
The EGF Domains of MUC4 Oncomucin Mediate HER2 Binding Affinity and Promote Pancreatic Cancer Cell Tumorigenesis

 , , , , ,
, , , , ,  , , ,
, , ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Cell Lines and Cell Culture
2.2. Generation of GST-EGF Fusion Proteins
2.3. Generation and Activity of eGFP-MUC4β, eGFP-MUC4βΔTM, and eGFP-MUC4ΔEGFs Fusion Proteins
2.4. GST Pull-Down Assay
2.5. Immunoprecipitation of HER2
2.6. SDS-PAGE and Western-Blotting
2.7. PLA Assay
2.8. MUC4EGFs/HER2 Interaction Studies Using Microscale Thermophoresis
2.9. Cell Proliferation Assay
2.10. Cell Migration and Invasion Assays
2.11. Computational Methods: Homology Modeling of Human MUC4EGF1/HER2 and MUC4EGF2/HER2C Complexes
2.12. Molecular Dynamics (MD) Simulations
2.13. Proteome Array Studies
2.14. Subcutaneous Xenografts in Scid Mice
2.15. Statistics
3. Results
3.1. MUC4EGF1 and MUC4EGF2 Drive the Binding Affinity with HER2 and Mediate Both Cell Proliferation and Migration of Human Pancreatic Cancer Cells
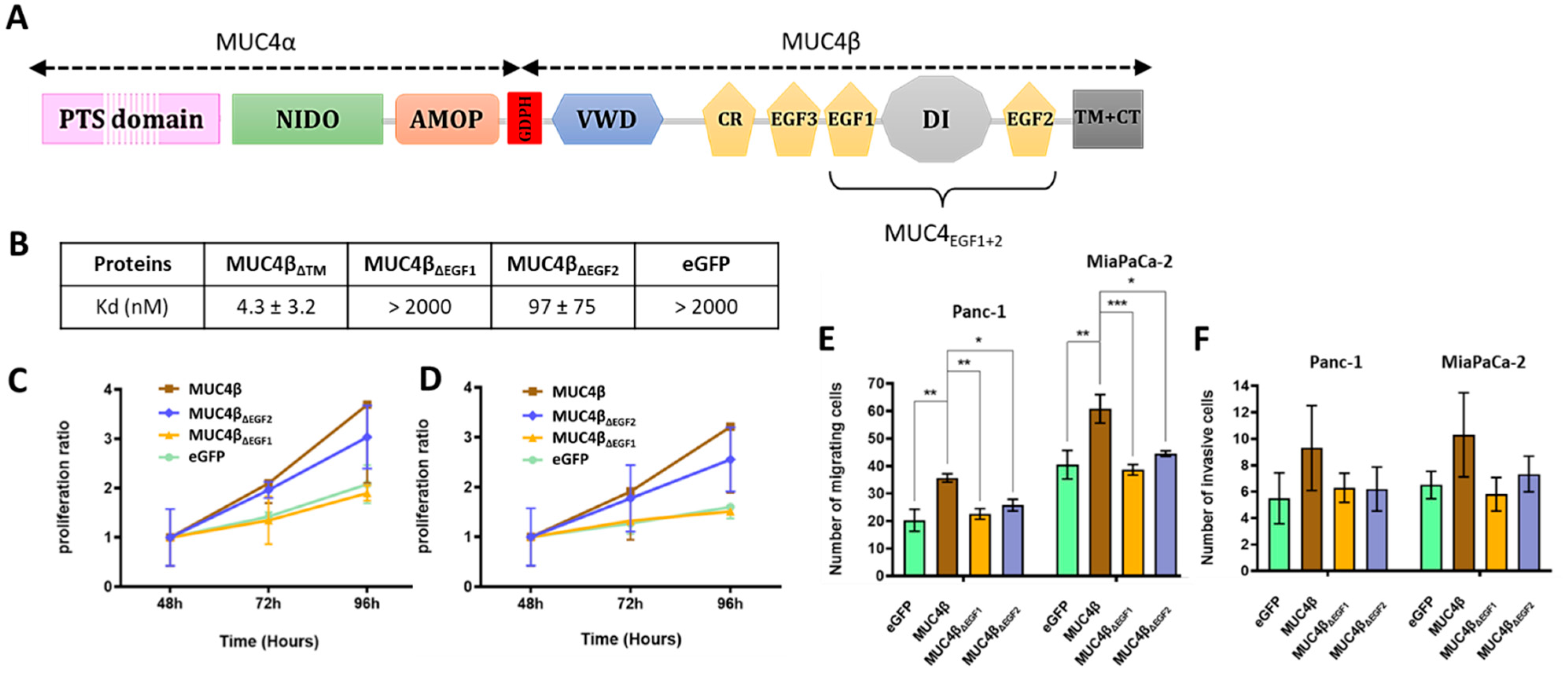
3.2. MUC4EGFs Domains Physically Interact with HER2
3.3. Molecular Dynamics (MD) Simulations of MUC4EGF1/HER2 and MUC4EGF2/HER2 Complex Models Led to Virtual Binding Hotspot Identification
3.4. Cluster Mutation Decreases In Vitro Proliferation of Human Pancreatic Cancer Cells and Binding Affinity of the Mutated MUC4EGF Domains
3.5. MUC4EGF Domains and Their Mutants Affect In Vitro Proliferation, Migration, and Invasion Properties of Human Pancreatic Cancer Cells
3.6. MUC4EGF Domains Enhance Pancreatic Tumor Growth In Vivo
3.7. MUC4EGF1+2 Domains Are Involved in Intracellular Signaling Pathway Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carraway, K.L.; Theodoropoulos, G.; Kozloski, G.A.; Carothers Carraway, C.A. Muc4/MUC4 functions and regulation in cancer. Future Oncol. 2009, 5, 1631–1640. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, K.; Rauth, S.; Marimuthu, S.; Kumar, S.; Batra, S.K. Unraveling mucin domains in cancer and metastasis: When protectors become predators. Cancer Metastasis Rev. 2020, 39, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, N.; Van Seuningen, I. The membrane-bound mucins: From cell signalling to transcriptional regulation and expression in epithelial cancers. Biochimie 2010, 92, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kufe, D.W. Mucins in cancer: Function, prognosis and therapy. Nat. Rev. Cancer. 2009, 9, 874–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, P.; Choi, A.H.; Deng, Z.; Yang, Y.; Zhao, J.; Wang, Y.; Hardwidge, P.R.; Zhu, G. Cell membrane-anchored MUC4 promotes tumorigenicity in epithelial carcinomas. Oncotarget 2017, 8, 14147–14157. [Google Scholar] [CrossRef] [Green Version]
- Torres, M.P.; Chakraborty, S.; Souchek, J.; Batra, S.K. Mucin-based targeted pancreatic cancer therapy. Curr. Pharm. Des. 2012, 18, 2472–2481. [Google Scholar] [CrossRef]
- Ménard, S.; Pupa, S.M.; Campiglio, M.; Tagliabue, E. Biologic and therapeutic role of HER2 in cancer. Oncogene 2003, 22, 6570–6578. [Google Scholar] [CrossRef] [Green Version]
- Liberelle, M.; Jonckheere, N.; Melnyk, P.; Van Seuningen, I.; Lebègue, N. EGF-Containing Membrane-Bound Mucins: A Hidden ErbB2 Targeting Pathway? J. Med. Chem. 2020, 63, 5074–5088. [Google Scholar] [CrossRef]
- Jonckheere, N.; Van Seuningen, I. The membrane-bound mucins: How large O-glycoproteins play key roles in epithelial cancers and hold promise as biological tools for gene-based and immunotherapies. Crit. Rev. Oncog. 2008, 14, 177–196. [Google Scholar] [CrossRef]
- Jonckheere, N.; Skrypek, N.; Merlin, J.; Dessein, A.F.; Dumont, P.; Leteurtre, E.; Harris, A.; Desseyn, J.L.; Susini, C.; Frénois, F.; et al. The mucin MUC4 and its membrane partner ErbB2 regulate biological properties of human CAPAN-2 pancreatic cancer cells via different signalling pathways. PLoS ONE 2012, 7, e32232. [Google Scholar] [CrossRef] [Green Version]
- Meric-Bernstam, F.; Johnson, A.M.; Dumbrava, E.E.I.; Raghav, K.; Balaji, K.; Bhatt, M.; Murthy, R.K.; Rodon, J.; Piha-Paul, S.A. Advances in HER2-Targeted Therapy: Novel Agents and Opportunities Beyond Breast and Gastric Cancer. Clin. Cancer Res. 2019, 25, 2033–2041. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.Y.; Bang, Y.J. HER2-targeted therapies—A role beyond breast cancer. Nat. Rev. Clin. Oncol. 2020, 17, 33–48. [Google Scholar] [CrossRef]
- Kumar, R.; George, B.; Campbell, M.R.; Verma, N.; Paul, A.M.; Melo-Alvim, C.; Ribeiro, L.; Pillai, M.R.; da Costa, L.M.; Moasser, M.M. HER family in cancer progression: From discovery to 2020 and beyond. Adv. Cancer Res. 2020, 147, 109–160. [Google Scholar] [PubMed]
- Thery, J.C.; Spano, J.P.; Azria, D.; Raymond, E.; Penault Llorca, F. Resistance to human epidermal growth factor receptor type 2-targeted therapies. Eur. J. Cancer. 2014, 50, 892–901. [Google Scholar] [CrossRef]
- Desai, M.D.; Saroya, B.S.; Lockhart, A.C. Investigational therapies targeting the ErbB (EGFR, HER2, HER3, HER4) family in GI cancers. Expert Opin. Investig. Drugs 2013, 22, 341–356. [Google Scholar] [CrossRef]
- Gautam, S.K.; Kumar, S.; Cannon, A.; Hall, B.; Bhatia, R.; Nasser, M.W.; Mahapatra, S.; Batra, S.K.; Jain, M. MUC4 mucin- a therapeutic target for pancreatic ductal adenocarcinoma. Expert Opin. Ther. Targets 2017, 21, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Han, S.H.; Ryu, K.H.; Kwon, A.Y. The Prognostic Impact of HER2 Genetic and Protein Expression in Pancreatic Carcinoma-HER2 Protein and Gene in Pancreatic Cancer. Diagnostics 2021, 11, 653. [Google Scholar] [CrossRef] [PubMed]
- Assenat, E.; Azria, D.; Mollevi, C.; Guimbaud, R.; Tubiana-Mathieu, N.; Smith, D.; Delord, J.P.; Samalin, E.; Portales, F.; Larbouret, C.; et al. Dual targeting of EGFR/EGFR and HER2 with cetuximab and trastuzumab in patients with metastatic pancreatic cancer after gemcitabine failure: Results of the “THERAPY” phase 1–2 trial. Oncotarget 2015, 6, 12796–12808. [Google Scholar] [CrossRef] [Green Version]
- Nagy, P.; Friedländer, E.; Tanner, M.; Kapanen, A.I.; Carraway, K.L.; Isola, J.; Jovin, T.M. Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res. 2005, 65, 473–482. [Google Scholar]
- Elster, N.; Collins, D.M.; Toomey, S.; Crown, J.; Eustace, A.J.; Hennessy, B.T. HER2-family signalling mechanisms, clinical implications and targeting in breast cancer. Breast Cancer Res. Treat. 2015, 149, 5–15. [Google Scholar] [CrossRef]
- Menyhárt, O.; Santarpia, L.; Győrffy, B. A Comprehensive Outline of Trastuzumab Resistance Biomarkers in HER2 Overexpressing Breast Cancer. Curr. Cancer Drug Targets 2015, 15, 665–683. [Google Scholar] [CrossRef]
- Nahta, R.; Yu, D.; Hung, M.C.; Hortobagyi, G.N.; Esteva, F.J. Mechanisms of disease: Understanding resistance to HER2-targeted therapy in human breast cancer. Nat. Clin. Pract. Oncol. 2006, 3, 269–280. [Google Scholar] [CrossRef]
- Jonckheere, N.; Skrypek, N.; Frénois, F.; Van Seuningen, I. Membrane-bound mucin modular domains: From structure to function. Biochimie. 2013, 95, 1077–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoup, N.; Liberelle, M.; Schulz, C.; Vasseur, R.; Magnez, R.; Thuru, X.; Melnyk, P.; Renault, N.; Jonckheere, N.; Lebegue, N.; et al. The EGF domains of MUC4 oncomucin interact with HER2 and mediate tumorigenic activity of cancer cells represent new potential therapeutic targets. FASEB J. 2021. [Google Scholar] [CrossRef]
- Liberelle, M.; Magnez, R.; Thuru, X.; Bencheikh, Y.; Ravez, S.; Quenon, C.; Drucbert, A.S.; Foulon, C.; Melnyk, P.; Van Seuningen, I.; et al. MUC4-ErbB2 Oncogenic Complex: Binding studies using Microscale Thermophoresis. Sci. Rep. 2020, 10, 6539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Amrani, M.; Corfiotti, F.; Corvaisier, M.; Vasseur, R.; Fulbert, M.; Skrzypczyk, C.; Deshorgues, A.C.; Gnemmi, V.; Tulasne, D.; Lahdaoui, F.; et al. Gemcitabine-induced epithelial-mesenchymal transition-like changes sustain chemoresistance of pancreatic cancer cells of mesenchymal-like phenotype. Mol. Carcinog. 2019, 58, 1985–1997. [Google Scholar] [CrossRef] [PubMed]
- Skrypek, N.; Duchêne, B.; Hebbar, M.; Leteurtre, E.; van Seuningen, I.; Jonckheere, N. The MUC4 mucin mediates gemcitabine resistance of human pancreatic cancer cells via the Concentrative Nucleoside Transporter family. Oncogene 2013, 32, 1714–1723. [Google Scholar] [CrossRef] [Green Version]
- Khavrutskii, L.; Yeh, J.; Timofeeva, O.; Tarasov, S.G.; Pritt, S.; Stefanisko, K.; Tarasova, N. Protein purification-free method of binding affinity determination by microscale thermophoresis. J. Vis. Exp. 2013, 78, 50541. [Google Scholar] [CrossRef] [Green Version]
- Piessen, G.; Jonckheere, N.; Vincent, A.; Hémon, B.; Ducourouble, M.P.; Copin, M.C.; Mariette, C.; Van Seuningen, I. Regulation of the human mucin MUC4 by taurodeoxycholic and taurochenodeoxycholic bile acids in oesophageal cancer cells is mediated by hepatocyte nuclear factor 1alpha. Biochem. J. 2007, 402, 81–91. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory. Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comp. Phys. Comm. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory. Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Drubay, V.; Skrypek, N.; Cordiez, L.; Vasseur, R.; Schulz, C.; Boukrout, N.; Duchêne, B.; Coppin, L.; Van Seuningen, I.; Jonckheere, N. TGF-βRII Knock-down in Pancreatic Cancer Cells Promotes Tumor Growth and Gemcitabine Resistance. Importance of STAT3 Phosphorylation on S727. Cancers 2018, 10, 254. [Google Scholar] [CrossRef] [Green Version]
- Alvarado, D.; Klein, D.E.; Lemmon, M.A. ErbB2 resembles an autoinhibited invertebrate epidermal growth factor receptor. Nature 2009, 461, 287–291. [Google Scholar] [CrossRef] [Green Version]
- Shilo, B.Z. Regulating the dynamics of EGF receptor signaling in space and time. Development 2005, 132, 4017–4027. [Google Scholar] [CrossRef] [Green Version]
- Garrett, T.P.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Zhu, H.J.; Walker, F.; Frenkel, M.J.; Hoyne, P.A.; et al. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell 2002, 110, 763–773. [Google Scholar] [CrossRef] [Green Version]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Rakers, C.; Bermudez, M.; Keller, B.G.; Mortier, J.; Wolber, G. Computational close up on protein–protein interactions: How to unravel the invisible using molecular dynamics simulations? WIREs Comput. Mol. Sci. 2015, 5, 345–359. [Google Scholar] [CrossRef]
- Yogurtcu, O.N.; Erdemli, S.B.; Nussinov, R.; Turkay, M.; Keskin, O. Restricted mobility of conserved residues in protein-protein interfaces in molecular simulations. Biophys. J. 2008, 94, 3475–3485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esha Sehanobish, E.; Dow, B.A.; Davidson, V.L. Analytical methods for assessing the effects of site-directed mutagenesis on protein–cofactor and protein–protein functional relationships. In In Vitro Mutagenesis. Methods in Molecular Biology; Reeves, A., Ed.; Humana Press: New York, NY, USA, 2017; Volume 1498, pp. 421–438. [Google Scholar]
- Ivanov, A.A.; Khuri, F.R.; Fu, H. Targeting protein-protein interactions as an anticancer strategy. Trends Pharmacol. Sci. 2013, 34, 393–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funes, M.; Miller, J.K.; Lai, C.; Carraway, K.L., III; Sweeney, C. The mucin Muc4 potentiates neuregulin signaling by increasing the cell-surface populations of ErbB2 and ErbB3. J. Biol. Chem. 2006, 281, 19310–19319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordoli, L.; Kiefer, F.; Arnold, K.; Benkert, P.; Battey, J.; Schwede, T. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 2009, 4, 1–13. [Google Scholar] [CrossRef]
- Cho, H.S.; Mason, K.; Ramyar, K.X.; Stanley, A.M.; Gabelli, S.B.; Denney, D.W., Jr.; Leahy, D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003, 421, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Xu, L.; Zheng, S.; Luo, X.; Shen, J.; Jiang, H.; Liu, X.; Zhou, M. Computational analysis of molecular basis of 1:1 interactions of NRG-1beta wild-type and variants with ErbB3 and ErbB4. Proteins 2005, 59, 742–756. [Google Scholar] [CrossRef] [PubMed]
- Engler, D.A.; Campion, S.R.; Hauser, M.R.; Cook, J.S.; Niyogi, S.K. Critical functional requirement for the guanidinium group of the arginine 41 side chain of human epidermal growth factor as revealed by mutagenic inactivation and chemical reactivation. J. Biol. Chem. 1992, 267, 2274–2281. [Google Scholar] [CrossRef]
- Jonckheere, N.; Van Seuningen, I. Integrative analysis of the cancer genome atlas and cancer cell lines encyclopedia large-scale genomic databases: MUC4/MUC16/MUC20 signature is associated with poor survival in human carcinomas. J. Transl. Med. 2018, 16, 259. [Google Scholar] [CrossRef]
- Jonckheere, N.; Auwercx, J.; Hadj Bachir, E.; Coppin, L.; Boukrout, N.; Vincent, A.; Neve, B.; Gautier, M.; Treviño, V.; Van Seuningen, I. Unsupervised Hierarchical Clustering of Pancreatic Adenocarcinoma Dataset from TCGA Defines a Mucin Expression Profile that Impacts Overall Survival. Cancers 2020, 12, 3309. [Google Scholar] [CrossRef]
- Zhi, X.; Tao, J.; Xie, K.; Zhu, Y.; Li, Z.; Tang, J.; Wang, W.; Xu, H.; Zhang, J.; Xu, Z. MUC4-induced nuclear translocation of β-catenin: A novel mechanism for growth, metastasis and angiogenesis in pancreatic cancer. Cancer Lett. 2014, 346, 104–113. [Google Scholar] [CrossRef]
- Chen, S.H.; Hung, W.C.; Wang, P.; Paul, C.; Konstantopoulos, K. Mesothelin binding to CA125/MUC16 promotes pancreatic cancer cell motility and invasion via MMP-7 activation. Sci. Rep. 2013, 3, 1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, S.B.; Luu, Y.; Shekels, L.L.; Batra, S.K.; Kandarian, B.; Evans, D.B.; Zaworski, P.G.; Wolfe, C.L.; Heinrikson, R.L. Activity of recombinant cysteine-rich domain proteins derived from the membrane-bound MUC17/Muc3 family mucins. Biochim. Biophys. Acta. 2010, 1800, 629–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.; Sikander, M.; Ebeling, M.C.; Ganju, A.; Kumari, S.; Yallapu, M.M.; Hafeez, B.B.; Ise, T.; Nagata, S.; Zafar, N.; et al. MUC13 interaction with receptor tyrosine kinase HER2 drives pancreatic ductal adenocarcinoma progression. Oncogene 2017, 36, 491–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoup, N.; Liberelle, M.; Schulz, C.; Cavdarli, S.; Vasseur, R.; Magnez, R.; Lahdaoui, F.; Skrypek, N.; Peretti, F.; Frénois, F.; et al. The EGF Domains of MUC4 Oncomucin Mediate HER2 Binding Affinity and Promote Pancreatic Cancer Cell Tumorigenesis. Cancers 2021, 13, 5746. https://doi.org/10.3390/cancers13225746
Stoup N, Liberelle M, Schulz C, Cavdarli S, Vasseur R, Magnez R, Lahdaoui F, Skrypek N, Peretti F, Frénois F, et al. The EGF Domains of MUC4 Oncomucin Mediate HER2 Binding Affinity and Promote Pancreatic Cancer Cell Tumorigenesis. Cancers. 2021; 13(22):5746. https://doi.org/10.3390/cancers13225746
Chicago/Turabian StyleStoup, Nicolas, Maxime Liberelle, Céline Schulz, Sumeyye Cavdarli, Romain Vasseur, Romain Magnez, Fatima Lahdaoui, Nicolas Skrypek, Fabien Peretti, Frédéric Frénois, and et al. 2021. "The EGF Domains of MUC4 Oncomucin Mediate HER2 Binding Affinity and Promote Pancreatic Cancer Cell Tumorigenesis" Cancers 13, no. 22: 5746. https://doi.org/10.3390/cancers13225746