
CAR T Cell Therapy’s Potential for Pediatric Brain Tumors


Abstract
:Simple Summary
Abstract

1. Introduction
2. Pediatric Brain Tumor Heterogeneity
2.1. Heterogeneity of Pediatric Brain Tumors
2.2. CAR Targets
2.2.1. Antigen Prerequisite for CNS Tumor CAR T Cell Targeting
2.2.2. Associated CAR Target for Pediatric CNS Tumors
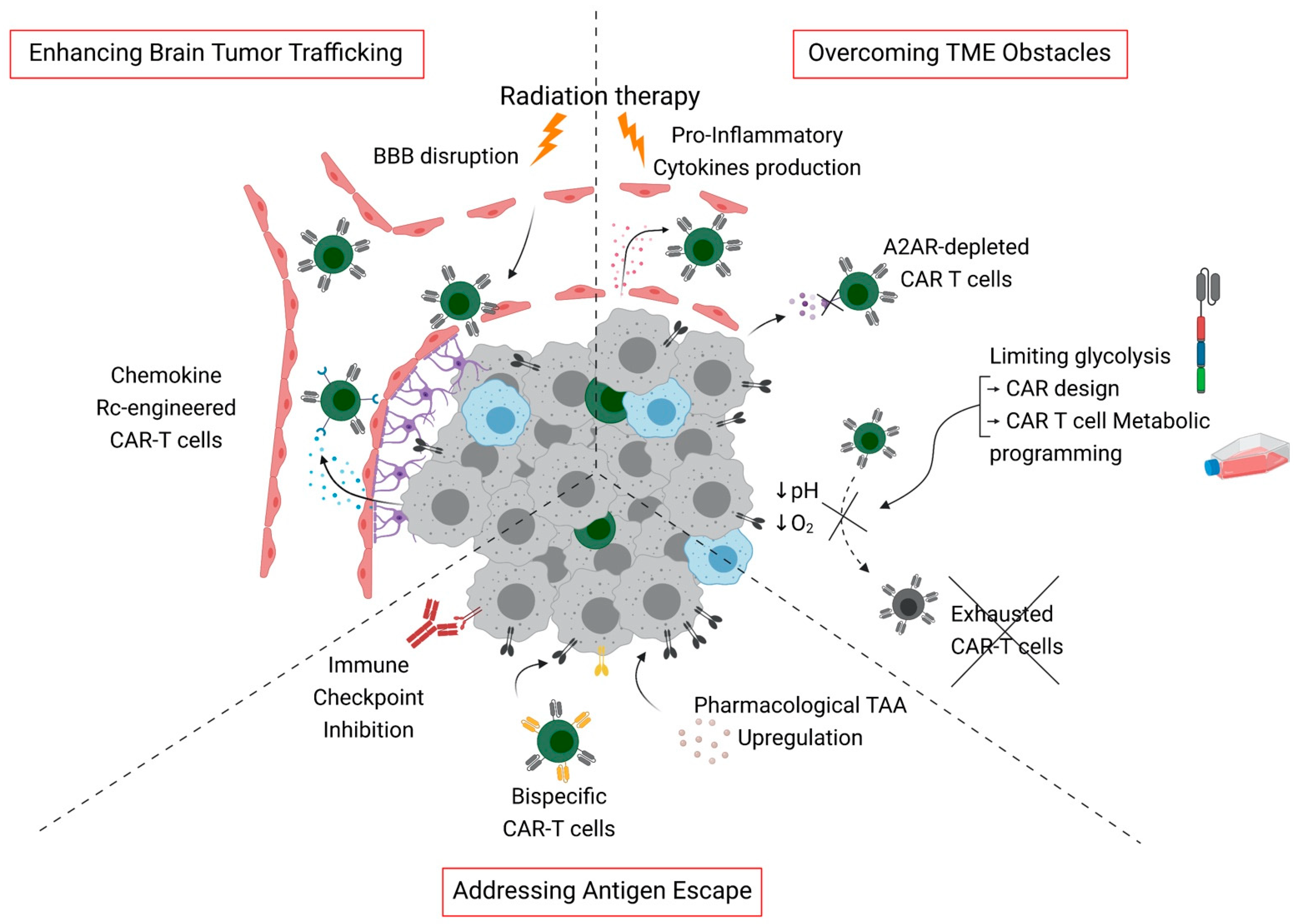
2.2.3. Addressing Antigen Escape
3. CAR T Cell Homing in the Brain Tumor Site
3.1. Lymphocytes’ Access to the Brain
3.2. Route of Administration for CAR T Cells for Brain Tumor Therapy
3.3. Strategies for Enhanced Brain Tumor Trafficking
4. The Antagonistic Pediatric Brain Tumor Microenvironment
4.1. Subgroup-Specific Immune Microenvironment in Pediatric Brain Tumors
4.2. Metabolic Barriers
4.3. Tumor-Derived Immunosuppressive Factors
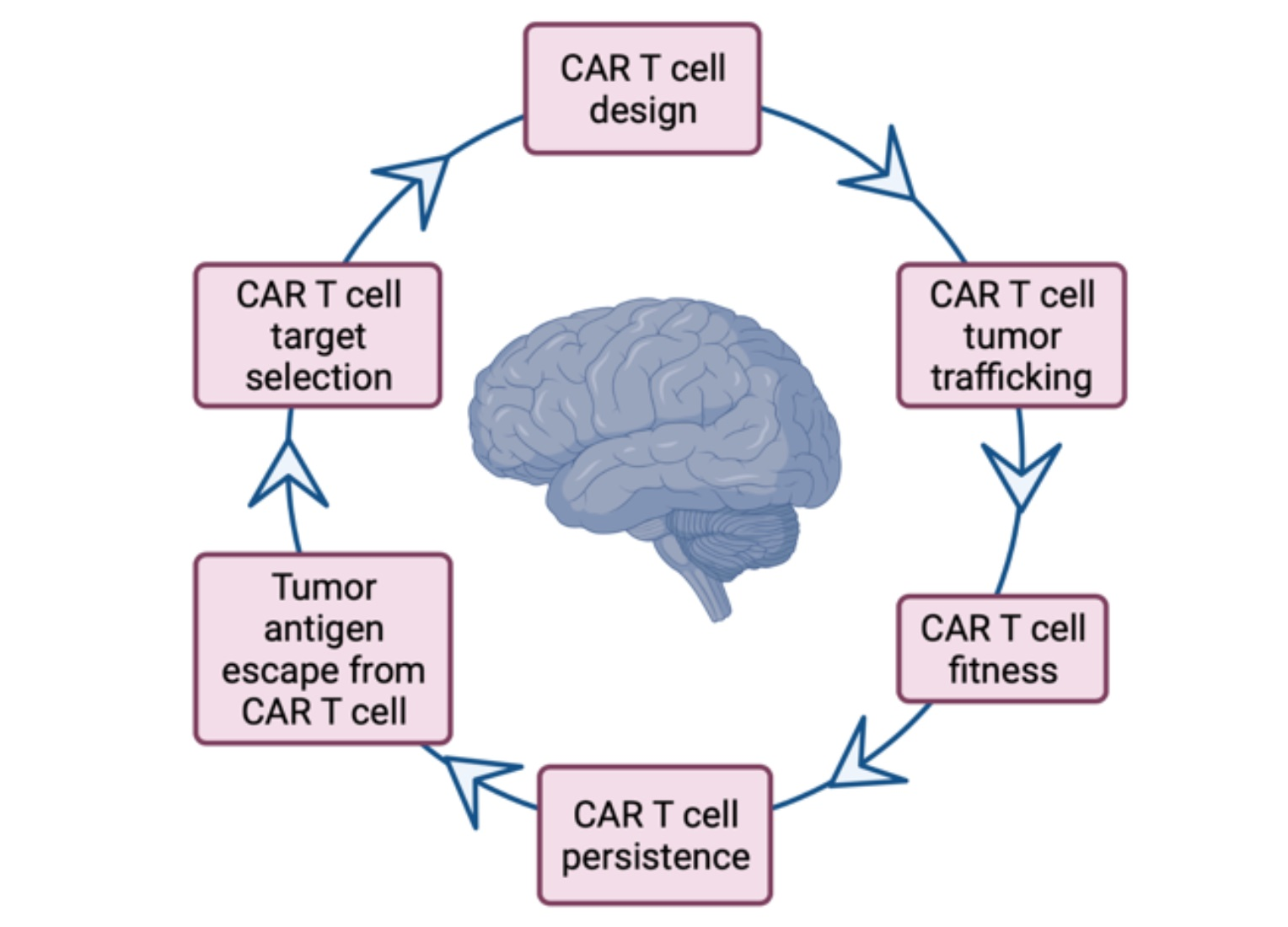
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patel, R.R.; Ramkissoon, S.H.; Ross, J.; Weintraub, L. Tumor mutational burden and driver mutations: Characterizing the genomic landscape of pediatric brain tumors. Pediatr. Blood Cancer 2020, 67, e28338. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouladi, M.; Stewart, C.F.; Blaney, S.M.; Onar-Thomas, A.; Schaiquevich, P.; Packer, R.J.; Goldman, S.; Geyer, J.R.; Gajjar, A.; Kun, L.E.; et al. A molecular biology and phase II trial of lapatinib in children with refractory CNS malignancies: A pediatric brain tumor consortium study. J. Neuro-Oncol. 2013, 114, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Packer, R.J.; Zhou, T.; Holmes, E.; Vezina, G.; Gajjar, A. Survival and secondary tumors in children with medulloblastoma receiving radiotherapy and adjuvant chemotherapy: Results of Children’s Oncology Group trial A9961. Neuro-Oncology 2012, 15, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Gunn, M.E.; Lähdesmäki, T.; Malila, N.; Arola, M.; Grönroos, M.; Matomäki, J.; Lähteenmäki, P.M. Late morbidity in long-term survivors of childhood brain tumors: A nationwide registry-based study in Finland. Neuro-Oncology 2014, 17, 747–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, S.M.; Fei, W.; Mitra, N.; Shinohara, E.T. Late causes of death in children treated for CNS malignancies. J. Neuro-Oncol. 2013, 115, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Pule, M.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ /CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Savoldo, B.; Ramos, C.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor–modified T cells in lymphoma patients. J. Clin. Investig. 2011, 121, 1822–1826. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2018, 20, 31–42. [Google Scholar] [CrossRef]
- Mian, A.; Hill, B.T. Brexucabtagene autoleucel for the treatment of relapsed/refractory mantle cell lymphoma. Expert Opin. Biol. Ther. 2021, 21, 435–441. [Google Scholar] [CrossRef]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2018, 25, 625–638. [Google Scholar] [CrossRef] [Green Version]
- Parker, K.R.; Migliorini, D.; Perkey, E.; Yost, K.E.; Bhaduri, A.; Bagga, P.; Haris, M.; Wilson, N.E.; Liu, F.; Gabunia, K.; et al. Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell 2020, 183, 126–142.e17. [Google Scholar] [CrossRef] [PubMed]
- Schaft, N. The Landscape of CAR-T Cell Clinical Trials against Solid Tumors—A Comprehensive Overview. Cancers 2020, 12, 2567. [Google Scholar] [CrossRef]
- Schmidts, A.; Maus, M.V. Making CAR T Cells a Solid Option for Solid Tumors. Front. Immunol. 2018, 9, 2593. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Clubb, J.D.; Chen, Y.Y. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell 2020, 38, 473–488. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Wrensch, M.; Minn, Y.; Chew, T.; Bondy, M.; Berger, M.S. Epidemiology of primary brain tumors: Current concepts and review of the literature. Neuro-Oncology 2002, 4, 278–299. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.T.W.; Kieran, M.W.; Bouffet, E.; Alexandrescu, S.; Bandopadhayay, P.; Bornhorst, M.; Ellison, D.; Fangusaro, J.; Fisher, M.J.; Foreman, N.; et al. Pediatric low-grade gliomas: Next biologically driven steps. Neuro-Oncology 2017, 20, 160–173. [Google Scholar] [CrossRef]
- Merchant, T.E.; Pollack, I.F.; Loeffler, J.S. Brain Tumors Across the Age Spectrum: Biology, Therapy, and Late Effects. Semin. Radiat. Oncol. 2010, 20, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Sturm, D.; Bender, S.; Jones, D.T.W.; Lichter, P.; Grill, J.; Becher, O.; Hawkins, C.; Majewski, J.; Jones, C.; Costello, J.F.; et al. Paediatric and adult glioblastoma: Multiform (epi)genomic culprits emerge. Nat. Rev. Cancer 2014, 14, 92–107. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Abou-Antoun, T.; Hale, J.S.; Lathia, J.D.; Dombrowski, S.M. Brain Cancer Stem Cells in Adults and Children: Cell Biology and Therapeutic Implications. Neurotherapeutics 2017, 14, 372–384. [Google Scholar] [CrossRef]
- Hussein, D.; Punjaruk, W.; Storer, L.C.; Shaw, L.; Othman, R.T.; Peet, A.; Miller, S.; Bandopadhyay, G.; Heath, R.; Kumari, R.; et al. Pediatric brain tumor cancer stem cells: Cell cycle dynamics, DNA repair, and etoposide extrusion. Neuro-Oncology 2010, 13, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Petralia, F.; Tignor, N.; Reva, B.; Koptyra, M.; Chowdhury, S.; Rykunov, D.; Krek, A.; Ma, W.; Zhu, Y.; Ji, J.; et al. Integrated Proteogenomic Characterization across Major Histological Types of Pediatric Brain Cancer. Cell 2020, 183, 1962–1985.e31. [Google Scholar] [CrossRef]
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.; Hargrave, D.; Holland, E.C.; et al. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019, 16, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Ris, M.D.; Packer, R.; Goldwein, J.; Jones-Wallace, D.; Boyett, J.M. Intellectual Outcome after Reduced-Dose Radiation Therapy Plus Adjuvant Chemotherapy for Medulloblastoma: A Children’s Cancer Group Study. J. Clin. Oncol. 2001, 19, 3470–3476. [Google Scholar] [CrossRef] [PubMed]
- Silber, J.H.; Radcliffe, J.; Peckham, V.; Perilongo, G.; Kishnani, P.; Fridman, M.; Goldwein, J.W.; Meadows, A.T. Whole-brain irradiation and decline in intelligence: The influence of dose and age on IQ score. J. Clin. Oncol. 1992, 10, 1390–1396. [Google Scholar] [CrossRef]
- Grimm, S.A.; Chamberlain, M.C. Brainstem Glioma: A Review. Curr. Neurol. Neurosci. Rep. 2013, 13, 346. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, N.A.P.; Vitanza, N.A.; Crane, C.A. Immunotherapy for brain tumors: Understanding early successes and limitations. Expert Rev. Neurother. 2018, 18, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Gallitto, M.; Lazarev, S.; Wasserman, I.; Stafford, J.M.; Wolden, S.L.; Terezakis, S.A.; Bindra, R.S.; Bakst, R.L. Role of Radiation Therapy in the Management of Diffuse Intrinsic Pontine Glioma: A Systematic Review. Adv. Radiat. Oncol. 2019, 4, 520–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Corrigendum: Estimation of clinical trial success rates and related parameters. Biostatistics 2018, 20, 366. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Mount, C.; Majzner, R.G.; Sundaresh, S.; Arnold, E.P.; Kadapakkam, M.; Haile, S.; Labanieh, L.; Hulleman, E.; Woo, P.J.; Rietberg, S.P.; et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas. Nat. Med. 2018, 24, 572–579. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Theruvath, J.; Sotillo, E.; Mount, C.W.; Graef, C.M.; Delaidelli, A.; Heitzeneder, S.; Labanieh, L.; Dhingra, S.; Leruste, A.; Majzner, R.G.; et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat. Med. 2020, 26, 712–719. [Google Scholar] [CrossRef]
- Haydar, D.; Houke, H.; Chiang, J.; Yi, Z.; Odé, Z.; Caldwell, K.; Zhu, X.; Mercer, K.S.; Stripay, J.L.; Shaw, T.I.; et al. Cell-surface antigen profiling of pediatric brain tumors: B7-H3 is consistently expressed and can be targeted via local or systemic CAR T-cell delivery. Neuro-Oncology 2020, 23, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef] [PubMed]
- Richman, S.A.; Nunez-Cruz, S.; Moghimi, B.; Li, L.; Gershenson, Z.T.; Mourelatos, Z.; Barrett, D.M.; Grupp, S.A.; Milone, M.C. High-Affinity GD2-Specific CAR T Cells Induce Fatal Encephalitis in a Preclinical Neuroblastoma Model. Cancer Immunol. Res. 2017, 6, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Weber, E.W.; Lynn, R.C.; Xu, P.; Mackall, C.L. Neurotoxicity Associated with a High-Affinity GD2 CAR—Letter. Cancer Immunol. Res. 2018, 6, 494–495. [Google Scholar] [CrossRef] [Green Version]
- Moghimi, B.; Muthugounder, S.; Jambon, S.; Tibbetts, R.; Hung, L.; Bassiri, H.; Hogarty, M.D.; Barrett, D.M.; Shimada, H.; Asgharzadeh, S. Preclinical assessment of the efficacy and specificity of GD2-B7H3 SynNotch CAR-T in metastatic neuroblastoma. Nat. Commun. 2021, 12, 511. [Google Scholar] [CrossRef]
- Lammie, G.; Cheung, N.; Gerald, W.; Rosenblum, M.; Cordoncardo, C. Ganglioside GD(2) expression in the human nervous-system and in neuroblastomas-An immunohistochemical study. Int. J. Oncol. 1993, 3, 909–915. [Google Scholar] [CrossRef]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 Antibody with GM-CSF, Interleukin-2, and Isotretinoin for Neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nellan, A.; Rota, C.; Majzner, R.; Lester-McCully, C.M.; Griesinger, A.M.; Levy, J.M.M.; Foreman, N.K.; Warren, K.E.; Lee, D.W. Durable regression of Medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J. Immunother. Cancer 2018, 6, 30. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced with a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Seidman, A.; Hudis, C.; Pierri, M.K.; Shak, S.; Paton, V.; Ashby, M.; Murphy, M.; Stewart, S.J.; Keefe, D. Cardiac Dysfunction in the Trastuzumab Clinical Trials Experience. J. Clin. Oncol. 2002, 20, 1215–1221. [Google Scholar] [CrossRef]
- Donovan, L.K.; Delaidelli, A.; Joseph, S.K.; Bielamowicz, K.; Fousek, K.; Holgado, B.L.; Manno, A.; Srikanthan, D.; Gad, A.Z.; Van Ommeren, R.; et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat. Med. 2020, 26, 720–731. [Google Scholar] [CrossRef]
- Wykosky, J.; Gibo, D.M.; Stanton, C.; Debinski, W. EphA2 as a Novel Molecular Marker and Target in Glioblastoma Multiforme. Mol. Cancer Res. 2005, 3, 541–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor–Modified Virus-Specific T Cells for Progressive Glioblastoma. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.; et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [Green Version]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.-F.; Orange, J.S.; Sumazin, P.; Man, T.-K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncology 2017, 20, 506–518. [Google Scholar] [CrossRef]
- Kailayangiri, S.; Altvater, B.; Lesch, S.; Balbach, S.; Göttlich, C.; Kühnemundt, J.; Mikesch, J.-H.; Schelhaas, S.; Jamitzky, S.; Meltzer, J.; et al. EZH2 Inhibition in Ewing Sarcoma Upregulates GD2 Expression for Targeting with Gene-Modified T Cells. Mol. Ther. 2019, 27, 933–946. [Google Scholar] [CrossRef]
- Van den Bijgaart, R.J.E.; Kroesen, M.; Brok, I.C.; Reijnen, D.; Wassink, M.; Boon, L.; Hoogerbrugge, P.M.; Adema, G.J. Anti-GD2 antibody and Vorinostat immunocombination therapy is highly effective in an aggressive orthotopic neuroblastoma model. OncoImmunology 2020, 9, 1817653. [Google Scholar] [CrossRef]
- Bijgaart, R.J.V.D.; Kroesen, M.; Wassink, M.; Brok, I.C.; Kers-Rebel, E.D.; Boon, L.; Heise, T.; van Scherpenzeel, M.; Lefeber, D.J.; Boltje, T.J.; et al. Combined sialic acid and histone deacetylase (HDAC) inhibitor treatment up-regulates the neuroblastoma antigen GD2. J. Biol. Chem. 2019, 294, 4437–4449. [Google Scholar] [CrossRef] [PubMed]
- Banik, D.; Moufarrij, S.; Villagra, A. Immunoepigenetics Combination Therapies: An Overview of the Role of HDACs in Cancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 2241. [Google Scholar] [CrossRef] [Green Version]
- Hong, L.K.; Chen, Y.; Smith, C.C.; Montgomery, S.A.; Vincent, B.G.; Dotti, G.; Savoldo, B. CD30-Redirected Chimeric Antigen Receptor T Cells Target CD30+ and CD30− Embryonal Carcinoma via Antigen-Dependent and Fas/FasL Interactions. Cancer Immunol. Res. 2018, 6, 1274–1287. [Google Scholar] [CrossRef] [Green Version]
- DeSelm, C.; Palomba, M.L.; Yahalom, J.; Hamieh, M.; Eyquem, J.; Rajasekhar, V.K.; Sadelain, M. Low-Dose Radiation Conditioning Enables CAR T Cells to Mitigate Antigen Escape. Mol. Ther. 2018, 26, 2542–2552. [Google Scholar] [CrossRef] [Green Version]
- Chuntova, P.; Downey, K.; Hegde, B.; Almeida, N.D.; Okada, H. Genetically Engineered T-Cells for Malignant Glioma: Overcoming the Barriers to Effective Immunotherapy. Front. Immunol. 2019, 9, 3062. [Google Scholar] [CrossRef] [Green Version]
- Wekerle, H.; Schwab, M.; Linington, C.; Meyermann, R. Antigen presentation in the peripheral nervous system: Schwann cells present endogenous myelin autoantigens to lymphocytes. Eur. J. Immunol. 1986, 16, 1551–1557. [Google Scholar] [CrossRef]
- Hickey, W.F. Migration of Hematogenous Cells through the Blood-Brain Barrier and the Initiation of CNS Inflammation. Brain Pathol. 1991, 1, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.V.; McGavern, D.B. Immune Surveillance of the CNS following Infection and Injury. Trends Immunol. 2015, 36, 637–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelhardt, B.; Ransohoff, R.M. Capture, crawl, cross: The T cell code to breach the blood-brain barriers. Trends Immunol. 2012, 33, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Krakowski, M.L.; Owens, T. The central nervous system environment controls effector CD4+ T cell cytokine profile in experimental allergic encephalomyelitis. Eur. J. Immunol. 1997, 27, 2840–2847. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Wolburg-Buchholz, K.; Wolburg, H. Involvement of the Choroid Plexus in Central Nervous System Inflammation. Microsc. Res. Tech. 2001, 52, 112–129. [Google Scholar] [CrossRef]
- Reboldi, A.; Coisne, C.; Baumjohann, D.; Benvenuto, F.; Bottinelli, D.; Lira, S.A.; Uccelli, A.; Lanzavecchia, A.; Engelhardt, B.; Sallusto, F. C-C chemokine receptor 6–regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol. 2009, 10, 514–523. [Google Scholar] [CrossRef]
- Fisher, Y.; Strominger, I.; Biton, S.; Nemirovsky, A.; Baron, R.; Monsonego, A. Th1 Polarization of T Cells Injected into the Cerebrospinal Fluid Induces Brain Immunosurveillance. J. Immunol. 2013, 192, 92–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.; Laramy, J.K.; et al. Is the blood–brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology 2017, 20, 184–191. [Google Scholar] [CrossRef]
- Mainprize, T.; Lipsman, N.; Huang, Y.; Meng, Y.; Bethune, A.; Ironside, S.; Heyn, C.; Alkins, R.; Trudeau, M.; Sahgal, A.; et al. Blood-Brain Barrier Opening in Primary Brain Tumors with Non-invasive MR-Guided Focused Ultrasound: A Clinical Safety and Feasibility Study. Sci. Rep. 2019, 9, 321. [Google Scholar] [CrossRef] [Green Version]
- Petrecca, K.; Guiot, M.-C.; Panet-Raymond, V.; Souhami, L. Failure pattern following complete resection plus radiotherapy and temozolomide is at the resection margin in patients with glioblastoma. J. Neuro-Oncol. 2012, 111, 19–23. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Wicha, M.S.; Zou, N.N.M.S.W.W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newick, K.; Moon, E.; Albelda, S.M. Chimeric antigen receptor T-cell therapy for solid tumors. Mol. Ther.-Oncolytics 2016, 3, 16006. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.W.; Kim, Y.H.; Oh, S.Y.; Suh, K.W.; Lee, G.; Yoon, J.E.; Park, S.S.; Lee, Y.; Park, Y.J.; Kim, H.S.; et al. Senescent Tumor Cells Build a Cytokine Shield in Colorectal Cancer. Adv. Sci. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.T.; Sun, W.; Hussain, S.F.; DeAngulo, G.; Prabhu, S.S.; Heimberger, A.B. Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol. Immunother. 2007, 57, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, A.; De Angelis, B.; Rooney, C.M.; Zhang, L.; Mahendravada, A.; Foster, A.E.; Heslop, H.E.; Brenner, M.K.; Dotti, G.; Savoldo, B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 2009, 113, 6392–6402. [Google Scholar] [CrossRef] [Green Version]
- Sandgren, J.; Holm, S.; Marino, A.M.; Asmundsson, J.; Grillner, P.; Nistér, M.; de Ståhl, T.D. Whole Exome- and mRNA-Sequencing of an AT/RT Case Reveals Few Somatic Mutations and Several Deregulated Signalling Pathways in the Context ofSMARCB1Deficiency. BioMed Res. Int. 2015, 2015, 862039. [Google Scholar] [CrossRef] [Green Version]
- Schüller, U.; Koch, A.; Hartmann, W.; Garrè, M.L.; Goodyer, C.G.; Cama, A.; Sörensen, N.; Wiestler, O.D.; Pietsch, T. Subtype-specific expression and genetic alterations of the chemokinereceptor geneCXCR4 in medulloblastomas. Int. J. Cancer 2005, 117, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Dono, A.; Zhu, P.; Zorofchian, S.; Takayasu, T.; Quezado, M.M.; Rios, A.; Powers, A.; Esquenazi, Y.; Ballester, L.Y. Variable expression of CXCR4 in molecular subtypes of infiltrating gliomas. Clin. Neuropathol. 2021, 40, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Golan, H.; Shukrun, R.; Caspi, R.; Vax, E.; Pode-Shakked, N.; Goldberg, S.; Pleniceanu, O.; Bar-Lev, D.D.; Mark-Danieli, M.; Pri-Chen, S.; et al. In Vivo Expansion of Cancer Stemness Affords Novel Cancer Stem Cell Targets: Malignant Rhabdoid Tumor as an Example. Stem Cell Rep. 2018, 11, 795–810. [Google Scholar] [CrossRef] [Green Version]
- Murty, S.; Haile, S.T.; Beinat, C.; Aalipour, A.; Alam, I.S.; Murty, T.; Shaffer, T.M.; Patel, C.B.; Graves, E.E.; Mackall, C.L.; et al. Intravital imaging reveals synergistic effect of CAR T-cells and radiation therapy in a preclinical immunocompetent glioblastoma model. OncoImmunology 2020, 9, 1757360. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, X.; Gao, L.; Wang, Y.; Guo, Y.; Xing, B.; Ma, W. Classification of pediatric gliomas based on immunological profiling: Implications for immunotherapy strategies. Mol. Ther.-Oncolytics 2020, 20, 34–47. [Google Scholar] [CrossRef]
- Griesinger, A.M.; Birks, D.K.; Donson, A.M.; Amani, V.; Hoffman, L.M.; Waziri, A.; Wang, M.; Handler, M.H.; Foreman, N. Characterization of Distinct Immunophenotypes across Pediatric Brain Tumor Types. J. Immunol. 2013, 191, 4880–4888. [Google Scholar] [CrossRef] [Green Version]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Zi, Z.; Jin, Y.; Li, G.; Shao, K.; Cai, Q.; Ma, X.; Wei, F. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol. Immunother. 2018, 68, 365–377. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; Van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Bockmayr, M.; Mohme, M.; Klauschen, F.; Winkler, B.; Budczies, J.; Rutkowski, S.; Schüller, U. Subgroup-specific immune and stromal microenvironment in medulloblastoma. OncoImmunology 2018, 7, e1462430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Z.L.; Lorenzini, M.H.; Chen, X.; Tran, U.; Bangayan, N.J.; Chen, Y.Y. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat. Chem. Biol. 2018, 14, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R.; et al. Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov. 2017, 7, 1238–1247. [Google Scholar] [CrossRef] [Green Version]
- Witt, D.A.; Donson, A.M.; Amani, V.; Moreira, D.C.; Sanford, B.; Hoffman, L.M.; Handler, M.H.; Levy, J.M.M.; Jones, K.L.; Nellan, A.; et al. Specific expression of PD-L1 in RELA-fusion supratentorial ependymoma: Implications for PD-1-targeted therapy. Pediatr. Blood Cancer 2018, 65, e26960. [Google Scholar] [CrossRef]
- Pavlova, N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Altman, B.; Stine, Z.E.; Dang, C. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Zhang, L.; Romero, P. Metabolic Control of CD8+ T Cell Fate Decisions and Antitumor Immunity. Trends Mol. Med. 2018, 24, 30–48. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Pearce, E.L. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat. Immunol. 2016, 17, 364–368. [Google Scholar] [CrossRef] [Green Version]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.; Muranski, P.; Karoly, E.D.; et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Investig. 2013, 123, 4479–4488. [Google Scholar] [CrossRef]
- Zhao, Z.; Condomines, M.; van der Stegen, S.J.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Nabe, S.; Yamada, T.; Suzuki, J.; Toriyama, K.; Yasuoka, T.; Kuwahara, M.; Shiraishi, A.; Takenaka, K.; Yasukawa, M.; Yamashita, M. Reinforce the antitumor activity of CD 8 + T cells via glutamine restriction. Cancer Sci. 2018, 109, 3737–3750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crompton, J.G.; Sukumar, M.; Roychoudhuri, R.; Clever, D.; Gros, A.; Eil, R.L.; Tran, E.; Hanada, K.-I.; Yu, Z.; Palmer, D.; et al. Akt Inhibition Enhances Expansion of Potent Tumor-Specific Lymphocytes with Memory Cell Characteristics. Cancer Res. 2014, 75, 296–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilipow, K.; Scamardella, E.; Puccio, S.; Gautam, S.; De Paoli, F.; Mazza, E.M.C.; De Simone, G.; Polletti, S.; Buccilli, M.; Zanon, V.; et al. Antioxidant metabolism regulates CD8+ T memory stem cell formation and antitumor immunity. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Vijayan, D.; Young, A.; Teng, M.W.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Beavis, P.; Henderson, M.A.; Giuffrida, L.; Mills, J.K.; Sek, K.; Cross, R.S.; Davenport, A.J.; John, L.B.; Mardiana, S.; Slaney, C.Y.; et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J. Clin. Investig. 2017, 127, 929–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockwell, J.; Jakova, E.; Cayabyab, F.S. Adenosine A1 and A2A Receptors in the Brain: Current Research and Their Role in Neurodegeneration. Molecules 2017, 22, 676. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.-M.; Oh, M.-H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; Tam, A.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef]
- Cao, N.; Li, S.; Wang, Z.; Ahmed, K.M.; Degnan, M.E.; Fan, M.; Dynlacht, J.R.; Li, J.J. NF-κB-MediatedHER2Overexpression inRadiation-Adaptive Resistance. Radiat. Res. 2009, 171, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanpouille-Box, C.; Diamond, J.M.; Pilones, K.A.; Zavadil, J.; Babb, J.; Formenti, S.C.; Barcellos-Hoff, M.H.; DeMaria, S. TGFβ Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Res. 2015, 75, 2232–2242. [Google Scholar] [CrossRef] [Green Version]
- Kaur, P.; Asea, A. Radiation-induced effects and the immune system in cancer. Front. Oncol. 2012, 2, 191. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Gliomas, Grade 4 |
|---|
| Around 50% of pediatric tumors |
| Pediatric-type diffuse high-grade gliomas |
| Diffuse midline glioma, H3K27-altered |
| Diffuse hemispheric glioma, H3 G34 mutant |
| Diffuse pediatric-type high-grade glioma, H3 wildtype and IDH wildtype |
| Infant-type hemispheric glioma |
| Embryonal Tumors, Grade 4 |
| Around 20% of CNS pediatric tumors |
| Medulloblastomas, molecularly defined |
| Medulloblastoma, WNT-activated |
| Medulloblastoma, SHH-activated and TP53 wildtype |
| Medulloblastoma, SHH-activated and TP53 mutant |
| Medulloblastoma, non-WNT/non-SHH |
| Medulloblastomas, histologically defined |
| Atypical teratoid/rhabdoid tumor |
| Ependymal Tumors, Grade 2, 3 |
| Around 10% of CNS pediatric tumors |
| Supratentorial ependymoma |
| Supratentorial ependymoma, ZFTA-fusion-positive |
| Posterior fossa ependymoma |
| Posterior fossa ependymoma, group PFA |
| Posterior fossa ependymoma, group PFB |
| Spinal ependymoma |
| Spinal ependymoma, MYCN-amplified |
| Clinical Trial Identifier | Phase | Target | Delivery 2 | Disease | Age | Status |
|---|---|---|---|---|---|---|
| NCT03500991 | I | HER2 | IT, IC | ATRT, ependymoma, GBM, and medulloblastoma | One year to twenty-six years | Recruiting |
| NCT04185038 | I | B7H3 | IT, IC | ATRT, DMG, DIPG, and medulloblastoma | One year to twenty-six years | Recruiting |
| NCT04661384 | I | IL13Rα2 | ICV | Ependymoma, GBM, and medulloblastoma | Eighteen years and older | Recruiting |
| NCT04196413 | I | GD2 | IV | DMG H3K27M mutant DIPG H3K27M mutant | Two years to thirty years | Recruiting |
| NCT03638167 | I | EGFR | IT, IC | ATRT Ependymoma GBM Medulloblastoma | Fifteen years to twenty-six years | Recruiting |
| NCT04099797 | I | GD2-CR7 | IV | DIPG HGG | One year to eighteen years | Recruiting |
| NCT02208362 | I | IL13Rα2 | IT, IC | GBM | Twelve years to seventy-five years | Recruiting |
| NCT04510051 | I | IL13Ra2 | ICV | Brain neoplasms | Four years to twenty-five years | Recruiting |
| NCT04903080 | I | HER2 | IV | Refractory or recurrent ependymoma | One year to twenty-two years | Not yet recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, P.; Galopin, N.; Bonérandi, E.; Clémenceau, B.; Fougeray, S.; Birklé, S. CAR T Cell Therapy’s Potential for Pediatric Brain Tumors. Cancers 2021, 13, 5445. https://doi.org/10.3390/cancers13215445
Thomas P, Galopin N, Bonérandi E, Clémenceau B, Fougeray S, Birklé S. CAR T Cell Therapy’s Potential for Pediatric Brain Tumors. Cancers. 2021; 13(21):5445. https://doi.org/10.3390/cancers13215445
Chicago/Turabian StyleThomas, Pauline, Natacha Galopin, Emma Bonérandi, Béatrice Clémenceau, Sophie Fougeray, and Stéphane Birklé. 2021. "CAR T Cell Therapy’s Potential for Pediatric Brain Tumors" Cancers 13, no. 21: 5445. https://doi.org/10.3390/cancers13215445
APA StyleThomas, P., Galopin, N., Bonérandi, E., Clémenceau, B., Fougeray, S., & Birklé, S. (2021). CAR T Cell Therapy’s Potential for Pediatric Brain Tumors. Cancers, 13(21), 5445. https://doi.org/10.3390/cancers13215445