
Targeting Oncogenic KRAS in Non-Small-Cell Lung Cancer



Abstract
:Simple Summary
Abstract
1. Introduction
2. Genetic Alterations of KRAS in Human Cancers
3. Multiple Faces of KRAS-Mutated NSCLC
4. Oncogenic KRAS Regulates the Tumor Microenvironment (TME)
5. Covalent KRAS G12C Inhibitors for KRAS-Mutated NSCLC
6. Resistance Mechanisms for Anti-KRAS-G12C Therapies
7. Combined Therapies Involving Targeting of Oncogenic KRAS plus Other Targeted Drugs for KRAS-Mutated NSCLC
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Herbst, R.S.; Garon, E.B.; Kim, D.W.; Cho, B.C.; Gervais, R.; Perez-Gracia, J.L.; Han, J.Y.; Majem, M.; Forster, M.D.; Monnet, I.; et al. Five Year Survival Update From KEYNOTE-010: Pembrolizumab Versus Docetaxel for Previously Treated, Programmed Death-Ligand 1-Positive Advanced NSCLC. J. Thorac. Oncol. 2021, 16, 1718–1732. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, I.; Morita, S.; Tashiro, N.; Imamura, F.; Inoue, A.; Seto, T.; Yamamoto, N.; Ohe, Y.; Nakagawa, K.; Fukuoka, M. Real world treatment and outcomes in EGFR mutation-positive non-small cell lung cancer: Long-term follow-up of a large patient cohort. Lung Cancer 2018, 117, 14–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteban, L.M.; Vicario-Abejon, C.; Fernandez-Salguero, P.; Fernandez-Medarde, A.; Swaminathan, N.; Yienger, K.; Lopez, E.; Malumbres, M.; McKay, R.; Ward, J.M.; et al. Targeted genomic disruption of H-ras and N-ras, individually or in combination, reveals the dispensability of both loci for mouse growth and development. Mol. Cell Biol. 2001, 21, 1444–1452. [Google Scholar] [CrossRef] [Green Version]
- Koera, K.; Nakamura, K.; Nakao, K.; Miyoshi, J.; Toyoshima, K.; Hatta, T.; Otani, H.; Aiba, A.; Katsuki, M. K-ras is essential for the development of the mouse embryo. Oncogene 1997, 15, 1151–1159. [Google Scholar] [CrossRef] [Green Version]
- Potenza, N.; Vecchione, C.; Notte, A.; De Rienzo, A.; Rosica, A.; Bauer, L.; Affuso, A.; De Felice, M.; Russo, T.; Poulet, R.; et al. Replacement of K-Ras with H-Ras supports normal embryonic development despite inducing cardiovascular pathology in adult mice. EMBO Rep. 2005, 6, 432–437. [Google Scholar] [CrossRef] [Green Version]
- Dance, M.; Montagner, A.; Salles, J.P.; Yart, A.; Raynal, P. The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell Signal. 2008, 20, 453–459. [Google Scholar] [CrossRef]
- Ran, H.; Tsutsumi, R.; Araki, T.; Neel, B.G. Sticking It to Cancer with Molecular Glue for SHP2. Cancer Cell 2016, 30, 194–196. [Google Scholar] [CrossRef] [Green Version]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Suda, K.; Tomizawa, K.; Mitsudomi, T. Biological and clinical significance of KRAS mutations in lung cancer: An oncogenic driver that contrasts with EGFR mutation. Cancer Metastasis Rev. 2010, 29, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589. [Google Scholar] [CrossRef] [Green Version]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Koga, T.; Suda, K.; Fujino, T.; Ohara, S.; Hamada, A.; Nishino, M.; Chiba, M.; Shimoji, M.; Takemoto, T.; Arita, T.; et al. KRAS Secondary Mutations That Confer Acquired Resistance to KRAS G12C Inhibitors, Sotorasib and Adagrasib, and Overcoming Strategies: Insights From In Vitro Experiments. J. Thorac. Oncol. 2021, 16, 1321–1332. [Google Scholar] [CrossRef]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical Acquired Resistance to KRAS(G12C) Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS-MAPK Reactivation. Cancer Discov. 2021, 11, 1913–1922. [Google Scholar] [CrossRef]
- Addeo, A.; Banna, G.L.; Friedlaender, A. KRAS G12C Mutations in NSCLC: From Target to Resistance. Cancers 2021, 13, 2541. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uprety, D.; Adjei, A.A. KRAS: From undruggable to a druggable Cancer Target. Cancer Treat. Rev. 2020, 89, 102070. [Google Scholar] [CrossRef]
- AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.C.; Qiu, W.; Juang, C.S.; Mansukhani, M.M.; Halmos, B.; Su, G.H. Mutant allele specific imbalance in oncogenes with copy number alterations: Occurrence, mechanisms, and potential clinical implications. Cancer Lett. 2017, 384, 86–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soh, J.; Okumura, N.; Lockwood, W.W.; Yamamoto, H.; Shigematsu, H.; Zhang, W.; Chari, R.; Shames, D.S.; Tang, X.; MacAulay, C.; et al. Oncogene mutations, copy number gains and mutant allele specific imbalance (MASI) frequently occur together in tumor cells. PLoS ONE 2009, 4, e7464. [Google Scholar] [CrossRef] [PubMed]
- Chiosea, S.I.; Sherer, C.K.; Jelic, T.; Dacic, S. KRAS mutant allele-specific imbalance in lung adenocarcinoma. Mod. Pathol. 2011, 24, 1571–1577. [Google Scholar] [CrossRef] [Green Version]
- Villaruz, L.C.; Socinski, M.A.; Cunningham, D.E.; Chiosea, S.I.; Burns, T.F.; Siegfried, J.M.; Dacic, S. The prognostic and predictive value of KRAS oncogene substitutions in lung adenocarcinoma. Cancer 2013, 119, 2268–2274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, E.M.; Gaude, E.; Turrell, F.K.; Frezza, C.; Martins, C.P. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 2016, 531, 110–113. [Google Scholar] [CrossRef] [Green Version]
- Nakatani, K.; Yamaoka, T.; Ohba, M.; Fujita, K.I.; Arata, S.; Kusumoto, S.; Taki-Takemoto, I.; Kamei, D.; Iwai, S.; Tsurutani, J.; et al. KRAS and EGFR Amplifications Mediate Resistance to Rociletinib and Osimertinib in Acquired Afatinib-Resistant NSCLC Harboring Exon 19 Deletion/T790M in EGFR. Mol. Cancer Ther. 2019, 18, 112–126. [Google Scholar] [CrossRef] [Green Version]
- Ihle, N.T.; Byers, L.A.; Kim, E.S.; Saintigny, P.; Lee, J.J.; Blumenschein, G.R.; Tsao, A.; Liu, S.; Larsen, J.E.; Wang, J.; et al. Effect of KRAS oncogene substitutions on protein behavior: Implications for signaling and clinical outcome. J. Natl. Cancer Inst. 2012, 104, 228–239. [Google Scholar] [CrossRef]
- Judd, J.; Abdel Karim, N.; Khan, H.; Naqash, A.R.; Baca, Y.; Xiu, J.; VanderWalde, A.M.; Mamdani, H.; Raez, L.E.; Nagasaka, M.; et al. Characterization of KRAS Mutation Subtypes in Non-small Cell Lung Cancer. Mol. Cancer Ther. 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Nadal, E.; Chen, G.; Prensner, J.R.; Shiratsuchi, H.; Sam, C.; Zhao, L.; Kalemkerian, G.P.; Brenner, D.; Lin, J.; Reddy, R.M.; et al. KRAS-G12C mutation is associated with poor outcome in surgically resected lung adenocarcinoma. J. Thorac. Oncol. 2014, 9, 1513–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.A.; Sima, C.S.; Shen, R.; Kass, S.; Gainor, J.; Shaw, A.; Hames, M.; Iams, W.; Aston, J.; Lovly, C.M.; et al. Prognostic impact of KRAS mutation subtypes in 677 patients with metastatic lung adenocarcinomas. J. Thorac. Oncol. 2015, 10, 431–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheffler, M.; Ihle, M.A.; Hein, R.; Merkelbach-Bruse, S.; Scheel, A.H.; Siemanowski, J.; Bragelmann, J.; Kron, A.; Abedpour, N.; Ueckeroth, F.; et al. K-ras Mutation Subtypes in NSCLC and Associated Co-occuring Mutations in Other Oncogenic Pathways. J. Thorac. Oncol. 2019, 14, 606–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, W.J.; Daemen, A.; Cheng, J.H.; Long, J.E.; Cooper, J.E.; Wang, B.E.; Tran, C.; Singh, M.; Gnad, F.; Modrusan, Z.; et al. Kras mutant genetically engineered mouse models of human cancers are genomically heterogeneous. Proc. Natl. Acad. Sci. USA 2017, 114, E10947–E10955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordiak, J.; Szemraj, J.; Grabska-Kobylecka, I.; Bialasiewicz, P.; Braun, M.; Kordek, R.; Nowak, D. Intratumor heterogeneity and tissue distribution of KRAS mutation in non-small cell lung cancer: Implications for detection of mutated KRAS oncogene in exhaled breath condensate. J. Cancer Res. Clin. Oncol. 2019, 145, 241–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Motohashi, H. NRF2 addiction in cancer cells. Cancer Sci. 2018, 109, 900–911. [Google Scholar] [CrossRef] [Green Version]
- Best, S.A.; Ding, S.; Kersbergen, A.; Dong, X.; Song, J.Y.; Xie, Y.; Reljic, B.; Li, K.; Vince, J.E.; Rathi, V.; et al. Distinct initiating events underpin the immune and metabolic heterogeneity of KRAS-mutant lung adenocarcinoma. Nat. Commun. 2019, 10, 4190. [Google Scholar] [CrossRef]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sanchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Daemen, A.; Nickles, D.; Jeon, S.M.; Foreman, O.; Sudini, K.; Gnad, F.; Lajoie, S.; Gour, N.; Mitzner, W.; et al. NRF2 Activation Promotes Aggressive Lung Cancer and Associates with Poor Clinical Outcomes. Clin. Cancer Res. 2021, 27, 877–888. [Google Scholar] [CrossRef]
- Adachi, Y.; Kimura, R.; Hirade, K.; Ebi, H. Escaping KRAS: Gaining Autonomy and Resistance to KRAS Inhibition in KRAS Mutant Cancers. Cancers 2021, 13, 5081. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Sunaga, N. Role of Immunotherapy for Oncogene-Driven Non-Small Cell Lung Cancer. Cancers 2018, 10, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamarsheh, S.; Gross, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef]
- Caetano, M.S.; Zhang, H.; Cumpian, A.M.; Gong, L.; Unver, N.; Ostrin, E.J.; Daliri, S.; Chang, S.H.; Ochoa, C.E.; Hanash, S.; et al. IL6 Blockade Reprograms the Lung Tumor Microenvironment to Limit the Development and Progression of K-ras-Mutant Lung Cancer. Cancer Res. 2016, 76, 3189–3199. [Google Scholar] [CrossRef] [Green Version]
- Sunaga, N.; Imai, H.; Shimizu, K.; Shames, D.S.; Kakegawa, S.; Girard, L.; Sato, M.; Kaira, K.; Ishizuka, T.; Gazdar, A.F.; et al. Oncogenic KRAS-induced interleukin-8 overexpression promotes cell growth and migration and contributes to aggressive phenotypes of non-small cell lung cancer. Int. J. Cancer. 2012, 130, 1733–1744. [Google Scholar] [CrossRef] [PubMed]
- Schalper, K.A.; Carleton, M.; Zhou, M.; Chen, T.; Feng, Y.; Huang, S.P.; Walsh, A.M.; Baxi, V.; Pandya, D.; Baradet, T.; et al. Elevated serum interleukin-8 is associated with enhanced intratumor neutrophils and reduced clinical benefit of immune-checkpoint inhibitors. Nat. Med. 2020, 26, 688–692. [Google Scholar] [CrossRef]
- Zhong, L.; Roybal, J.; Chaerkady, R.; Zhang, W.; Choi, K.; Alvarez, C.A.; Tran, H.; Creighton, C.J.; Yan, S.; Strieter, R.M.; et al. Identification of secreted proteins that mediate cell-cell interactions in an in vitro model of the lung cancer microenvironment. Cancer Res. 2008, 68, 7237–7245. [Google Scholar] [CrossRef] [Green Version]
- Marazioti, A.; Lilis, I.; Vreka, M.; Apostolopoulou, H.; Kalogeropoulou, A.; Giopanou, I.; Giotopoulou, G.A.; Krontira, A.C.; Iliopoulou, M.; Kanellakis, N.I.; et al. Myeloid-derived interleukin-1beta drives oncogenic KRAS-NF-kappaBeta addiction in malignant pleural effusion. Nat. Commun. 2018, 9, 672. [Google Scholar] [CrossRef] [Green Version]
- Liclican, E.L.; Walser, T.C.; Hazra, S.; Krysan, K.; Park, S.J.; Pagano, P.C.; Gardner, B.K.; Larsen, J.E.; Minna, J.D.; Dubinett, S.M. Loss of miR125a expression in a model of K-ras-dependent pulmonary premalignancy. Cancer Prev. Res. 2014, 7, 845–855. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Aref, A.R.; Cohoon, T.J.; Barbie, T.U.; Imamura, Y.; Yang, S.; Moody, S.E.; Shen, R.R.; Schinzel, A.C.; Thai, T.C.; et al. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 2014, 4, 452–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldinucci, D.; Borghese, C.; Casagrande, N. The CCL5/CCR5 Axis in Cancer Progression. Cancers 2020, 12, 1765. [Google Scholar] [CrossRef]
- Lan, B.; Ma, C.; Zhang, C.; Chai, S.; Wang, P.; Ding, L.; Wang, K. Association between PD-L1 expression and driver gene status in non-small-cell lung cancer: A meta-analysis. Oncotarget 2018, 9, 7684–7699. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Sunaga, N.; Kyoichi, K.; Tsukagoshi, Y.; Osaki, T.; Sakurai, R.; Hisada, T.; Girard, L.; Minna, J.D. Oncogenic KRAS mutations induce PD-L1 overexpression through MAPK pathway activation in non-small cell lung cancer cells. Cancer Res. 2016, 76, Abstract nr 4028. [Google Scholar] [CrossRef]
- Sumimoto, H.; Takano, A.; Teramoto, K.; Daigo, Y. RAS-Mitogen-Activated Protein Kinase Signal Is Required for Enhanced PD-L1 Expression in Human Lung Cancers. PLoS ONE 2016, 11, e0166626. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.A.; de Carne Trecesson, S.; Rana, S.; Zecchin, D.; Moore, C.; Molina-Arcas, M.; East, P.; Spencer-Dene, B.; Nye, E.; Barnouin, K.; et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity 2017, 47, 1083–1099.e6. [Google Scholar] [CrossRef] [Green Version]
- Lastwika, K.J.; Wilson, W., 3rd; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Parra, E.R.; Villalobos, P.; Zhang, J.; Behrens, C.; Mino, B.; Swisher, S.; Sepesi, B.; Weissferdt, A.; Kalhor, N.; Heymach, J.V.; et al. Immunohistochemical and Image Analysis-Based Study Shows that Several Immune Checkpoints are Co-expressed in Non-Small Cell Lung Carcinoma Tumors. J. Thorac. Oncol. 2018, 13, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer. 2020, 19, 116. [Google Scholar] [CrossRef]
- Zdanov, S.; Mandapathil, M.; Abu Eid, R.; Adamson-Fadeyi, S.; Wilson, W.; Qian, J.; Carnie, A.; Tarasova, N.; Mkrtichyan, M.; Berzofsky, J.A.; et al. Mutant KRAS Conversion of Conventional T Cells into Regulatory T Cells. Cancer Immunol. Res. 2016, 4, 354–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunaga, N.; Shames, D.S.; Girard, L.; Peyton, M.; Larsen, J.E.; Imai, H.; Soh, J.; Sato, M.; Yanagitani, N.; Kaira, K.; et al. Knockdown of oncogenic KRAS in non-small cell lung cancers suppresses tumor growth and sensitizes tumor cells to targeted therapy. Mol. Cancer Ther. 2011, 10, 336–346. [Google Scholar] [CrossRef] [Green Version]
- Le, X.; Negrao, M.V.; Reuben, A.; Federico, L.; Diao, L.; McGrail, D.; Nilsson, M.; Robichaux, J.; Munoz, I.G.; Patel, S.; et al. Characterization of the Immune Landscape of EGFR-Mutant NSCLC Identifies CD73/Adenosine Pathway as a Potential Therapeutic Target. J. Thorac. Oncol. 2021, 16, 583–600. [Google Scholar] [CrossRef]
- Shen, X.; Zhao, Y.; Liu, G.; Zhou, H.L.; Fan, J.; Zhang, L.; Li, Y.L.; Wang, Y.; Liang, J.; Xu, Z.X. Upregulation of programmed death ligand 1 by liver kinase B1 and its implication in programmed death 1 blockade therapy in non-small cell lung cancer. Life Sci. 2020, 256, 117923. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.Y.; Zhong, W.Z.; Zhang, X.C.; Su, J.; Xie, Z.; Liu, S.Y.; Tu, H.Y.; Chen, H.J.; Sun, Y.L.; Zhou, Q.; et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef] [Green Version]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to Tumor Angiogenesis from Innate Immune Cells within the Tumor Microenvironment: Implications for Immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef] [PubMed]
- Mortara, L.; Benest, A.V.; Bates, D.O.; Noonan, D.M. Can the co-dependence of the immune system and angiogenesis facilitate pharmacological targeting of tumours? Curr. Opin. Pharmacol. 2017, 35, 66–74. [Google Scholar] [CrossRef]
- Ott, P.A.; Hodi, F.S.; Buchbinder, E.I. Inhibition of Immune Checkpoints and Vascular Endothelial Growth Factor as Combination Therapy for Metastatic Melanoma: An Overview of Rationale, Preclinical Evidence, and Initial Clinical Data. Front. Oncol. 2015, 5, 202. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.; Ishida, T.; Oyama, T.; Ran, S.; Kravtsov, V.; Nadaf, S.; Carbone, D.P. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood 1998, 92, 4150–4166. [Google Scholar] [CrossRef] [PubMed]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Terme, M.; Pernot, S.; Marcheteau, E.; Sandoval, F.; Benhamouda, N.; Colussi, O.; Dubreuil, O.; Carpentier, A.F.; Tartour, E.; Taieb, J. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 2013, 73, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Konishi, T.; Huang, C.L.; Adachi, M.; Taki, T.; Inufusa, H.; Kodama, K.; Kohno, N.; Miyake, M. The K-ras gene regulates vascular endothelial growth factor gene expression in non-small cell lung cancers. Int. J. Oncol. 2000, 16, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Rak, J.; Mitsuhashi, Y.; Bayko, L.; Filmus, J.; Shirasawa, S.; Sasazuki, T.; Kerbel, R.S. Mutant ras oncogenes upregulate VEGF/VPF expression: Implications for induction and inhibition of tumor angiogenesis. Cancer Res. 1995, 55, 4575–4580. [Google Scholar]
- Pore, N.; Jiang, Z.; Gupta, A.; Cerniglia, G.; Kao, G.D.; Maity, A. EGFR tyrosine kinase inhibitors decrease VEGF expression by both hypoxia-inducible factor (HIF)-1-independent and HIF-1-dependent mechanisms. Cancer Res. 2006, 66, 3197–3204. [Google Scholar] [CrossRef] [Green Version]
- West, H.; Cappuzzo, F.; Reck, M.; Mok, T.; Jotte, R.M.; Nishio, M.; Kim, E.; Morris, S.; Shankar, G.; Zou, W.; et al. IMpower150: A post hoc analysis of efficacy outcomes in patients with KRAS, STK11 and KEAP1 mutations. Ann. Oncol. 2020, 31 (Suppl. 4), S817–S818. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Stock-Martineau, S.; Magner, K.; Jao, K.; Wheatley-Price, P. Challenges of Immunotherapy in Stage IV Non-Small-Cell Lung Cancer. JCO Oncol. Pract. 2021, 17, 465–471. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Govindan, R.; Dy, G.K.; Shapiro, G.; Bauml, J.; Schuler, M.H.; Addeo, A.; Kato, T.; Besse, B.; et al. Overall survival and exploratory subgroup analyses from the phase 2 CodeBreaK 100 trial evaluating sotorasib in pretreated KRAS p.G12C mutated non-small cell lung cancer. J. Clin. Oncol. 2021, 39, 9003. [Google Scholar] [CrossRef]
- Riely, G.J.; Ou, S.-H.I.; Rybkin, I.; Spira, A.; Papadopoulos, K.; Sabari, J.K.; Johnson, M.; Heist, R.S.; Bazhenova, L.; Barve, M.; et al. KRYSTAL-1: Activity and preliminary pharmacodynamic (PD) analysis of adagrasib (MRTX849) in patients (Pts) with advanced non-small cell lung cancer (NSCLC) harboring KRAS G12C mutation. J. Thorac. Oncol. 2021, 16, S751–S752. [Google Scholar] [CrossRef]
- Briere, D.M.; Li, S.; Calinisan, A.; Sudhakar, N.; Aranda, R.; Hargis, L.; Peng, D.H.; Deng, J.; Engstrom, L.D.; Hallin, J.; et al. The KRAS(G12C) Inhibitor MRTX849 Reconditions the Tumor Immune Microenvironment and Sensitizes Tumors to Checkpoint Inhibitor Therapy. Mol. Cancer Ther. 2021, 20, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell. 2020, 37, 471–484. [Google Scholar] [CrossRef]
- Turajlic, S.; Sottoriva, A.; Graham, T.; Swanton, C. Resolving genetic heterogeneity in cancer. Nat. Rev. Genet. 2019, 20, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Zhao, C.; Li, J.; Su, C.; Chen, X.; Ren, S.; Li, X.; Zhou, C. Clinical features and therapeutic options in non-small cell lung cancer patients with concomitant mutations of EGFR, ALK, ROS1, KRAS or BRAF. Cancer Med. 2019, 8, 2858–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef]
- Xu, W.; Wang, Z.; Zhang, W.; Qian, K.; Li, H.; Kong, D.; Li, Y.; Tang, Y. Mutated K-ras activates CDK8 to stimulate the epithelial-to-mesenchymal transition in pancreatic cancer in part via the Wnt/beta-catenin signaling pathway. Cancer Lett. 2015, 356, 613–627. [Google Scholar] [CrossRef]
- Zhang, L.; Ma, T.; Brozick, J.; Babalola, K.; Budiu, R.; Tseng, G.; Vlad, A.M. Effects of Kras activation and Pten deletion alone or in combination on MUC1 biology and epithelial-to-mesenchymal transition in ovarian cancer. Oncogene 2016, 35, 5010–5020. [Google Scholar] [CrossRef] [Green Version]
- Konen, J.M.; Rodriguez, B.L.; Padhye, A.; Ochieng, J.K.; Gibson, L.; Diao, L.; Fowlkes, N.W.; Fradette, J.J.; Peng, D.H.; Cardnell, R.J.; et al. Dual Inhibition of MEK and AXL Targets Tumor Cell Heterogeneity and Prevents Resistant Outgrowth Mediated by the Epithelial-to-Mesenchymal Transition in NSCLC. Cancer Res. 2021, 81, 1398–1412. [Google Scholar] [CrossRef]
- Adachi, Y.; Ito, K.; Hayashi, Y.; Kimura, R.; Tan, T.Z.; Yamaguchi, R.; Ebi, H. Epithelial-to-Mesenchymal Transition is a Cause of Both Intrinsic and Acquired Resistance to KRAS G12C Inhibitor in KRAS G12C-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 5962–5973. [Google Scholar] [CrossRef]
- Young, A.; Lou, D.; McCormick, F. Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov. 2013, 3, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Sunaga, N.; Miura, Y.; Tsukagoshi, Y.; Kasahara, N.; Masuda, T.; Sakurai, R.; Kaira, K.; Hisada, T. Dual inhibition of MEK and p38 impairs tumor growth in KRAS-mutated non-small cell lung cancer. Oncol. Lett. 2019, 17, 3569–3575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamran, S.; Seyedrezazadeh, E.; Shanehbandi, D.; Asadi, M.; Zafari, V.; Shekari, N.; Namvar, L.; Zarredar, H. Combination Therapy with KRAS and P38alpha siRNA Suppresses Colorectal Cancer Growth and Development in SW480 Cell Line. J. Gastrointest. Cancer. 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Zarredar, H.; Pashapour, S.; Farajnia, S.; Ansarin, K.; Baradaran, B.; Ahmadzadeh, V.; Safari, F. Targeting the KRAS, p38alpha, and NF-kappaB in lung adenocarcinoma cancer cells: The effect of combining RNA interferences with a chemical inhibitor. J. Cell Biochem. 2019, 120, 10670–10677. [Google Scholar] [CrossRef]
- Engelman, J.A.; Chen, L.; Tan, X.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.; et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef] [Green Version]
- Yao, W.; Yue, P.; Zhang, G.; Owonikoko, T.K.; Khuri, F.R.; Sun, S.Y. Enhancing therapeutic efficacy of the MEK inhibitor, MEK162, by blocking autophagy or inhibiting PI3K/Akt signaling in human lung cancer cells. Cancer Lett. 2015, 364, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Qu, G.P.; Shi, M.; Wang, D.; Wu, J.H.; Wang, P.; Gong, M.L.; Zhang, Z.J. Dual targeting of MEK and PI3K effectively controls the proliferation of human EGFR-TKI resistant non-small cell lung carcinoma cell lines with different genetic backgrounds. BMC Pulm. Med. 2021, 21, 208. [Google Scholar] [CrossRef] [PubMed]
- Kitai, H.; Ebi, H.; Tomida, S.; Floros, K.V.; Kotani, H.; Adachi, Y.; Oizumi, S.; Nishimura, M.; Faber, A.C.; Yano, S. Epithelial-to-Mesenchymal Transition Defines Feedback Activation of Receptor Tyrosine Kinase Signaling Induced by MEK Inhibition in KRAS-Mutant Lung Cancer. Cancer Discov. 2016, 6, 754–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misale, S.; Fatherree, J.P.; Cortez, E.; Li, C.; Bilton, S.; Timonina, D.; Myers, D.T.; Lee, D.; Gomez-Caraballo, M.; Greenberg, M.; et al. KRAS G12C NSCLC Models Are Sensitive to Direct Targeting of KRAS in Combination with PI3K Inhibition. Clin. Cancer Res. 2019, 25, 796–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, D.L.; Haderk, F.; Bivona, T.G. Allosteric SHP2 inhibitors in cancer: Targeting the intersection of RAS, resistance, and the immune microenvironment. Curr. Opin. Chem. Biol. 2021, 62, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ruess, D.A.; Heynen, G.J.; Ciecielski, K.J.; Ai, J.; Berninger, A.; Kabacaoglu, D.; Gorgulu, K.; Dantes, Z.; Wormann, S.M.; Diakopoulos, K.N.; et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med. 2018, 24, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lu, H.; Wang, H.; Loo, A.; Zhang, X.; Yang, G.; Kowal, C.; Delach, S.; Wang, Y.; Goldoni, S.; et al. Combinations with Allosteric SHP2 Inhibitor TNO155 to Block Receptor Tyrosine Kinase Signaling. Clin. Cancer Res. 2021, 27, 342–354. [Google Scholar] [CrossRef]
- Mohi, M.G.; Neel, B.G. The role of Shp2 (PTPN11) in cancer. Curr. Opin. Genet. Dev. 2007, 17, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.J.; Hahn, W.C. Synthetic Lethal Vulnerabilities in KRAS-Mutant Cancers. Cold Spring Harb. Perspect. Med. 2018, 8, a031518. [Google Scholar] [CrossRef]
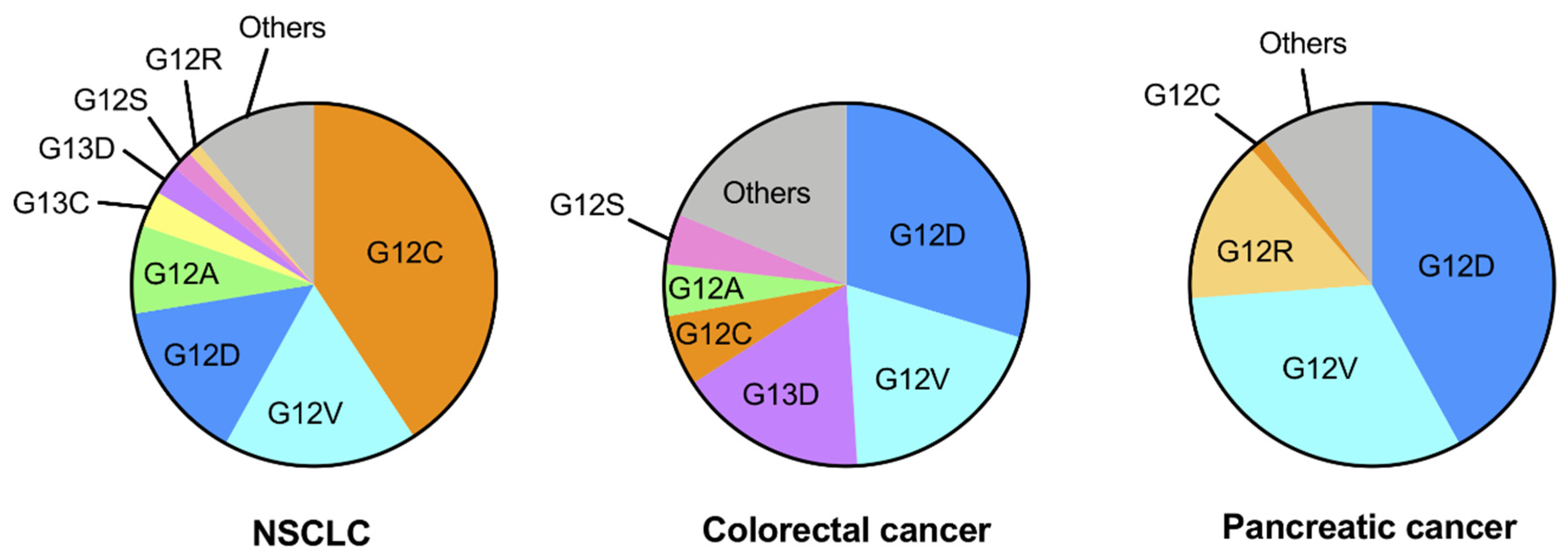
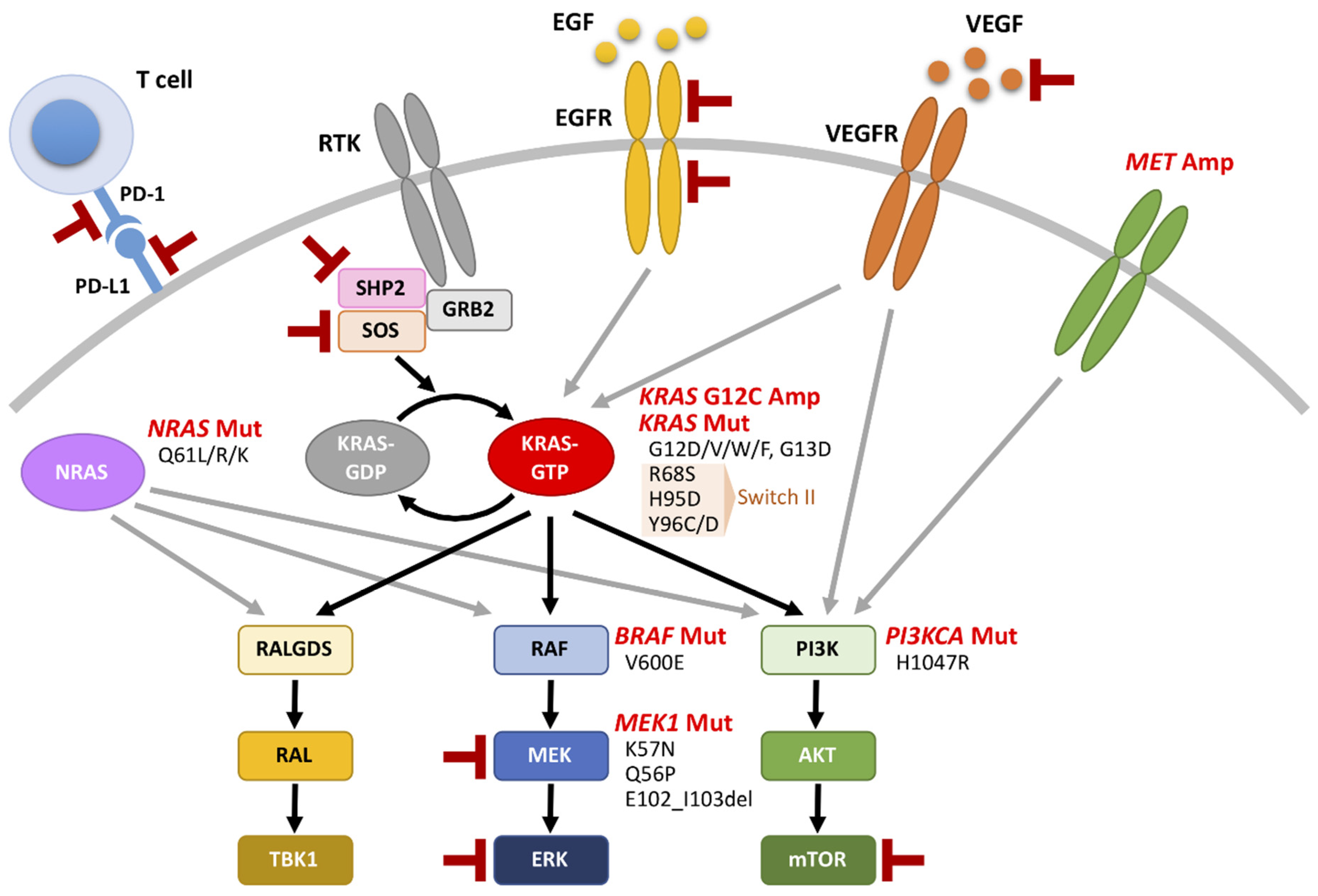

{kind=link}
{kind=link}
| Drug | Sponsor | NCT Number | Trial Name | Phase | Disease or Condition | Combination Therapy * |
|---|---|---|---|---|---|---|
| AMG510 (sotorasib) | Amgen | NCT03600883 | CodeBreaK 100 | 1/2 | Advanced solid tumors | PD-1 or PD-L1 antibodies |
| NCT04185883 | CodeBreaK 101 | 1/2 | Advanced solid tumors | AMG404 (PD-1 antibody) | ||
| Trametinib (MEK inhibitor) | ||||||
| RMC-4630 (SHP2 inhibitor) | ||||||
| Afatinib (EGFR-TKI) | ||||||
| Pembrolizumab (PD-1 antibody) | ||||||
| Chemotherapy (CBDCA, PEM, DTX) | ||||||
| Atezolizumab (PD-L1 antibody) | ||||||
| Everolimus (mTOR inhibitor) | ||||||
| Palbociclib (CDK4/6 inhibitor) | ||||||
| MVASI® (VEGF antibody) | ||||||
| TNO155 (SHP2 inhibitor) | ||||||
| Revolution Medicines | NCT05054725 | 2 | Advanced NSCLC | RMC-4630 (SHP2 inhibitor) | ||
| MRTX849 (adagrasib) | Mirati | NCT03785249 | KRYSTAL 1 | 1/2 | Advanced solid tumors | Pembrolizumab (PD-1 antibody) |
| Afatinib (EGFR-TKI) | ||||||
| NCT04613596 | KRYSTAL 7 | 2 | Advanced NSCLC | Pembrolizumab (PD-1 antibody) | ||
| NCT04975256 | KRYSTAL 14 | 1 | Advanced solid tumors | BI1701963 (SOS1 inhibitor) | ||
| GDC-6036 | Roche/Genentech | NCT04449874 | 1 | Advanced solid tumors | Atezolizumab (PD-L1 antibody) | |
| Bevacizumab (VEGF antibody) | ||||||
| Erlotinib (EGFR-TKI) | ||||||
| GDC-1971 (SHP2 inhibitor) | ||||||
| D-1553 | InventisBio | NCT04585035 | 1/2 | Advanced solid tumors | Standard treatment | |
| JDQ443 | Novartis | NCT04699188 | 1/2 | Advanced solid tumors | TNO155 (SHP2 inhibitor) | |
| Spartalizumab (PD-1 antibody) | ||||||
| LY3537982 | Eli Lilly | NCT04956640 | 1 | Advanced solid tumors | Abemaciclib (CDK4/6 inhibitor) | |
| Erlotinib (EGFR-TKI) | ||||||
| Sintilimab (PD-1 antibody) | ||||||
| Temuterkib (ERK inhibitor) | ||||||
| LY3295668 (Aurora kinase inhibitor) | ||||||
| Cetuximab (EGFR antibody) | ||||||
| BI1823911 | Boehringer | NCT04973163 | 1 | Advanced solid tumors | BI1701963 (SOS1 inhibitor) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sunaga, N.; Miura, Y.; Kasahara, N.; Sakurai, R. Targeting Oncogenic KRAS in Non-Small-Cell Lung Cancer. Cancers 2021, 13, 5956. https://doi.org/10.3390/cancers13235956
Sunaga N, Miura Y, Kasahara N, Sakurai R. Targeting Oncogenic KRAS in Non-Small-Cell Lung Cancer. Cancers. 2021; 13(23):5956. https://doi.org/10.3390/cancers13235956
Chicago/Turabian StyleSunaga, Noriaki, Yosuke Miura, Norimitsu Kasahara, and Reiko Sakurai. 2021. "Targeting Oncogenic KRAS in Non-Small-Cell Lung Cancer" Cancers 13, no. 23: 5956. https://doi.org/10.3390/cancers13235956
APA StyleSunaga, N., Miura, Y., Kasahara, N., & Sakurai, R. (2021). Targeting Oncogenic KRAS in Non-Small-Cell Lung Cancer. Cancers, 13(23), 5956. https://doi.org/10.3390/cancers13235956