
Targeting Metabolism to Control Immune Responses in Cancer and Improve Checkpoint Blockade Immunotherapy


Abstract
:Simple Summary
Abstract
1. Introduction
2. Pro- and Antitumor Immune Responses
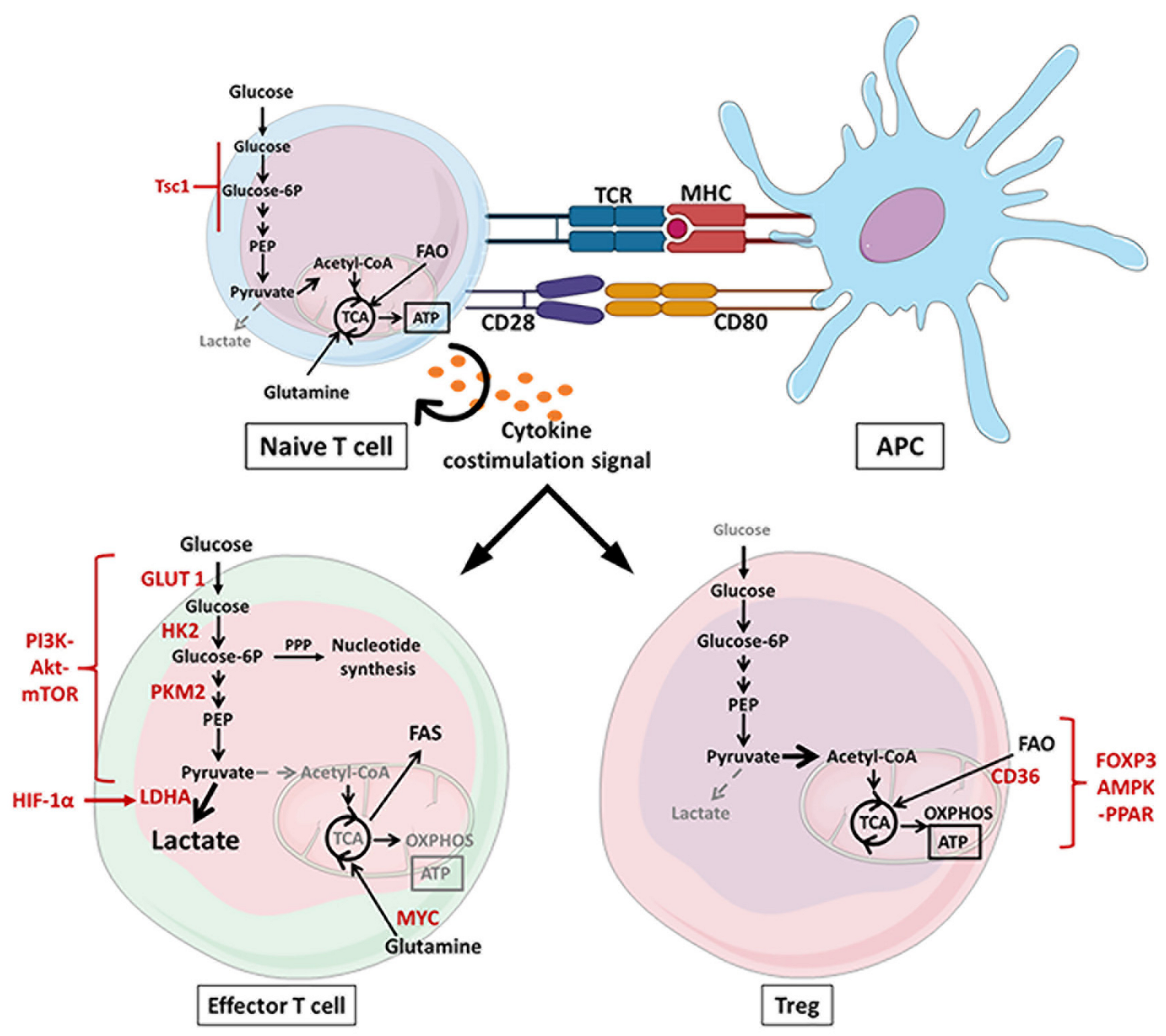
2.1. Metabolic Reprograming of T Cells
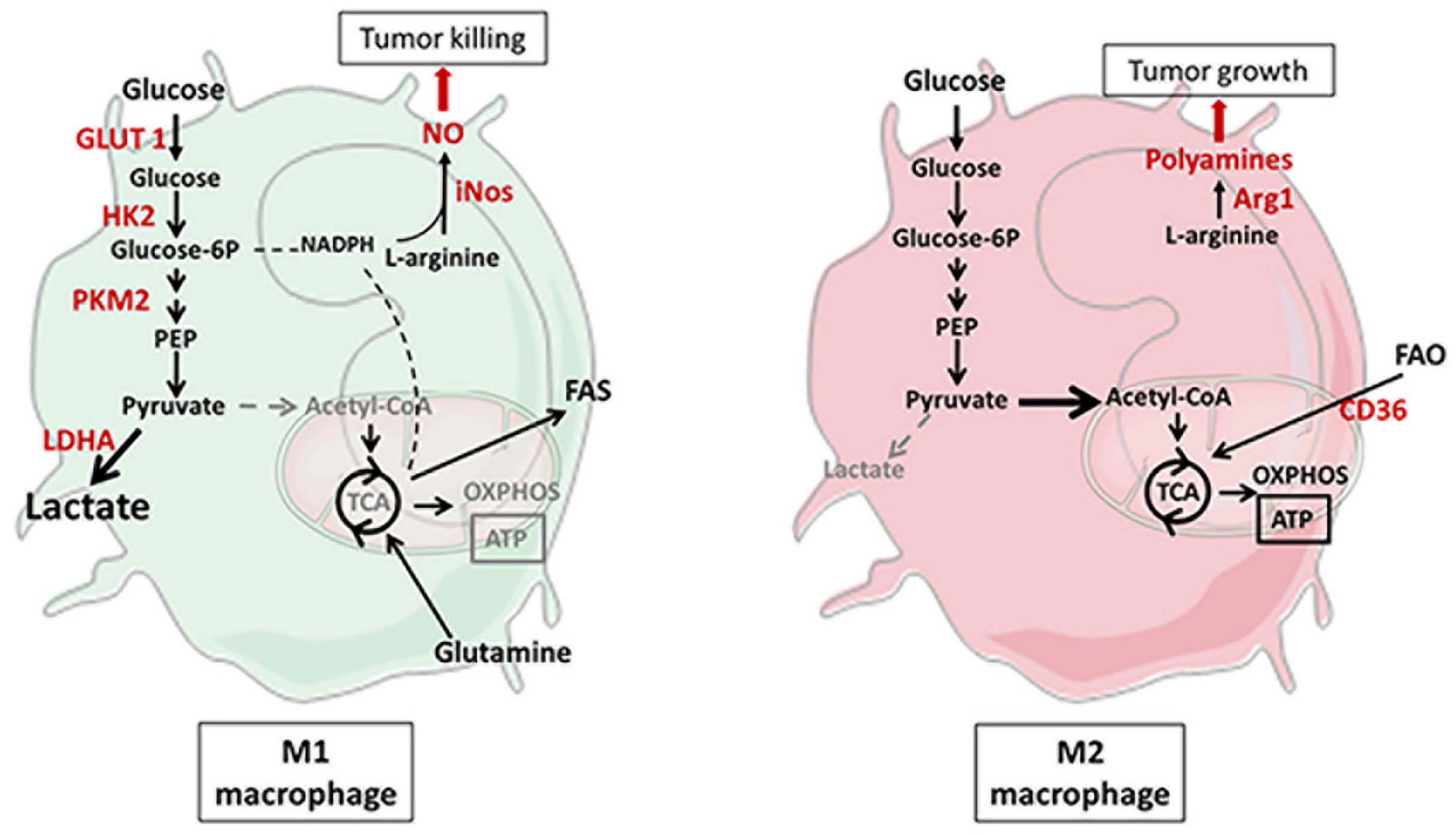
2.2. Metabolic Reprogramming of Macrophages
3. Metabolic Variation in the TME and Consequences on Immunity
3.1. Glucose–Lactate–Oxygen
3.2. Lipids
3.3. Amino Acids
3.3.1. Tryptophan
3.3.2. Arginine
3.3.3. Other Amino Acids
3.4. Adenosine
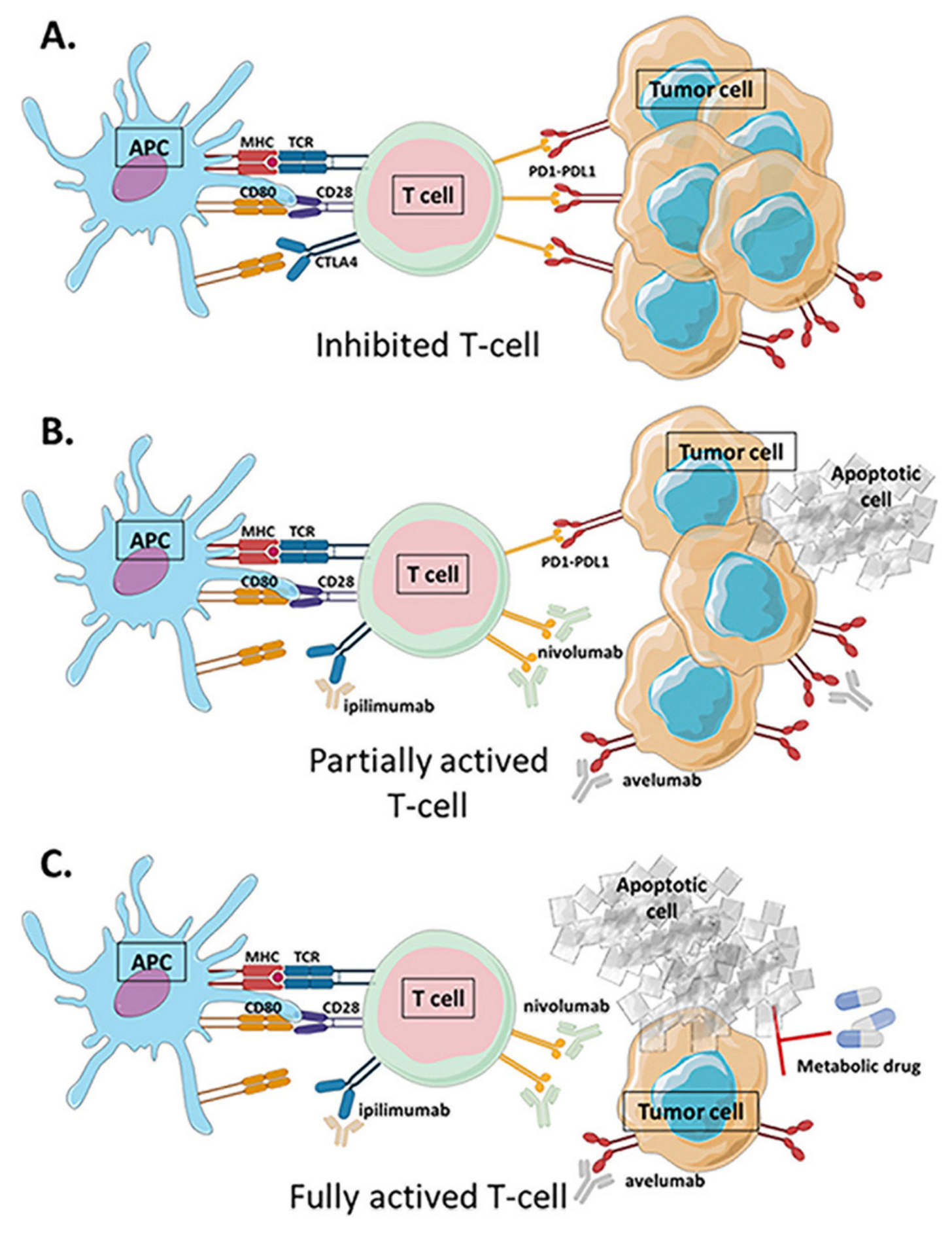
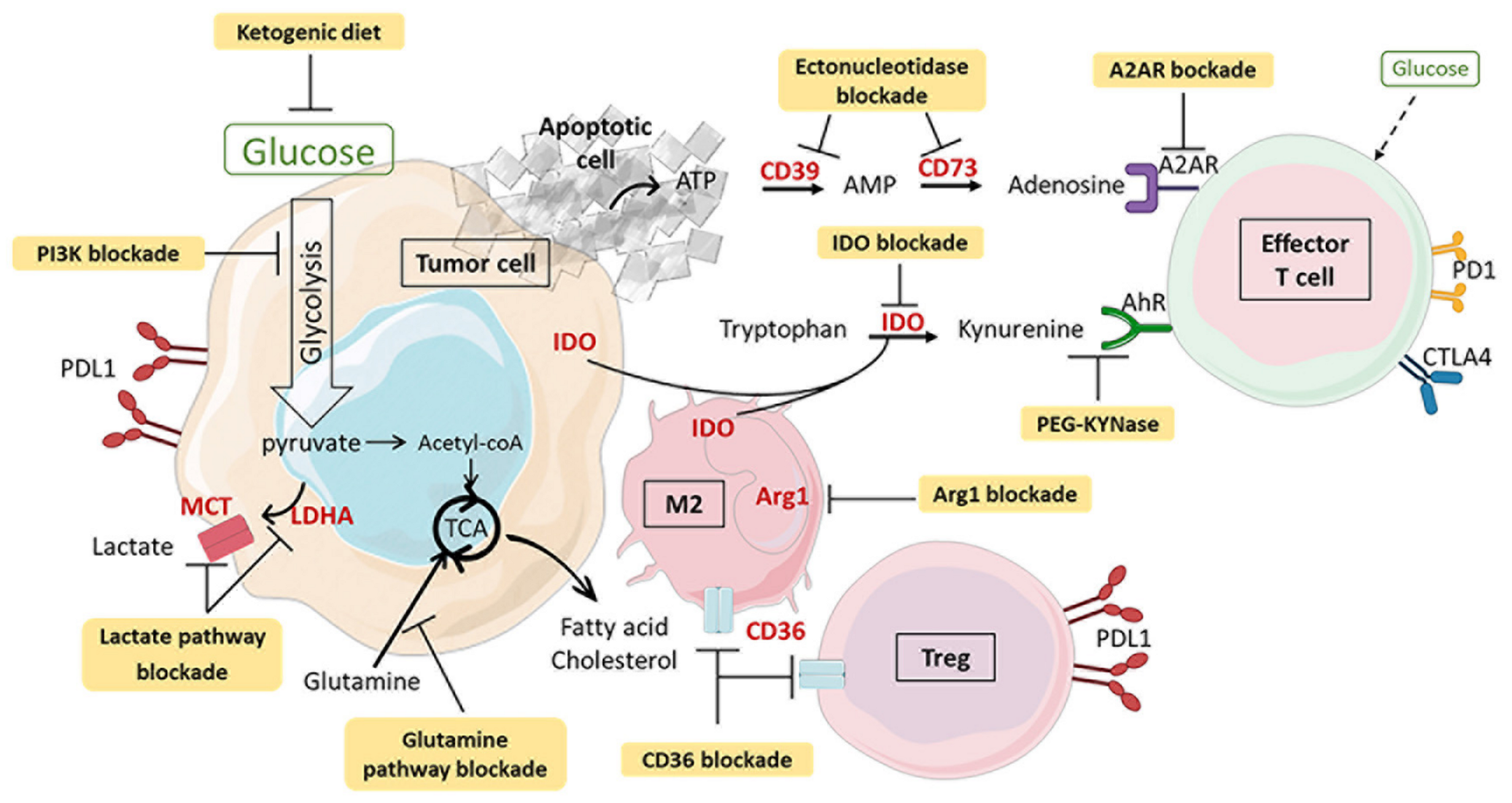
4. Combination of Metabolic Intervention and Immunotherapy
4.1. PI3K Inhibitors
4.2. Lactate Metabolism Modulation
4.3. AMPK Activation
4.4. Tryptophan–Kynurenine Pathway Blockade
4.5. Glutamine Pathway Inhibitors
4.6. Hypoxia and Adenosine Signaling Blockade
4.7. Dietary Modifications and Microbiota Modulation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, C.S.; Metallo, C.M. Mitochondria as Biosynthetic Factories for Cancer Proliferation. Cancer Metab. 2015, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Amoedo, N.D.; Sarlak, S.; Obre, E.; Esteves, P.; Bégueret, H.; Kieffer, Y.; Rousseau, B.; Dupis, A.; Izotte, J.; Bellance, N.; et al. Targeting the Mitochondrial Trifunctional Protein Restrains Tumor Growth in Oxidative Lung Carcinomas. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Robey, I.F.; Lien, A.D.; Welsh, S.J.; Baggett, B.K.; Gillies, R.J. Hypoxia-Inducible Factor-1α and the Glycolytic Phenotype in Tumors. Neoplasia 2005, 7, 324–330. [Google Scholar] [CrossRef] [Green Version]
- Dibble, C.C.; Manning, B.D. Signal Integration by MTORC1 Coordinates Nutrient Input with Biosynthetic Output. Nat. Cell Biol. 2013, 15, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Leiphrakpam, P.D.; Chowdhury, S.; Wang, J.; Black, J.D.; Are, C. The Role and Therapeutic Implications of PI3K Signaling Pathway in Cancer. J. Surg. Oncol. 2021, 123, 39–41. [Google Scholar] [CrossRef]
- Leone, R.D.; Powell, J.D. Metabolism of Immune Cells in Cancer. Nat. Rev. Cancer 2020, 20, 516–531. [Google Scholar] [CrossRef]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Andrejeva, G.; Rathmell, J.C. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab. 2017, 26, 49–70. [Google Scholar] [CrossRef] [Green Version]
- Riera-Domingo, C.; Audigé, A.; Granja, S.; Cheng, W.-C.; Ho, P.-C.; Baltazar, F.; Stockmann, C.; Mazzone, M. Immunity, Hypoxia, and Metabolism–the Ménage à Trois of Cancer: Implications for Immunotherapy. Physiol. Rev. 2019, 100, 1–102. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Immunobiology of Cancer Immunosurveillance and Immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer Immunoediting and Resistance to T Cell-Based Immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Franco, F.; Jaccard, A.; Romero, P.; Yu, Y.-R.; Ho, P.-C. Metabolic and Epigenetic Regulation of T-Cell Exhaustion. Nat. Metab. 2020, 2, 1001–1012. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-Cell Exhaustion in the Tumor Microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 377, 1345–1356. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wenes, M.; Romero, P.; Huang, S.C.-C.; Fendt, S.-M.; Ho, P.-C. Navigating Metabolic Pathways to Enhance Antitumour Immunity and Immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 425–441. [Google Scholar] [CrossRef]
- Ceredig, R.; Rolink, T. A Positive Look at Double-Negative Thymocytes. Nat. Rev. Immunol. 2002, 2, 888–897. [Google Scholar] [CrossRef]
- Finlay, D. Regulation of Glucose Metabolism in T Cells: New Insight into the Role of Phosphoinositide 3-Kinases. Front. Immunol. 2012, 3, 247. [Google Scholar] [CrossRef] [Green Version]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T Cell Metabolism Drives Immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacIver, N.J.; Michalek, R.D.; Rathmell, J.C. Metabolic Regulation of T Lymphocytes. Annu. Rev. Immunol. 2013, 31, 259–283. [Google Scholar] [CrossRef] [Green Version]
- Newton, R.H.; Shrestha, S.; Sullivan, J.M.; Yates, K.B.; Compeer, E.B.; Ron-Harel, N.; Blazar, B.R.; Bensinger, S.J.; Haining, W.N.; Dustin, M.L.; et al. Maintenance of CD4 T Cell Fitness through Regulation of Foxo1. Nat. Immunol. 2018, 19, 838–848. [Google Scholar] [CrossRef]
- Hamilton, S.E.; Jameson, S.C. CD8 T Cell Quiescence Revisited. Trends Immunol. 2012, 33, 224–230. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Liu, Y.; Chen, C.; Ikenoue, T.; Qiao, Y.; Li, C.-S.; Li, W.; Guan, K.-L.; Liu, Y.; Zheng, P. The Tuberous Sclerosis Complex–Mammalian Target of Rapamycin Pathway Maintains the Quiescence and Survival of Naive T Cells. J. Immunol. 2011, 187, 1106–1112. [Google Scholar] [CrossRef]
- Yang, K.; Neale, G.; Green, D.R.; He, W.; Chi, H. The Tumor Suppressor Tsc1 Enforces Quiescence of Naive T Cells to Promote Immune Homeostasis and Function. Nat. Immunol. 2011, 12, 888–897. [Google Scholar] [CrossRef]
- Chapman, N.M.; Boothby, M.R.; Chi, H. Metabolic Coordination of T Cell Quiescence and Activation. Nat. Rev. Immunol. 2020, 20, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Shrestha, S.; Zeng, H.; Karmaus, P.W.F.; Neale, G.; Vogel, P.; Guertin, D.A.; Lamb, R.F.; Chi, H. T Cell Exit from Quiescence and Differentiation into Th2 Cells Depend on Raptor-MTORC1-Mediated Metabolic Reprogramming. Immunity 2013, 39, 1043–1056. [Google Scholar] [CrossRef] [Green Version]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 Signaling Pathway Regulates Glucose Metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef] [Green Version]
- DiToro, D.; Winstead, C.J.; Pham, D.; Witte, S.; Andargachew, R.; Singer, J.R.; Wilson, C.G.; Zindl, C.L.; Luther, R.J.; Silberger, D.J.; et al. Differential IL-2 Expression Defines Developmental Fates of Follicular versus Nonfollicular Helper T Cells. Science 2018, 361. [Google Scholar] [CrossRef]
- Uche, U.U.; Piccirillo, A.R.; Kataoka, S.; Grebinoski, S.J.; D’Cruz, L.M.; Kane, L.P. PIK3IP1/TrIP Restricts Activation of T Cells through Inhibition of PI3K/Akt. J. Exp. Med. 2018, 215, 3165–3179. [Google Scholar] [CrossRef] [Green Version]
- Hawse, W.F.; Sheehan, R.P.; Miskov-Zivanov, N.; Menk, A.V.; Kane, L.P.; Faeder, J.R.; Morel, P.A. Cutting Edge: Differential Regulation of PTEN by TCR, Akt, and FoxO1 Controls CD4+ T Cell Fate Decisions. J. Immunol. 2015, 194, 4615–4619. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; He, Y.; Zhou, S.; Cao, Y.; Li, Y.; Bi, Y.; Liu, G. HIF1α-Dependent Metabolic Signals Control the Differentiation of Follicular Helper T Cells. Cells 2019, 8, 1450. [Google Scholar] [CrossRef] [Green Version]
- Palazon, A.; Tyrakis, P.A.; Macias, D.; Veliça, P.; Rundqvist, H.; Fitzpatrick, S.; Vojnovic, N.; Phan, A.T.; Loman, N.; Hedenfalk, I.; et al. An HIF-1α/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell 2017, 32, 669–683.e5. [Google Scholar] [CrossRef] [Green Version]
- Menk, A.V.; Scharping, N.E.; Moreci, R.S.; Zeng, X.; Guy, C.; Salvatore, S.; Bae, H.; Xie, J.; Young, H.A.; Wendell, S.G.; et al. Early TCR Signaling Induces Rapid Aerobic Glycolysis Enabling Distinct Acute T Cell Effector Functions. Cell Rep. 2018, 22, 1509–1521. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.O.; Wolf, M.M.; Madden, M.Z.; Andrejeva, G.; Sugiura, A.; Contreras, D.C.; Maseda, D.; Liberti, M.V.; Paz, K.; Kishton, R.J.; et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell 2018, 175, 1780–1795.e19. [Google Scholar] [CrossRef] [Green Version]
- Carr, E.L.; Kelman, A.; Wu, G.S.; Gopaul, R.; Senkevitch, E.; Aghvanyan, A.; Turay, A.M.; Frauwirth, K.A. Glutamine Uptake and Metabolism Are Coordinately Regulated by ERK/MAPK during T Lymphocyte Activation. J. Immunol. 2010, 185, 1037–1044. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.-H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.-C. Inflammatory T Cell Responses Rely on Amino Acid Transporter ASCT2 Facilitation of Glutamine Uptake and MTORC1 Kinase Activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef] [Green Version]
- Klysz, D.; Tai, X.; Robert, P.A.; Craveiro, M.; Cretenet, G.; Oburoglu, L.; Mongellaz, C.; Floess, S.; Fritz, V.; Matias, M.I.; et al. Glutamine-Dependent α-Ketoglutarate Production Regulates the Balance between T Helper 1 Cell and Regulatory T Cell Generation. Sci. Signal. 2015, 8, ra97. [Google Scholar] [CrossRef] [Green Version]
- Ma, E.H.; Bantug, G.; Griss, T.; Condotta, S.; Johnson, R.M.; Samborska, B.; Mainolfi, N.; Suri, V.; Guak, H.; Balmer, M.L.; et al. Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab. 2017, 25, 345–357. [Google Scholar] [CrossRef]
- Cluxton, D.; Petrasca, A.; Moran, B.; Fletcher, J.M. Differential Regulation of Human Treg and Th17 Cells by Fatty Acid Synthesis and Glycolysis. Front. Immunol. 2019, 10, 115. [Google Scholar] [CrossRef] [Green Version]
- Berod, L.; Friedrich, C.; Nandan, A.; Freitag, J.; Hagemann, S.; Harmrolfs, K.; Sandouk, A.; Hesse, C.; Castro, C.N.; Bähre, H.; et al. De Novo Fatty Acid Synthesis Controls the Fate between Regulatory T and T Helper 17 Cells. Nat. Med. 2014, 20, 1327–1333. [Google Scholar] [CrossRef]
- Lee, J.; Walsh, M.C.; Hoehn, K.L.; James, D.E.; Wherry, E.J.; Choi, Y. Regulator of Fatty Acid Metabolism, Acetyl Coenzyme A Carboxylase 1, Controls T Cell Immunity. J. Immunol. 2014, 192, 3190–3199. [Google Scholar] [CrossRef] [Green Version]
- Gabryšová, L.; Christensen, J.R.; Wu, X.; Kissenpfennig, A.; Malissen, B.; O’Garra, A. Integrated T-Cell Receptor and Costimulatory Signals Determine TGF-β-Dependent Differentiation and Maintenance of Foxp3+ Regulatory T Cells. Eur. J. Immunol. 2011, 41, 1242–1248. [Google Scholar] [CrossRef]
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J.; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e7. [Google Scholar] [CrossRef] [Green Version]
- Strauss, L.; Whiteside, T.L.; Knights, A.; Bergmann, C.; Knuth, A.; Zippelius, A. Selective Survival of Naturally Occurring Human CD4+CD25+Foxp3+ Regulatory T Cells Cultured with Rapamycin. J. Immunol. 2007, 178, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Franco, F.; Tsui, Y.-C.; Xie, X.; Trefny, M.P.; Zappasodi, R.; Mohmood, S.R.; Fernández-García, J.; Tsai, C.-H.; Schulze, I.; et al. CD36-Mediated Metabolic Adaptation Supports Regulatory T Cell Survival and Function in Tumors. Nat. Immunol. 2020, 21, 298–308. [Google Scholar] [CrossRef]
- Ma, E.H.; Poffenberger, M.C.; Wong, A.H.-T.; Jones, R.G. The Role of AMPK in T Cell Metabolism and Function. Curr. Opin. Immunol. 2017, 46, 45–52. [Google Scholar] [CrossRef]
- Blagih, J.; Coulombe, F.; Vincent, E.E.; Dupuy, F.; Galicia-Vázquez, G.; Yurchenko, E.; Raissi, T.C.; van der Windt, G.J.W.; Viollet, B.; Pearce, E.L.; et al. The Energy Sensor AMPK Regulates T Cell Metabolic Adaptation and Effector Responses In Vivo. Immunity 2015, 42, 41–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, M.D.; O’Sullivan, D.; Klein Geltink, R.I.; Curtis, J.D.; Chang, C.-H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.W.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicoli, F.; Papagno, L.; Frere, J.J.; Cabral-Piccin, M.P.; Clave, E.; Gostick, E.; Toubert, A.; Price, D.A.; Caputo, A.; Appay, V. Naïve CD8+ T-Cells Engage a Versatile Metabolic Program Upon Activation in Humans and Differ Energetically From Memory CD8+ T-Cells. Front. Immunol. 2018, 9, 736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simula, L.; Pacella, I.; Colamatteo, A.; Procaccini, C.; Cancila, V.; Bordi, M.; Tregnago, C.; Corrado, M.; Pigazzi, M.; Barnaba, V.; et al. Drp1 Controls Effective T Cell Immune-Surveillance by Regulating T Cell Migration, Proliferation, and CMyc-Dependent Metabolic Reprogramming. Cell Rep. 2018, 25, 3059–3073.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Ley, K. M1 and M2 Macrophages: The Chicken and the Egg of Immunity. JIN 2014, 6, 716–726. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, S.; Jeon, R.; Vuckovic, I.; Jiang, X.; Lerman, A.; Folmes, C.D.; Dzeja, P.D.; Herrmann, J. Interferon Gamma Induces Reversible Metabolic Reprogramming of M1 Macrophages to Sustain Cell Viability and Pro-Inflammatory Activity. EBioMedicine 2018, 30, 303–316. [Google Scholar] [CrossRef] [Green Version]
- Penny, H.L.; Sieow, J.L.; Adriani, G.; Yeap, W.H.; Ee, P.S.C.; Luis, B.S.; Lee, B.; Lee, T.; Mak, S.Y.; Ho, Y.S.; et al. Warburg Metabolism in Tumor-Conditioned Macrophages Promotes Metastasis in Human Pancreatic Ductal Adenocarcinoma. OncoImmunology 2016, 5, e1191731. [Google Scholar] [CrossRef]
- Bingle, L.; Lewis, C.E.; Corke, K.P.; Reed, M.W.R.; Brown, N.J. Macrophages Promote Angiogenesis in Human Breast Tumour Spheroids in Vivo. Br. J. Cancer 2006, 94, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Xie, M.; Yang, M.; Yu, Y.; Zhu, S.; Hou, W.; Kang, R.; Lotze, M.T.; Billiar, T.R.; Wang, H.; et al. PKM2 Regulates the Warburg Effect and Promotes HMGB1 Release in Sepsis. Nat. Commun. 2014, 5, 4436. [Google Scholar] [CrossRef]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic Reprogramming of Macrophages: Glucose transporter 1 (glut1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef] [Green Version]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.R.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.M.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.W.; et al. Pyruvate Kinase M2 Regulates Hif-1α Activity and IL-1β Induction and Is a Critical Determinant of the Warburg Effect in LPS-Activated Macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Wang, Y.; Dong, L.; He, Y.; Liu, R.; Yang, Q.; Cao, Y.; Wang, Y.; Jia, A.; Bi, Y.; et al. Regulations of Glycolytic Activities on Macrophages Functions in Tumor and Infectious Inflammation. Front. Cell Infect. Microbiol. 2020, 10, 287. [Google Scholar] [CrossRef]
- Tan, Z.; Xie, N.; Cui, H.; Moellering, D.R.; Abraham, E.; Thannickal, V.J.; Liu, G. Pyruvate Dehydrogenase Kinase 1 Participates in Macrophage Polarization via Regulating Glucose Metabolism. J. Immunol. 2015, 194, 6082–6089. [Google Scholar] [CrossRef]
- Wang, T.; Liu, H.; Lian, G.; Zhang, S.-Y.; Wang, X.; Jiang, C. HIF1α-Induced Glycolysis Metabolism is Essential to the Activation of Inflammatory Macrophages. Mediat. Inflamm. 2017, 2017, e9029327. [Google Scholar] [CrossRef] [Green Version]
- Galván-Peña, S.; O’Neill, L.A.J. Metabolic Reprograming in Macrophage Polarization. Front. Immunol. 2014, 5, 420. [Google Scholar] [CrossRef] [Green Version]
- Bailey, J.D.; Diotallevi, M.; Nicol, T.; McNeill, E.; Shaw, A.; Chuaiphichai, S.; Hale, A.; Starr, A.; Nandi, M.; Stylianou, E.; et al. Nitric Oxide Modulates Metabolic Remodeling in Inflammatory Macrophages through TCA Cycle Regulation and Itaconate Accumulation. Cell Rep. 2019, 28, 218–230.e7. [Google Scholar] [CrossRef] [Green Version]
- Anttila, M.; Voutilainen, K.; Merivalo, S.; Saarikoski, S.; Kosma, V. Prognostic Significance of INOS in Epithelial Ovarian Cancer. Gynecol. Oncol. 2007, 105, 97–103. [Google Scholar] [CrossRef]
- Puhakka, A.; Kinnula, V.; Näpänkangas, U.; Säily, M.; Koistinen, P.; Pääkkö, P.; Soini, Y. High Expression of Nitric Oxide Synthases is a Favorable Prognostic Sign in Non-Small Cell Lung Carcinoma. APMIS 2003, 111, 1137–1146. [Google Scholar] [CrossRef]
- Le, X.; Wei, D.; Huang, S.; Lancaster, J.R.; Xie, K. Nitric Oxide Synthase II Suppresses the Growth and Metastasis of Human Cancer Regardless of its Up-Regulation of Protumor Factors. Proc. Natl. Acad. Sci. USA 2005, 102, 8758–8763. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.C.-C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-Intrinsic Lysosomal Lipolysis Is Essential for Alternative Activation of Macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef] [Green Version]
- Tavakoli, S.; Zamora, D.; Ullevig, S.; Asmis, R. Bioenergetic Profiles Diverge During Macrophage Polarization: Implications for the Interpretation of 18F-FDG PET Imaging of Atherosclerosis. J. Nucl. Med. 2013, 54, 1661–1667. [Google Scholar] [CrossRef] [Green Version]
- Szanto, A.; Balint, B.L.; Nagy, Z.S.; Barta, E.; Dezso, B.; Pap, A.; Szeles, L.; Poliska, S.; Oros, M.; Evans, R.M.; et al. STAT6 Transcription Factor Is a Facilitator of the Nuclear Receptor PPARγ-Regulated Gene Expression in Macrophages and Dendritic Cells. Immunity 2010, 33, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.T.; Welch, J.S.; Ricote, M.; Binder, C.J.; Willson, T.M.; Kelly, C.; Witztum, J.L.; Funk, C.D.; Conrad, D.; Glass, C.K. Interleukin-4-Dependent Production of PPAR-γ Ligands in Macrophages by 12/15-Lipoxygenase. Nature 1999, 400, 378–382. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, H.; Mao, C.; Sun, M.; Dominah, G.; Chen, L.; Zhuang, Z. Fatty Acid Oxidation Contributes to IL-1β Secretion in M2 Macrophages and Promotes Macrophage-Mediated Tumor Cell Migration. Mol. Immunol. 2018, 94, 27–35. [Google Scholar] [CrossRef]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative Metabolism and PGC-1β Attenuate Macrophage-Mediated Inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Eagle, A.R.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-Specific PPARγ Controls Alternative Activation and Improves Insulin Resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Liu, L.; Gong, C.; Shi, H.; Zeng, Y.; Wang, X.; Zhao, Y.; Wei, Y. Prognostic Significance of Tumor-Associated Macrophages in Solid Tumor: A Meta-Analysis of the Literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czystowska-Kuzmicz, M.; Sosnowska, A.; Nowis, D.; Ramji, K.; Szajnik, M.; Chlebowska-Tuz, J.; Wolinska, E.; Gaj, P.; Grazul, M.; Pilch, Z.; et al. Small Extracellular Vesicles Containing Arginase-1 Suppress T-Cell Responses and Promote Tumor Growth in Ovarian Carcinoma. Nat. Commun. 2019, 10, 3000. [Google Scholar] [CrossRef]
- Ma, Z.; Lian, J.; Yang, M.; Wuyang, J.; Zhao, C.; Chen, W.; Liu, C.; Zhao, Q.; Lou, C.; Han, J.; et al. Overexpression of Arginase-1 Is an Indicator of Poor Prognosis in Patients with Colorectal Cancer. Pathol. Res. Pract. 2019, 215, 152383. [Google Scholar] [CrossRef]
- Grzywa, T.M.; Sosnowska, A.; Matryba, P.; Rydzynska, Z.; Jasinski, M.; Nowis, D.; Golab, J. Myeloid Cell-Derived Arginase in Cancer Immune Response. Front. Immunol. 2020, 11, 938. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.-C.; Smith, A.M.; Everts, B.; Colonna, M.; Pearce, E.L.; Schilling, J.D.; Pearce, E.J. Metabolic Reprogramming Mediated by the MTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity 2016, 45, 817–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covarrubias, A.J.; Aksoylar, H.I.; Yu, J.; Snyder, N.W.; Worth, A.J.; Iyer, S.S.; Wang, J.; Ben-Sahra, I.; Byles, V.; Polynne-Stapornkul, T.; et al. Akt-MTORC1 Signaling Regulates Acly to Integrate Metabolic Input to Control of Macrophage Activation. eLife 2016, 5, e11612. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, S.; Vuckovic, I.; Jeon, R.; Lerman, A.; Folmes, C.D.; Dzeja, P.P.; Herrmann, J. Glycolytic Stimulation Is Not a Requirement for M2 Macrophage Differentiation. Cell Metab. 2018, 28, 463–475.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.-R.; Imrichova, H.; Wang, H.; Chao, T.; Xiao, Z.; Gao, M.; Rincon-Restrepo, M.; Franco, F.; Genolet, R.; Cheng, W.-C.; et al. Disturbed Mitochondrial Dynamics in CD8 + TILs Reinforce T Cell Exhaustion. Nat. Immunol. 2020, 21, 1540–1551. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016, 45, 374–388. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekaran, S.; Sasaki, M.; Scharer, C.D.; Kissick, H.T.; Patterson, D.G.; Magliocca, K.R.; Seykora, J.T.; Sapkota, B.; Gutman, D.A.; Cooper, L.A.; et al. Phosphoinositide 3-Kinase Signaling Can Modulate MHC Class I and II Expression. Mol. Cancer Res. 2019, 17, 2395–2409. [Google Scholar] [CrossRef] [Green Version]
- Marijt, K.A.; Sluijter, M.; Blijleven, L.; Tolmeijer, S.H.; Scheeren, F.A.; van der Burg, S.H.; van Hall, T. Metabolic Stress in Cancer Cells Induces Immune Escape through a PI3K-Dependent Blockade of IFNγ Receptor Signaling. J. Immunother. Cancer 2019, 7, 152. [Google Scholar] [CrossRef] [Green Version]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 Alters T-Cell Metabolic Reprogramming by Inhibiting Glycolysis and Promoting Lipolysis and Fatty Acid Oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8+ T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 2017, 32, 377–391.e9. [Google Scholar] [CrossRef] [Green Version]
- Reinfeld, B.I.; Madden, M.Z.; Wolf, M.M.; Chytil, A.; Bader, J.E.; Patterson, A.R.; Sugiura, A.; Cohen, A.S.; Ali, A.; Do, B.T.; et al. Cell Programmed Nutrient Partitioning in the Tumor Microenvironment. Nature 2021, 593, 282–288. [Google Scholar] [CrossRef]
- Murdoch, C.; Giannoudis, A.; Lewis, C.E. Mechanisms Regulating the Recruitment of Macrophages into Hypoxic Areas of Tumors and Other Ischemic Tissues. Blood 2004, 104, 2224–2234. [Google Scholar] [CrossRef]
- Miar, A.; Arnaiz, E.; Bridges, E.; Beedie, S.; Cribbs, A.P.; Downes, D.J.; Beagrie, R.A.; Rehwinkel, J.; Harris, A.L. Hypoxia Induces Transcriptional and Translational Downregulation of the Type I IFN Pathway in Multiple Cancer Cell Types. Cancer Res. 2020, 80, 5245–5256. [Google Scholar] [CrossRef]
- Kim, J.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-Mediated Expression of Pyruvate Dehydrogenase Kinase: A Metabolic Switch Required for Cellular Adaptation to Hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Chaudhury, A.; Zhang, M.; Savoldo, B.; Metelitsa, L.S.; Rodgers, J.; Yustein, J.T.; Neilson, J.R.; Dotti, G. Glycolysis Determines Dichotomous Regulation of T Cell Subsets in Hypoxia. J. Clin. Investig. 2016, 126, 2678–2688. [Google Scholar] [CrossRef]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A Key Player in Antitumor Immune Response. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol.-Cell Physiol. 2015, 309, C569–C579. [Google Scholar] [CrossRef] [Green Version]
- El-Kenawi, A.; Gatenbee, C.; Robertson-Tessi, M.; Bravo, R.; Dhillon, J.; Balagurunathan, Y.; Berglund, A.; Vishvakarma, N.; Ibrahim-Hashim, A.; Choi, J.; et al. Acidity Promotes Tumour Progression by Altering Macrophage Phenotype in Prostate Cancer. Br. J. Cancer 2019, 121, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [Green Version]
- Quinn, W.J.; Jiao, J.; TeSlaa, T.; Stadanlick, J.; Wang, Z.; Wang, L.; Akimova, T.; Angelin, A.; Schäfer, P.M.; Cully, M.D.; et al. Lactate Limits T Cell Proliferation via the NAD(H) Redox State. Cell Rep. 2020, 33, 108500. [Google Scholar] [CrossRef]
- Zappasodi, R.; Serganova, I.; Cohen, I.J.; Maeda, M.; Shindo, M.; Senbabaoglu, Y.; Watson, M.J.; Leftin, A.; Maniyar, R.; Verma, S.; et al. CTLA-4 Blockade Drives Loss of T Reg Stability in Glycolysis-Low Tumours. Nature 2021, 591, 652–658. [Google Scholar] [CrossRef]
- Chen, F.; Chen, J.; Yang, L.; Liu, J.; Zhang, X.; Zhang, Y.; Tu, Q.; Yin, D.; Lin, D.; Wong, P.-P.; et al. Extracellular Vesicle-Packaged HIF-1α-Stabilizing LncRNA from Tumour-Associated Macrophages Regulates Aerobic Glycolysis of Breast Cancer Cells. Nat. Cell Biol. 2019, 21, 498–510. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, G.; Chu, H.; Wang, X.; Xiong, L.; Cai, G.; Liu, R.; Gao, H.; Tao, B.; Li, W.; et al. Macrophage-Associated PGK1 Phosphorylation Promotes Aerobic Glycolysis and Tumorigenesis. Mol. Cell 2018, 71, 201–215.e7. [Google Scholar] [CrossRef] [Green Version]
- Bacci, M.; Lorito, N.; Smiriglia, A.; Morandi, A. Fat and Furious: Lipid Metabolism in Antitumoral Therapy Response and Resistance. Trends Cancer 2021, 7, 198–213. [Google Scholar] [CrossRef]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. MTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823.e12. [Google Scholar] [CrossRef] [Green Version]
- Al-Khami, A.A.; Zheng, L.; Valle, L.D.; Hossain, F.; Wyczechowska, D.; Zabaleta, J.; Sanchez, M.D.; Dean, M.J.; Rodriguez, P.C.; Ochoa, A.C. Exogenous Lipid Uptake Induces Metabolic and Functional Reprogramming of Tumor-Associated Myeloid-Derived Suppressor Cells. OncoImmunology 2017, 6, e1344804. [Google Scholar] [CrossRef]
- Pacella, I.; Procaccini, C.; Focaccetti, C.; Miacci, S.; Timperi, E.; Faicchia, D.; Severa, M.; Rizzo, F.; Coccia, E.M.; Bonacina, F.; et al. Fatty Acid Metabolism Complements Glycolysis in the Selective Regulatory T Cell Expansion during Tumor Growth. Proc. Natl. Acad. Sci. USA 2018, 115, E6546–E6555. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.A.; Wei, J.; Nguyen, T.-L.M.; Shi, H.; Su, W.; Palacios, G.; Dhungana, Y.; Chapman, N.M.; Long, L.; Saravia, J.; et al. Lipid Signalling Enforces Treg Cell Functional Specialization in Tumours. Nature 2021, 591, 306–311. [Google Scholar] [CrossRef]
- Herber, D.L.; Cao, W.; Nefedova, Y.; Novitskiy, S.V.; Nagaraj, S.; Tyurin, V.A.; Corzo, A.; Cho, H.-I.; Celis, E.; Lennox, B.; et al. Lipid Accumulation and Dendritic Cell Dysfunction in Cancer. Nat. Med. 2010, 16, 880–886. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Chaudhary, O.; Rodríguez-Morales, P.; Sun, X.; Chen, D.; Zappasodi, R.; Xu, Z.; Pinto, A.F.M.; Williams, A.; Schulze, I.; et al. Uptake of Oxidized Lipids by the Scavenger Receptor CD36 Promotes Lipid Peroxidation and Dysfunction in CD8+ T Cells in Tumors. Immunity 2021, 54, 1561–1577.e7. [Google Scholar] [CrossRef]
- Ma, X.; Xiao, L.; Liu, L.; Ye, L.; Su, P.; Bi, E.; Wang, Q.; Yang, M.; Qian, J.; Yi, Q. CD36-Mediated Ferroptosis Dampens Intratumoral CD8+ T Cell Effector Function and Impairs Their Antitumor Ability. Cell Metab. 2021, 33, 1001–1012.e5. [Google Scholar] [CrossRef]
- Baek, A.E.; Yu, Y.-R.A.; He, S.; Wardell, S.E.; Chang, C.-Y.; Kwon, S.; Pillai, R.V.; McDowell, H.B.; Thompson, J.W.; Dubois, L.G.; et al. The Cholesterol Metabolite 27 Hydroxycholesterol Facilitates Breast Cancer Metastasis through Its Actions on Immune Cells. Nat. Commun. 2017, 8, 864. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.R.; Wardell, S.E.; Jasper, J.S.; Park, S.; Suchindran, S.; Howe, M.K.; Carver, N.J.; Pillai, R.V.; Sullivan, P.M.; Sondhi, V.; et al. 27-Hydroxycholesterol Links Hypercholesterolemia and Breast Cancer Pathophysiology. Science 2013, 342, 1094–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzo, T.; Prentice, B.M.; Anderson, K.G.; Raman, A.; Schalck, A.; Codreanu, G.S.; Nava Lauson, C.B.; Tiberti, S.; Raimondi, A.; Jones, M.A.; et al. Accumulation of Long-Chain Fatty Acids in the Tumor Microenvironment Drives Dysfunction in Intrapancreatic CD8+ T Cells. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Bi, E.; Lu, Y.; Su, P.; Huang, C.; Liu, L.; Wang, Q.; Yang, M.; Kalady, M.F.; Qian, J.; et al. Cholesterol Induces CD8+ T Cell Exhaustion in the Tumor Microenvironment. Cell Metab. 2019, 30, 143–156.e5. [Google Scholar] [CrossRef]
- Puccetti, P.; Fallarino, F.; Italiano, A.; Soubeyran, I.; MacGrogan, G.; Debled, M.; Velasco, V.; Bodet, D.; Eimer, S.; Veldhoen, M.; et al. Accumulation of an Endogenous Tryptophan-Derived Metabolite in Colorectal and Breast Cancers. PLoS ONE 2015, 10, e0122046. [Google Scholar] [CrossRef]
- Yu, C.-P.; Fu, S.-F.; Chen, X.; Ye, J.; Ye, Y.; Kong, L.-D.; Zhu, Z. The Clinicopathological and Prognostic Significance of IDO1 Expression in Human Solid Tumors: Evidence from a Systematic Review and Meta-Analysis. CPB 2018, 49, 134–143. [Google Scholar] [CrossRef]
- Katz, J.B.; Muller, A.J.; Prendergast, G.C. Indoleamine 2,3-Dioxygenase in T-Cell Tolerance and Tumoral Immune Escape. Immunol. Rev. 2008, 222, 206–221. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Liakopoulos, V.; Stefanidis, I. IDO Decreases Glycolysis and Glutaminolysis by Activating GCN2K, While It Increases Fatty Acid Oxidation by Activating AhR, Thus Preserving CD4+ T-cell Survival and Proliferation. Int. J. Mol. Med. 2018, 42, 557–568. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, X.; Dong, W.; Fang, Y.; Lv, J.; Zhang, T.; Fiskesund, R.; Xie, J.; Liu, J.; Yin, X.; et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8+ T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell 2018, 33, 480–494.e7. [Google Scholar] [CrossRef] [Green Version]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An Interaction between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [Green Version]
- Thaker, A.I.; Rao, M.S.; Bishnupuri, K.S.; Kerr, T.A.; Foster, L.; Marinshaw, J.M.; Newberry, R.D.; Stenson, W.F.; Ciorba, M.A. IDO1 Metabolites Activate β-Catenin Signaling to Promote Cancer Cell Proliferation and Colon Tumorigenesis in Mice. Gastroenterology 2013, 145, 416–425.e4. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.C.; Falcone, M.; Huerta Uribe, A.; Zhang, T.; Athineos, D.; Pietzke, M.; Vazquez, A.; Blyth, K.; Maddocks, O.D.K. Immune-Regulated IDO1-Dependent Tryptophan Metabolism Is Source of One-Carbon Units for Pancreatic Cancer and Stellate Cells. Mol. Cell 2021, 81, 2290–2302. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-Tumor Activity. Cell 2016, 167, 829–842.e13. [Google Scholar] [CrossRef] [Green Version]
- Zea, A.H.; Rodriguez, P.C.; Atkins, M.B.; Hernandez, C.; Signoretti, S.; Zabaleta, J.; McDermott, D.; Quiceno, D.; Youmans, A.; O’Neill, A.; et al. Arginase-Producing Myeloid Suppressor Cells in Renal Cell Carcinoma Patients: A Mechanism of Tumor Evasion. Cancer Res. 2005, 65, 3044–3048. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Jiang, H.; Liu, P.; Xie, N.; Fu, R.; Wang, H.; Liu, C.; Zhang, T.; Wang, H.; Shao, Z. Increased Myeloid-Derived Suppressor Cells in Patients with Myelodysplastic Syndromes Suppress CD8+ T Lymphocyte Function through the STAT3-ARG1 Pathway. Leuk. Lymphoma 2021, 62, 218–223. [Google Scholar] [CrossRef]
- Croce, M.; Damonte, P.; Morini, M.; Pigozzi, S.; Chiossone, L.; Vacca, P.; Bronte, V.; Barbieri, O.; Astigiano, S. Increased Arginase1 Expression in Tumor Microenvironment Promotes Mammary Carcinogenesis via Multiple Mechanisms. Carcinogenesis 2020, 41, 1695–1702. [Google Scholar] [CrossRef]
- Cao, Y.; Feng, Y.; Zhang, Y.; Zhu, X.; Jin, F. L-Arginine Supplementation Inhibits the Growth of Breast Cancer by Enhancing Innate and Adaptive Immune Responses Mediated by Suppression of MDSCs in Vivo. BMC Cancer 2016, 16, 343. [Google Scholar] [CrossRef] [Green Version]
- Satoh, Y.; Kotani, H.; Iida, Y.; Taniura, T.; Notsu, Y.; Harada, M. Supplementation of L-Arginine Boosts the Therapeutic Efficacy of Anticancer Chemoimmunotherapy. Cancer Sci. 2020, 111, 2248–2258. [Google Scholar] [CrossRef]
- Líndez, A.-A.M.I.; Dunand-Sauthier, I.; Conti, M.; Gobet, F.; Núñez, N.; Hannich, J.T.; Riezman, H.; Geiger, R.; Piersigilli, A.; Hahn, K.; et al. Mitochondrial Arginase-2 Is a Cell-autonomous Regulator of CD8+ T Cell Function and Antitumor Efficacy. JCI Insight 2020, 4, e132975. [Google Scholar] [CrossRef]
- Bian, Y.; Li, W.; Kremer, D.M.; Sajjakulnukit, P.; Li, S.; Crespo, J.; Nwosu, Z.C.; Zhang, L.; Czerwonka, A.; Pawłowska, A.; et al. Cancer SLC43A2 Alters T Cell Methionine Metabolism and Histone Methylation. Nature 2020, 585, 277–282. [Google Scholar] [CrossRef]
- Lappas, C.M.; Rieger, J.M.; Linden, J. A2A Adenosine Receptor Induction Inhibits IFN-γ Production in Murine CD4+ T Cells. J. Immunol. 2005, 174, 1073–1080. [Google Scholar] [CrossRef] [Green Version]
- Mastelic-Gavillet, B.; Rodrigo, B.N.; Décombaz, L.; Wang, H.; Ercolano, G.; Ahmed, R.; Lozano, L.E.; Ianaro, A.; Derré, L.; Valerio, M.; et al. Adenosine Mediates Functional and Metabolic Suppression of Peripheral and Tumor-Infiltrating CD8+ T Cells. J. Immunother. Cancer 2019, 7, 257. [Google Scholar] [CrossRef] [PubMed]
- Montalbán del Barrio, I.; Penski, C.; Schlahsa, L.; Stein, R.G.; Diessner, J.; Wöckel, A.; Dietl, J.; Lutz, M.B.; Mittelbronn, M.; Wischhusen, J.; et al. Adenosine-Generating Ovarian Cancer Cells Attract Myeloid Cells Which Differentiate into Adenosine-Generating Tumor Associated Macrophages—A Self-Amplifying, CD39- and CD73-Dependent Mechanism for Tumor Immune Escape. J. Immunother. Cancer 2016, 4, 49. [Google Scholar] [CrossRef] [Green Version]
- Cekic, C.; Day, Y.-J.; Sag, D.; Linden, J. Myeloid Expression of Adenosine A2A Receptor Suppresses T and NK Cell Responses in the Solid Tumor Microenvironment. Cancer Res. 2014, 74, 7250–7259. [Google Scholar] [CrossRef] [Green Version]
- Ohta, A.; Kini, R.; Ohta, A.; Subramanian, M.; Madasu, M.; Sitkovsky, M. The Development and Immunosuppressive Functions of CD4+ CD25+ FoxP3+ Regulatory T Cells Are under Influence of the Adenosine-A2A Adenosine Receptor Pathway. Front. Immunol. 2012, 3, 190. [Google Scholar] [CrossRef] [Green Version]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.-F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine Generation Catalyzed by CD39 and CD73 Expressed on Regulatory T Cells Mediates Immune Suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef] [Green Version]
- Sitkovsky, M.V.; Hatfield, S.; Abbott, R.; Belikoff, B.; Lukashev, D.; Ohta, A. Hostile, Hypoxia-A2-Adenosinergic Tumor Biology as the Next Barrier to the Tumor Immunologists. Cancer Immunol. Res. 2014, 2, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Nagate, Y.; Ezoe, S.; Fujita, J.; Okuzaki, D.; Motooka, D.; Ishibashi, T.; Ichii, M.; Tanimura, A.; Kurashige, M.; Morii, E.; et al. Ectonucleotidase CD39 Is Highly Expressed on ATLL Cells and Is Responsible for Their Immunosuppressive Function. Leukemia 2021, 35, 107–118. [Google Scholar] [CrossRef] [Green Version]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Wang, S.; Zhou, Q. The Resistance Mechanisms of Lung Cancer Immunotherapy. Front. Oncol. 2020, 10, 59. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell–Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borcoman, E.; De La Rochere, P.; Richer, W.; Vacher, S.; Chemlali, W.; Krucker, C.; Sirab, N.; Radvanyi, F.; Allory, Y.; Pignot, G.; et al. Inhibition of PI3K Pathway Increases Immune Infiltrate in Muscle-Invasive Bladder Cancer. Oncoimmunology 2019, 8, e1581556. [Google Scholar] [CrossRef] [PubMed]
- Deken, M.A.; Gadiot, J.; Jordanova, E.S.; Lacroix, R.; van Gool, M.; Kroon, P.; Pineda, C.; Foppen, M.H.G.; Scolyer, R.; Song, J.-Y.; et al. Targeting the MAPK and PI3K Pathways in Combination with PD1 Blockade in Melanoma. OncoImmunology 2016, 5, e1238557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, M.J.; Vignali, P.D.A.; Mullett, S.J.; Overacre-Delgoffe, A.E.; Peralta, R.M.; Grebinoski, S.; Menk, A.V.; Rittenhouse, N.L.; DePeaux, K.; Whetstone, R.D.; et al. Metabolic Support of Tumour-Infiltrating Regulatory T Cells by Lactic Acid. Nature 2021, 591, 645–651. [Google Scholar] [CrossRef]
- Diem, S.; Kasenda, B.; Spain, L.; Martin-Liberal, J.; Marconcini, R.; Gore, M.; Larkin, J. Serum Lactate Dehydrogenase as an Early Marker for Outcome in Patients Treated with Anti-PD-1 Therapy in Metastatic Melanoma. Br. J. Cancer 2016, 114, 256–261. [Google Scholar] [CrossRef]
- Kim, E.-Y.; Chung, T.-W.; Han, C.W.; Park, S.Y.; Park, K.H.; Jang, S.B.; Ha, K.-T. A Novel Lactate Dehydrogenase Inhibitor, 1-(Phenylseleno)-4-(Trifluoromethyl) Benzene, Suppresses Tumor Growth through Apoptotic Cell Death. Sci. Rep. 2019, 9, 3969. [Google Scholar] [CrossRef] [Green Version]
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Wang, Z.; Bristow, C.A.; Carugo, A.; et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018, 27, 977–987.e4. [Google Scholar] [CrossRef]
- Feng, Y.; Xiong, Y.; Qiao, T.; Li, X.; Jia, L.; Han, Y. Lactate Dehydrogenase A: A Key Player in Carcinogenesis and Potential Target in Cancer Therapy. Cancer Med. 2018, 7, 6124–6136. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Boily, G.; Izreig, S.; Griss, T.; Samborska, B.; Dong, Z.; Dupuy, F.; Chambers, C.; Fuerth, B.J.; Viollet, B.; et al. AMPK Is a Negative Regulator of the Warburg Effect and Suppresses Tumor Growth In Vivo. Cell Metab. 2013, 17, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Marini, C.; Salani, B.; Massollo, M.; Amaro, A.; Esposito, A.I.; Orengo, A.M.; Capitanio, S.; Emionite, L.; Riondato, M.; Bottoni, G.; et al. Direct Inhibition of Hexokinase Activity by Metformin at Least Partially Impairs Glucose Metabolism and Tumor Growth in Experimental Breast Cancer. Cell Cycle 2013, 12, 3490–3499. [Google Scholar] [CrossRef] [Green Version]
- Marini, C.; Bianchi, G.; Buschiazzo, A.; Ravera, S.; Martella, R.; Bottoni, G.; Petretto, A.; Emionite, L.; Monteverde, E.; Capitanio, S.; et al. Divergent Targets of Glycolysis and Oxidative Phosphorylation Result in Additive Effects of Metformin and Starvation in Colon and Breast Cancer. Sci. Rep. 2016, 6, 19569. [Google Scholar] [CrossRef]
- Qin, G.; Lian, J.; Huang, L.; Zhao, Q.; Liu, S.; Zhang, Z.; Chen, X.; Yue, D.; Li, L.; Li, F.; et al. Metformin Blocks Myeloid-Derived Suppressor Cell Accumulation through AMPK-DACH1-CXCL1 Axis. Oncoimmunology 2018, 7, e1442167. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Liang, G.; Yao, Z.; Zhang, J.; Liu, R.; Chen, H.; Zhou, Y.; Wu, H.; Yang, B.; He, Q. Metformin Prevents Cancer Metastasis by Inhibiting M2-like Polarization of Tumor Associated Macrophages. Oncotarget 2015, 6, 36441–36455. [Google Scholar] [CrossRef] [Green Version]
- Veeramachaneni, R.; Yu, W.; Newton, J.M.; Kemnade, J.O.; Skinner, H.D.; Sikora, A.G.; Sandulache, V.C. Metformin Generates Profound Alterations in Systemic and Tumor Immunity with Associated Antitumor Effects. J. Immunother. Cancer 2021, 9, e002773. [Google Scholar] [CrossRef]
- Eikawa, S.; Nishida, M.; Mizukami, S.; Yamazaki, C.; Nakayama, E.; Udono, H. Immune-Mediated Antitumor Effect by Type 2 Diabetes Drug, Metformin. Proc. Natl. Acad. Sci. USA 2015, 112, 1809–1814. [Google Scholar] [CrossRef] [Green Version]
- Cha, J.-H.; Yang, W.-H.; Xia, W.; Wei, Y.; Chan, L.-C.; Lim, S.-O.; Li, C.-W.; Kim, T.; Chang, S.-S.; Lee, H.-H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell 2018, 71, 606–620.e7. [Google Scholar] [CrossRef] [Green Version]
- Afzal, M.Z.; Mercado, R.R.; Shirai, K. Efficacy of Metformin in Combination with Immune Checkpoint Inhibitors (Anti-PD-1/Anti-CTLA-4) in Metastatic Malignant Melanoma. J. Immunother. Cancer 2018, 6, 64. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus Pembrolizumab versus Placebo plus Pembrolizumab in Patients with Unresectable or Metastatic Melanoma (ECHO-301/KEYNOTE-252): A Phase 3, Randomised, Double-Blind Study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-Dioxygenase Is a Critical Resistance Mechanism in Antitumor T Cell Immunotherapy Targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef]
- Triplett, T.A.; Garrison, K.C.; Marshall, N.; Donkor, M.; Blazeck, J.; Lamb, C.; Qerqez, A.; Dekker, J.D.; Tanno, Y.; Lu, W.-C.; et al. Reversal of Indoleamine 2,3-Dioxygenase–Mediated Cancer Immune Suppression by Systemic Kynurenine Depletion with a Therapeutic Enzyme. Nat. Biotechnol. 2018, 36, 758–764. [Google Scholar] [CrossRef]
- Dang, C.V. Glutaminolysis: Supplying Carbon or Nitrogen or Both for Cancer Cells? Cell Cycle 2010, 9, 3884–3886. [Google Scholar] [CrossRef] [Green Version]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [Green Version]
- Eagan, R.T.; Frytak, S.; Nichols, W.C.; Creagan, E.T.; Ingle, J.N. Phase II Study on DON in Patients with Previously Treated Advanced Lung Cancer. Cancer Treat. Rep. 1982, 66, 1665–1666. [Google Scholar]
- Lynch, G.; Kemeny, N.; Casper, E. Phase II Evaluation of DON (6-Diazo-5-Oxo-L-Norleucine) in Patients with Advanced Colorectal Carcinoma. Am. J. Clin. Oncol 1982, 5, 541–543. [Google Scholar] [CrossRef]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.-M.; Oh, M.-H.; Sun, I.-H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine Blockade Induces Divergent Metabolic Programs to Overcome Tumor Immune Evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.-K.; Park, M.; Lee, S.; Yun, J.W.; Lee, J.; Kim, J.S.; Cho, S.J.; Jeon, H.-J.; Lee, I.-K.; Choi, Y.-K.; et al. Inhibition of Glutamine Utilization Synergizes with Immune Checkpoint Inhibitor to Promote Antitumor Immunity. Mol. Cell 2020, 80, 592–606.e8. [Google Scholar] [CrossRef]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R.; et al. Immunological Mechanisms of the Antitumor Effects of Supplemental Oxygenation. Sci. Transl. Med. 2015, 7, 277ra30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Belikoff, B.; Schreiber, T.H.; Sethumadhavan, S.; Abbott, R.; Philbrook, P.; Thayer, M.; Shujia, D.; et al. Systemic Oxygenation Weakens the Hypoxia and Hypoxia Inducible Factor 1α-Dependent and Extracellular Adenosine-Mediated Tumor Protection. J. Mol. Med. 2014, 92, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Borodovsky, A.; Barbon, C.M.; Wang, Y.; Ye, M.; Prickett, L.; Chandra, D.; Shaw, J.; Deng, N.; Sachsenmeier, K.; Clarke, J.D.; et al. Small Molecule AZD4635 Inhibitor of A2AR Signaling Rescues Immune Cell Function Including CD103+ Dendritic Cells Enhancing Anti-Tumor Immunity. J. Immunother. Cancer 2020, 8, e000417. [Google Scholar] [CrossRef]
- Willingham, S.B.; Ho, P.Y.; Hotson, A.; Hill, C.; Piccione, E.C.; Hsieh, J.; Liu, L.; Buggy, J.J.; McCaffery, I.; Miller, R.A. A2AR Antagonism with CPI-444 Induces Antitumor Responses and Augments Efficacy to Anti–PD-(L)1 and Anti–CTLA-4 in Preclinical Models. Cancer Immunol. Res. 2018, 6, 1136–1149. [Google Scholar] [CrossRef] [Green Version]
- Stagg, J.; Divisekera, U.; Duret, H.; Sparwasser, T.; Teng, M.W.L.; Darcy, P.K.; Smyth, M.J. CD73-Deficient Mice Have Increased Antitumor Immunity and Are Resistant to Experimental Metastasis. Cancer Res. 2011, 71, 2892–2900. [Google Scholar] [CrossRef] [Green Version]
- Hay, C.M.; Sult, E.; Huang, Q.; Mulgrew, K.; Fuhrmann, S.R.; McGlinchey, K.A.; Hammond, S.A.; Rothstein, R.; Rios-Doria, J.; Poon, E.; et al. Targeting CD73 in the Tumor Microenvironment with MEDI9447. OncoImmunology 2016, 5, e1208875. [Google Scholar] [CrossRef]
- Yang, R.; Elsaadi, S.; Misund, K.; Abdollahi, P.; Vandsemb, E.N.; Moen, S.H.; Kusnierczyk, A.; Slupphaug, G.; Standal, T.; Waage, A.; et al. Conversion of ATP to Adenosine by CD39 and CD73 in Multiple Myeloma Can Be Successfully Targeted Together with Adenosine Receptor A2A Blockade. J. Immunother. Cancer 2020, 8, e000610. [Google Scholar] [CrossRef]
- Mimoto, F.; Tatsumi, K.; Shimizu, S.; Kadono, S.; Haraya, K.; Nagayasu, M.; Suzuki, Y.; Fujii, E.; Kamimura, M.; Hayasaka, A.; et al. Exploitation of Elevated Extracellular ATP to Specifically Direct Antibody to Tumor Microenvironment. Cell Rep. 2020, 33, 108542. [Google Scholar] [CrossRef]
- Sullivan, M.R.; Danai, L.V.; Lewis, C.A.; Chan, S.H.; Gui, D.Y.; Kunchok, T.; Dennstedt, E.A.; Vander Heiden, M.G.; Muir, A. Quantification of Microenvironmental Metabolites in Murine Cancers Reveals Determinants of Tumor Nutrient Availability. eLife 2019, 8, e44235. [Google Scholar] [CrossRef]
- Fontana, L.; Meyer, T.E.; Klein, S.; Holloszy, J.O. Long-Term Calorie Restriction Is Highly Effective in Reducing the Risk for Atherosclerosis in Humans. Proc. Natl. Acad. Sci. USA 2004, 101, 6659–6663. [Google Scholar] [CrossRef] [Green Version]
- Lv, M.; Zhu, X.; Wang, H.; Wang, F.; Guan, W. Roles of Caloric Restriction, Ketogenic Diet and Intermittent Fasting during Initiation, Progression and Metastasis of Cancer in Animal Models: A Systematic Review and Meta-Analysis. PLoS ONE 2014, 9, e115147. [Google Scholar] [CrossRef]
- Stafford, P.; Abdelwahab, M.G.; Kim, D.Y.; Preul, M.C.; Rho, J.M.; Scheck, A.C. The Ketogenic Diet Reverses Gene Expression Patterns and Reduces Reactive Oxygen Species Levels When Used as an Adjuvant Therapy for Glioma. Nutr. Metab. 2010, 7, 74. [Google Scholar] [CrossRef] [Green Version]
- Otto, C.; Kaemmerer, U.; Illert, B.; Muehling, B.; Pfetzer, N.; Wittig, R.; Voelker, H.U.; Thiede, A.; Coy, J.F. Growth of Human Gastric Cancer Cells in Nude Mice Is Delayed by a Ketogenic Diet Supplemented with Omega-3 Fatty Acids and Medium-Chain Triglycerides. BMC Cancer 2008, 8, 2061–2068. [Google Scholar] [CrossRef] [Green Version]
- Mavropoulos, J.C.; Buschemeyer, W.C.; Tewari, A.K.; Rokhfeld, D.; Pollak, M.; Zhao, Y.; Febbo, P.G.; Cohen, P.; Hwang, D.; Devi, G.; et al. The Effects of Varying Dietary Carbohydrate and Fat Content on Survival in a Murine LNCaP Prostate Cancer Xenograft Model. Cancer Prev. Res. 2009, 2, 557–565. [Google Scholar] [CrossRef] [Green Version]
- Martuscello, R.T.; Vedam-Mai, V.; McCarthy, D.J.; Schmoll, M.E.; Jundi, M.A.; Louviere, C.D.; Griffith, B.G.; Skinner, C.L.; Suslov, O.; Deleyrolle, L.P.; et al. A Supplemented High-Fat Low-Carbohydrate Diet for the Treatment of Glioblastoma. Clin. Cancer Res. 2016, 22, 2482–2495. [Google Scholar] [CrossRef] [Green Version]
- Seyfried, T.N.; Sanderson, T.M.; El-Abbadi, M.M.; McGowan, R.; Mukherjee, P. Role of Glucose and Ketone Bodies in the Metabolic Control of Experimental Brain Cancer. Br. J. Cancer 2003, 89, 1375–1382. [Google Scholar] [CrossRef] [Green Version]
- Lussier, D.M.; Woolf, E.C.; Johnson, J.L.; Brooks, K.S.; Blattman, J.N.; Scheck, A.C. Enhanced Immunity in a Mouse Model of Malignant Glioma Is Mediated by a Therapeutic Ketogenic Diet. BMC Cancer 2016, 16, 310. [Google Scholar] [CrossRef]
- Regulation of Kynurenine Metabolism by a Ketogenic Diet. J. Lipid Res. 2018, 59, 958–966. [CrossRef] [PubMed] [Green Version]
- Orillion, A.; Damayanti, N.P.; Shen, L.; Adelaiye-Ogala, R.; Affronti, H.; Elbanna, M.; Chintala, S.; Ciesielski, M.; Fontana, L.; Kao, C.; et al. Dietary Protein Restriction Reprograms Tumor-Associated Macrophages and Enhances Immunotherapy. Clin. Cancer Res. 2018, 24, 6383–6395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio-Patiño, C.; Bossowski, J.P.; De Donatis, G.M.; Mondragón, L.; Villa, E.; Aira, L.E.; Chiche, J.; Mhaidly, R.; Lebeaupin, C.; Marchetti, S.; et al. Low-Protein Diet Induces IRE1α-Dependent Anticancer Immunosurveillance. Cell Metab. 2018, 27, 828–842.e7. [Google Scholar] [CrossRef] [Green Version]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer Immunotherapy by CTLA-4 Blockade Relies on the Gut Microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Routy, B.; Chatelier, E.L.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut Microbiome Influences Efficacy of PD-1–Based Immunotherapy against Epithelial Tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Fu, L.; Li, Y.; Wang, W.; Gong, M.; Zhang, J.; Dong, X.; Huang, J.; Wang, Q.; Mackay, C.R.; et al. Gut Microbial Metabolites Facilitate Anticancer Therapy Efficacy by Modulating Cytotoxic CD8+ T Cell Immunity. Cell Metab. 2021, 33, 988–1000.e7. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut Microbiome Modulates Response to Anti–PD-1 Immunotherapy in Melanoma Patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Conlon, K.C.; Miljkovic, M.D.; Waldmann, T.A. Cytokines in the Treatment of Cancer. J. Interferon Cytokine Res. 2019, 39, 6–21. [Google Scholar] [CrossRef]
- Perica, K.; Varela, J.C.; Oelke, M.; Schneck, J. Adoptive T Cell Immunotherapy for Cancer. Rambam Maimonides Med. J. 2015, 6, e0004. [Google Scholar] [CrossRef]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic Cancer Vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Metabolic Drug/Strategy | Cellular Effect | Immune Outcome |
|---|---|---|---|
| PI3K | GSK2636771 BMK120 | Reduction of cancer cell glycolysis | Increase of lymphocyte infiltration and CTL cytotoxicity |
| Lactate | AZD3975 GSK2837808A 1-4-benzene | Decrease of lactate in the TME | Alteration of Treg activity and restoration of effector T cell functions |
| AMPK | metformin | Decrease of anabolic pathway activity | Increase of CD8+ infiltration and memory T cell differentiation |
| Tryptophan | PEGylated kynureninase | Degradation of kynurenine | Inhibition of Treg expansion |
| Glutamine | JHU083 CB-839 | Reduction of cancer cell glutaminolysis | Increase of effector T cell proliferation |
| Hypoxia | hyperoxia | Decrease of intratumor hypoxia | Increase of lymphocyte infiltration |
| Adenosine | ciforadenant AZD4635 AB928 MEDI9447 POM-1 ATP switch antibody | Reduction of cAMP levels in the TME | Enhanced cytotoxicity of antitumor cells |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luby, A.; Alves-Guerra, M.-C. Targeting Metabolism to Control Immune Responses in Cancer and Improve Checkpoint Blockade Immunotherapy. Cancers 2021, 13, 5912. https://doi.org/10.3390/cancers13235912
Luby A, Alves-Guerra M-C. Targeting Metabolism to Control Immune Responses in Cancer and Improve Checkpoint Blockade Immunotherapy. Cancers. 2021; 13(23):5912. https://doi.org/10.3390/cancers13235912
Chicago/Turabian StyleLuby, Angèle, and Marie-Clotilde Alves-Guerra. 2021. "Targeting Metabolism to Control Immune Responses in Cancer and Improve Checkpoint Blockade Immunotherapy" Cancers 13, no. 23: 5912. https://doi.org/10.3390/cancers13235912
APA StyleLuby, A., & Alves-Guerra, M.-C. (2021). Targeting Metabolism to Control Immune Responses in Cancer and Improve Checkpoint Blockade Immunotherapy. Cancers, 13(23), 5912. https://doi.org/10.3390/cancers13235912