.; Bull, D.; Million Women Study Collaborators. Hormonal Therapy for Menopause and Breast-Cancer Risk by Histological Type: A Cohort Study and Meta-Analysis. Lancet Oncol. 2006, 7, 910–918. [Google Scholar] [CrossRef]
- Lee, J.Y.; Schizas, M.; Geyer, F.C.; Selenica, P.; Piscuoglio, S.; Sakr, R.A.; Ng, C.K.Y.; Carniello, J.V.S.; Towers, R.; Giri, D.D.; et al. Lobular Carcinomas In Situ Display Intralesion Genetic Heterogeneity and Clonal Evolution in the Progression to Invasive Lobular Carcinoma. Clin. Cancer Res. 2019, 25, 674–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goss, P.E.; Reid, C.; Pintilie, M.; Lim, R.; Miller, N. Male Breast Carcinoma: A Review of 229 Patients Who Presented to the Princess Margaret Hospital during 40 Years: 1955–1996. Cancer 1999, 85, 629–639. [Google Scholar] [CrossRef]
- Piscuoglio, S.; Ng, C.K.Y.; Murray, M.P.; Guerini-Rocco, E.; Martelotto, L.G.; Geyer, F.C.; Bidard, F.-C.; Berman, S.; Fusco, N.; Sakr, R.A.; et al. The Genomic Landscape of Male Breast Cancers. Clin. Cancer Res. 2016, 22, 4045–4056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvadori, B.; Saccozzi, R.; Manzari, A.; Andreola, S.; Conti, R.A.; Cusumano, F.; Grassi, M. Prognosis of Breast Cancer in Males: An Analysis of 170 Cases. Eur. J. Cancer 1994, 30, 930–935. [Google Scholar] [CrossRef]
- van der Meer, D.; Barthorpe, S.; Yang, W.; Lightfoot, H.; Hall, C.; Gilbert, J.; Francies, H.E.; Garnett, M.J. Cell Model Passports—A Hub for Clinical, Genetic and Functional Datasets of Preclinical Cancer Models. Nucleic Acids Res. 2019, 47, D923–D929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
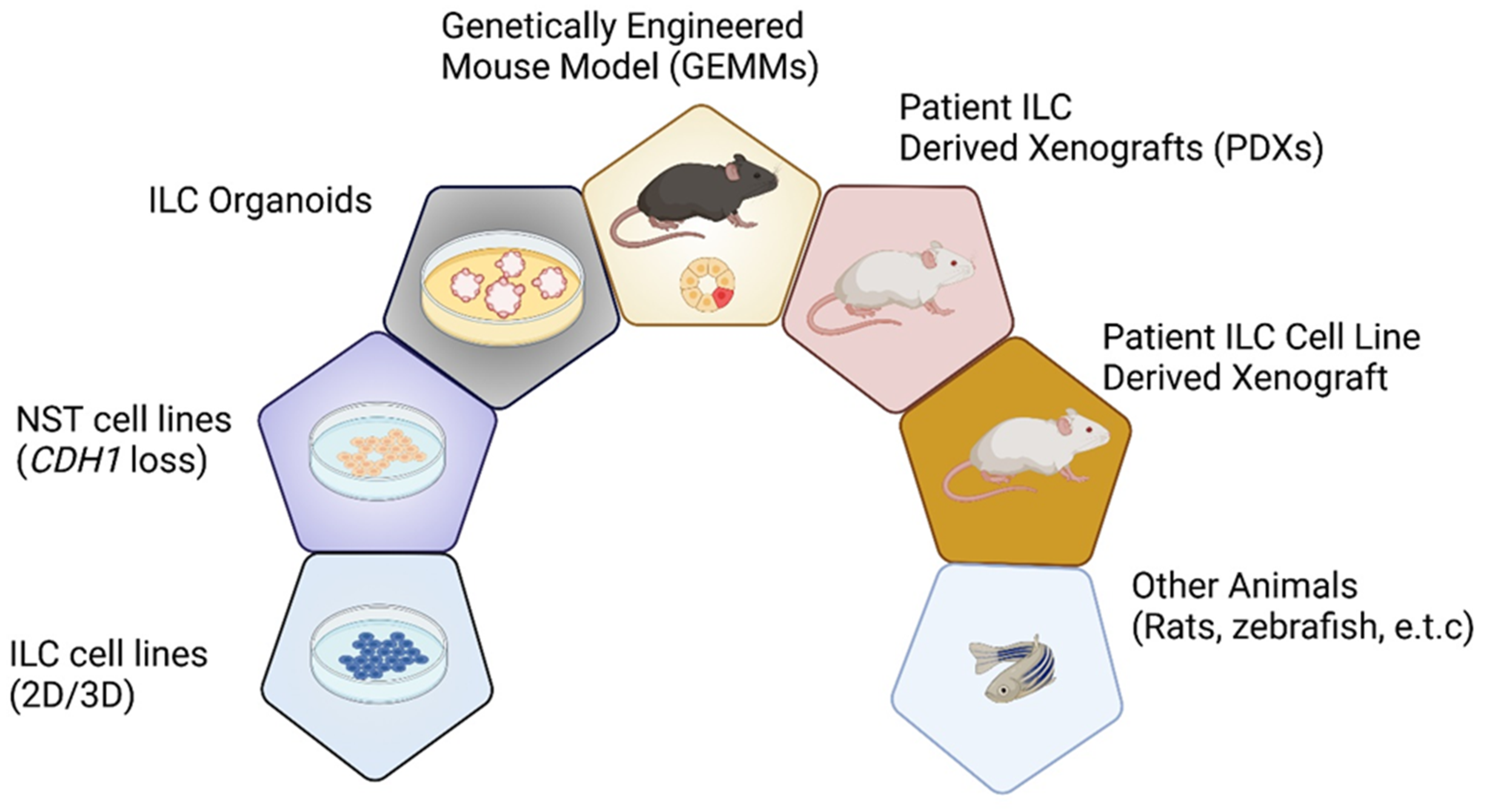
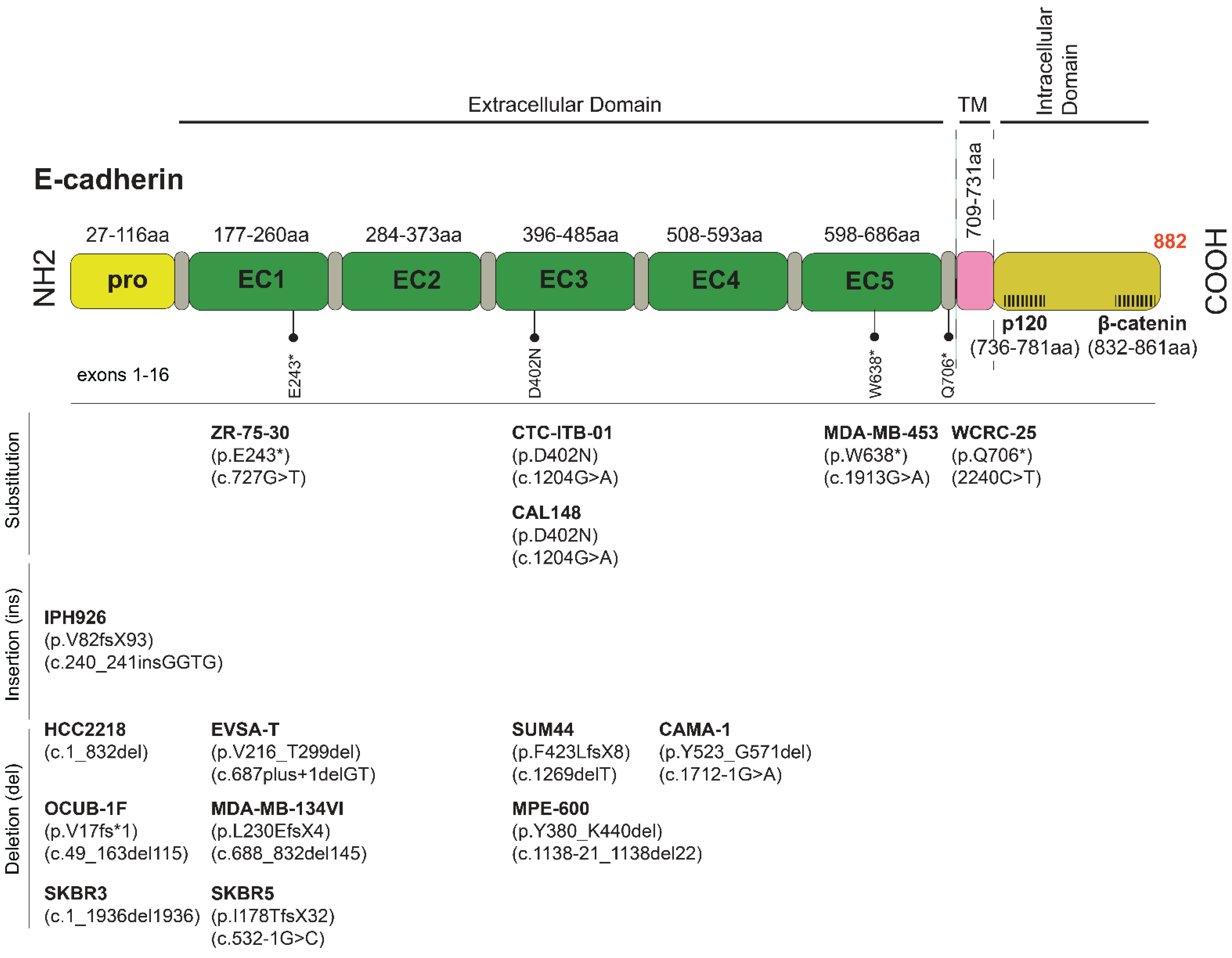
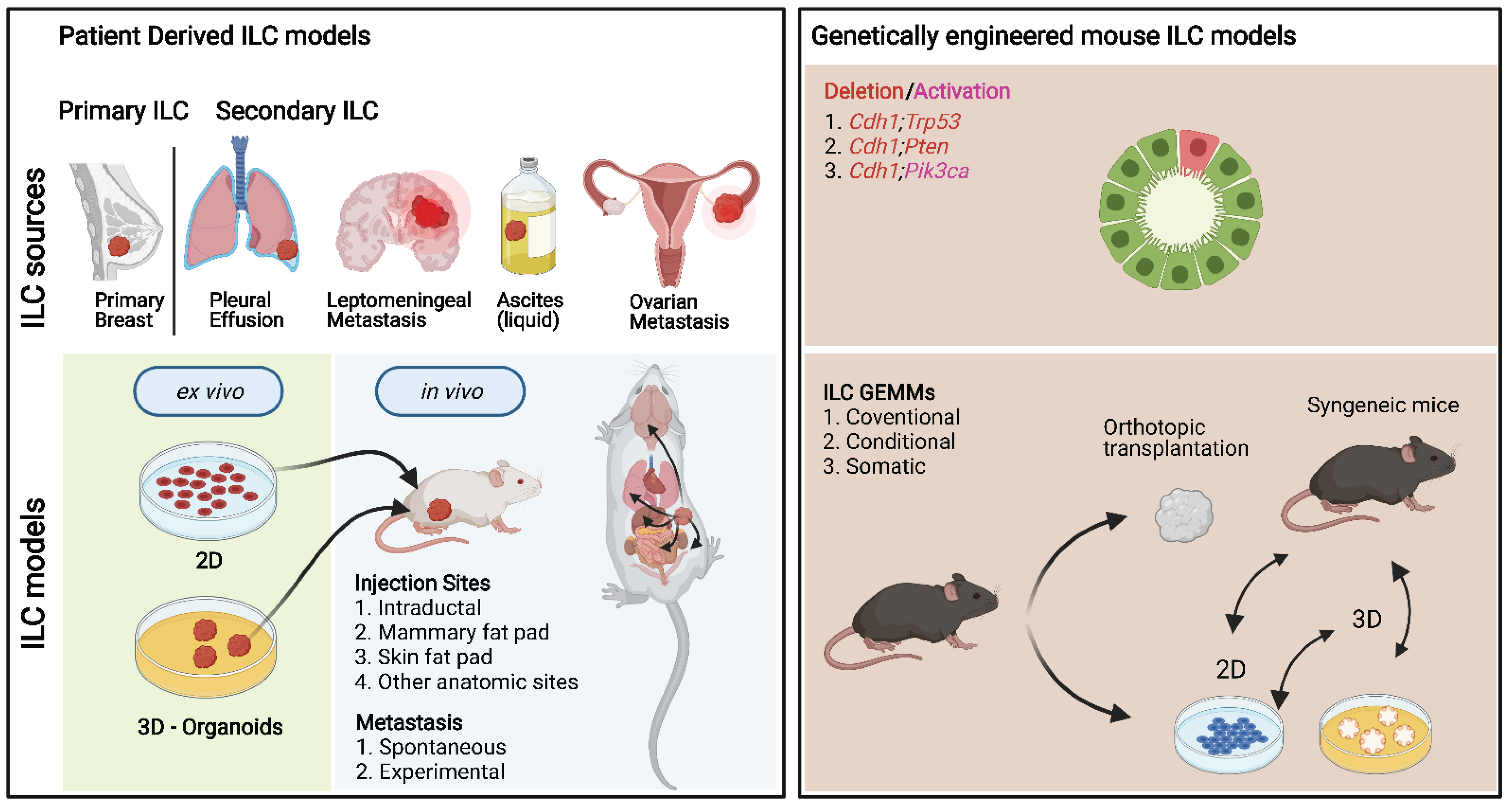
| Year | Model | Significance | Morphological Characteristics | Applications | PMID |
|---|---|---|---|---|---|
| 1974 | MDA-MB-134-VI | First ILC cell line - expansion/characterization (in vitro) | R, SF | P, EnR, HoR, DrR | 4412247 |
| 1978 | SUM-44PE | ILC cell line - expansion/characterization (in vitro) | R, SF | P, EnR, HoR, DrR | 8425198 |
| 2006 | Cdh1; Trp53 | First GEM ILC model Rational: CDH1 is an ILC hallmark | PILC | LCIS, P, TP, I, DrR, D, M | 17097565 |
| 2008 | SUM-44PE variants | Studies on endocrine resistance (in vitro) | R, SF | P, EnR | 18974135 |
| 2009 2012 | IPH-926 | First well characterized TN ILC cell line | R, SF | P, DrR | 19191266 22945757 |
| 2011 | ILC PDXs (cleared mammary fat pad) | First ILC PDXs | LCIS, PILC, SR, SF | P, TP, EnR, HoR, DrR, D, M | 22019887 |
| 2016 | Cdh1; Pten | GEM ILC model. Rational: PTEN loss found in ~10% of ILCs | cILC | LCIS, P, TP, I, EnR, HoR, DrR, D, M | 27524621 |
| 2016 2018 | ILC PDXs (murine milk ducts) | First intraductal ILC PDXs | LCIS, PILC, SR, SF | LCIS, P, TP, I, EnR, HoR, DrR, D, M | 30430577 19191266 |
| 2018 | Breast Cancer Organoids | First ILC organoids | SR | P, EnR, HoR, DrR | 29224780 |
| 2018 | Cdh1; Pik3ca | GEM ILC model. Rational: PIK3CA mutations found in ~40% of ILCs | cILC, SF, T | LCIS, P, TP, I, EnR, HoR, DrR, D, M | 30332649 |
| 2021 | ILC xenografts (murine milk ducts) | First intraductal SUM-44PE and MDA-MB-134-VI xenografts | LCIS, PILC, SR, SF | LCIS, P, TP, I, EnR, HoR, DrR, D, M | 33616307 |
| Name | Tissue | Tumor | Biomarker | E-Cadherin/CDH1 | Morphology | Ref. |
|---|---|---|---|---|---|---|
| ILC Cell Lines | ||||||
| SUM-44 PE | PE | ILC | ER+, PRlow, HER2− | p.F423LfsX8 | Rounded | [33] |
| IPH-926 | MA | ILC | ER−, PR−, HER2− | p.V82fsX93 | Rounded | [34] |
| MDA-MB-134-VI | PE | NST | ER+, PR−, HER2− | p.L230EfsX4 | Rounded | [33] |
| MDA-MB-330 | PE | ILC | ER+/−, PR−, HER2+ | wt | Rounded | [35] |
| UACC-3133 | PE | ILC | ERlow, PR−, HER2+ | n/a | n/a | [36] |
| MA-11 | BmM | ILC | ER−, PR−, HER2− | n/a | Rounded | [37] |
| WCRC-25 | PE | ILC | ER−, PR−, HER2− | p.Q706 * | Rounded | npy |
| ILC-Like Cell Lines | ||||||
| BCK4 | PE | ILC (mucinous) | ER+, PR+, HER2− | n/a | Rounded | [38] |
| MDA-MB-453 | PF | n/a | ER−, PR−, HER2+ | p.W638X | Rounded | [39] |
| MDA-MB-468 | PE | n/a | ER−, PR−, HER2− | wt | Rounded | [39] |
| CAMA-1 | PE | Solid | ER+, PR+, HER2− | p.Y523_G571del | Rounded | [39] |
| SK-BR-3 | PE | n/a | ER−, PR−, HER2+ | c.1_1936del1936 | Rounded | [35] |
| SK-BR-5 | n/a | n/a | ER−, PR−, HER2+ | p.I178TfsX32 | n/a | [40] |
| EVSA-T | AF | n/a | ER−, PR− | p.V216_T229del | Rounded | [41] |
| CAL-148 | PE | n/a | ER−, PR−, HER2− | D402N, deep deletion | n/a | [42] |
| ZR-75-30 | AF | NST | ER+, PR−, HER2+ | p.Glu243Ter-p.E243X | Rounded | [43] |
| HCC2218 | PBC | NST | ER−, PR−, HER2+ | c.1-832del | Rounded | [44] |
| 600MPE | PE | NST | n/a | p.Y380_K440del | Rounded | [45] |
| BT549 | n/a | Papillary | ER−, PR−, HER2− | n/a | Rounded | [46] |
| MA-11 | n/a | ILC and tubular | ER−, PR−, HER2− | wt | Rounded | [37] |
| OCUB-1F | PE | n/a | ER−, PR− | p.Val17fs*1 | Rounded | [47] |
| Name | Type | Clinical (Biomarker) | Organoids (Biomarkers) | E-Cadherin/CDH1 | Laboratory/Institute | Ref. |
|---|---|---|---|---|---|---|
| Human ILC Organoids (female) | ||||||
| T35 | ILC | ER+, PR+, HER2− | ER+, PR−, HER2+ | n/a | Prof. Hans Clevers/HI | [117] |
| T66 | ILC | ER+, PR+, HER2+ | ER+, PR+, HER2+ | n/a | Prof. Hans Clevers/HI | [117] |
| T74 | ILC (apocrine?) | ER+, PR+, HER2− | ER+, PR+, HER2− | n/a | Prof. Hans Clevers/HI | [117] |
| T105 | ILC | ER+, PR+, HER2− | ER+, PR+, HER2− | n/a | Prof. Hans Clevers/HI | [117] |
| P008 | ILC | ER+, PR−, HER2− | ER−, PR−, HER2− | p.(Ser180Tyr) | Prof. Clare Isacke/ICR | npy |
| KCL320 | ILC | ER+, PR+, HER2− | ER−, PR−, HER2− | splice variant g.68823627G>A | Prof. Clare Isacke/ICR | npy |
| Human ILC Organoids (male) | ||||||
| UDL-MBC6 | ILC | n/a | n/a | c.85del p.(His29fs) | Prof. Patrick WB Derksen/UMC Prof. Van Diest/UMC | npy |
| Mouse primary ILC | ||||||
| UDL-WEP9 | PILC | ER−, PR−, Her2− | ER−, PR−, Her2− | null | Prof. Patrick WB Derksen/UMC | npy |
| UDL-WEP10 | PILC | ER−, PR−, Her2− | ER−, PR−, Her2− | null | Prof. Patrick WB Derksen/ UMC | npy |
| Deletion/Activation | System | Primary Tumor | ER | Tumor Onset (Weeks) | Laboratory/Institute | Ref. |
|---|---|---|---|---|---|---|
| Cdh1; Tp53 | Tg | Pleomorphic ILC | Neg. | 20–32 | Prof. Jos Jonkers/NKI | [25] |
| Cdh1; Pten | Tg | Classical ILC-like features | Pos. | 8–16 | Prof. Jos Jonkers/NKI | [125] |
| Cdh1 and Pik3ca | Tg | Immune-related ILC-like | Pos. | 5–12 | Prof. Sean E. Egan/U of T | [126] |
| Cdh1 and Pten | CRISPR/Cas9 | Unknown-ILC histology | n/t | 28 | Prof. Jos Jonkers/NKI | [127] |
| Cdh1 and AKTE17K | GEMM-ESC | Typical ILC histology | n/t | n/s | Prof. Jos Jonkers/NKI | [127] |
| Cdh1 and Myh9 | CRISPR/Cas9 | Classical ILC-like features | n/t | n/s | Prof. Jos Jonkers/NKI | [128] |
| Cdh1; t-ASPP2 | GEMM-ESC | Classical ILC-like features | n/t | 9–15 | Prof. Jos Jonkers/NKI | [128,129] |
| Cdh1; Pten; t-ASPP2 | GEMM-ESC/Tg | Classical ILC-like features | n/t | 5–9 | Prof. Jos Jonkers/NKI | [129] |
| Cdh1; t-MYPT1 | GEMM-ESC | Classical ILC-like features | n/t | 10–16 | Prof. Jos Jonkers/NKI | [128,129] |
| Cdh1; Pten; t-MYPT1 | GEMM-ESC/Tg | Classical ILC-like features | n/t | 5–8 | Prof. Jos Jonkers/NKI | [129] |
| Cdh1; Trps1 | GEMM-ESC | Classical ILC-like features | n/t | 76 | Prof. Jos Jonkers/NKI | [130] |
| ILC Models | Major Experimental Pros | Major Experimental Cons | Ref. |
|---|---|---|---|
| Cell lines (in vitro) |
|
| [33,34,36,37,39,63] |
| Organoids |
|
| [117] |
| GEMMs |
|
| [25,125,126] |
| Cell lines (in vivo–xenografts) |
|
| [38,53,154,160,193] |
| PDXs |
|
| [34,153,154,160,163,172,174,175,176,178,179] |
| Implantation Sites | |||
| Subcutaneous (SM) |
|
| [172,174,175] |
| Mammary fat pad (SM) |
|
| [178,179] |
| Injection into the milk ducts (SM) |
|
| [154,160,176] |
| Tail vein (EM) |
|
| [207] |
| Intracardiac (EM) |
|
| [207] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sflomos, G.; Schipper, K.; Koorman, T.; Fitzpatrick, A.; Oesterreich, S.; Lee, A.V.; Jonkers, J.; Brunton, V.G.; Christgen, M.; Isacke, C.; et al. Atlas of Lobular Breast Cancer Models: Challenges and Strategic Directions. Cancers 2021, 13, 5396. https://doi.org/10.3390/cancers13215396
Sflomos G, Schipper K, Koorman T, Fitzpatrick A, Oesterreich S, Lee AV, Jonkers J, Brunton VG, Christgen M, Isacke C, et al. Atlas of Lobular Breast Cancer Models: Challenges and Strategic Directions. Cancers. 2021; 13(21):5396. https://doi.org/10.3390/cancers13215396
Chicago/Turabian StyleSflomos, George, Koen Schipper, Thijs Koorman, Amanda Fitzpatrick, Steffi Oesterreich, Adrian V. Lee, Jos Jonkers, Valerie G. Brunton, Matthias Christgen, Clare Isacke, and et al. 2021. "Atlas of Lobular Breast Cancer Models: Challenges and Strategic Directions" Cancers 13, no. 21: 5396. https://doi.org/10.3390/cancers13215396