
Targeting MERTK and AXL in EGFR Mutant Non-Small Cell Lung Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
2. Physiologic Roles for MERTK and AXL
3. Oncogenic Roles for MERTK and AXL
3.1. Roles in NSCLC
3.2. Functions in Cancer Cells
3.3. Signaling in Cancer Cells
3.4. Immune Regulatory Functions in the Tumor Microenvironment
4. Targeting TAM Kinases and EGFR in NSCLC
5. MERTK and AXL Inhibitors for Potential Use in NSCLC
5.1. Biological Agents
5.2. Small Molecule Inhibitors
5.3. Potential on-Target Toxicities Associated with MERTK and/or AXL Inhibition
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [Green Version]
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non-Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. [Google Scholar] [CrossRef]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef] [Green Version]
- Travis, W.D.; Brambilla, E.; Burke, A.P.; Marx, A.; Nicholson, A.G. Introduction to the 2015 World Health Organization Classification of Tumors of the Lung, Pleura, Thymus, and Heart. J. Thorac. Oncol. 2015, 10, 1240–1242. [Google Scholar] [CrossRef] [Green Version]
- Datta, D.; Lahiri, B. Preoperative evaluation of patients undergoing lung resection surgery. Chest 2003, 123, 2096–2103. [Google Scholar] [CrossRef] [Green Version]
- Group NM-aC; Arriagada, R.; Auperin, A.; Burdett, S.; Higgins, J.P.; Johnson, D.H.; Le Chevalier, T.; Le Pechoux, C.; Parmar, M.K.; Pignon, J.P.; et al. Adjuvant chemotherapy, with or without postoperative radiotherapy, in operable non-small-cell lung cancer: Two meta-analyses of individual patient data. Lancet 2010, 375, 1267–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labbe, C.; Anderson, M.; Simard, S.; Tremblay, L.; Laberge, F.; Vaillancourt, R.; Lacasse, Y. Wait times for diagnosis and treatment of lung cancer: A single-centre experience. Curr. Oncol. 2017, 24, 367–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pignon, J.P.; Tribodet, H.; Scagliotti, G.V.; Douillard, J.Y.; Shepherd, F.A.; Stephens, R.J.; Dunant, A.; Torri, V.; Rosell, R.; Seymour, L.; et al. Lung adjuvant cisplatin evaluation: A pooled analysis by the LACE Collaborative Group. J. Clin. Oncol. 2008, 26, 3552–3559. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.J.; Blyth, K.G. Predicting lung cancer recurrence from circulating tumour DNA. Commentary on ‘Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution’. Cell Death Differ. 2017, 24, 1473–1474. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Kocher, F.; Hilbe, W.; Seeber, A.; Pircher, A.; Schmid, T.; Greil, R.; Auberger, J.; Nevinny-Stickel, M.; Sterlacci, W.; Tzankov, A.; et al. Longitudinal analysis of 2293 NSCLC patients: A comprehensive study from the TYROL registry. Lung Cancer 2015, 87, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Group, N.M.-A.C. Chemotherapy in addition to supportive care improves survival in advanced non-small-cell lung cancer: A systematic review and meta-analysis of individual patient data from 16 randomized controlled trials. J. Clin. Oncol. 2008, 26, 4617–4625. [Google Scholar] [CrossRef]
- Carney, D.N. Lung cancer—time to move on from chemotherapy. N. Engl. J. Med. 2002, 346, 126–128. [Google Scholar] [CrossRef] [PubMed]
- Goldstraw, P.; Chansky, K.; Crowley, J.; Rami-Porta, R.; Asamura, H.; Eberhardt, W.E.; Nicholson, A.G.; Groome, P.; Mitchell, A.; Bolejack, V.; et al. The IASLC Lung Cancer Staging Project: Proposals for Revision of the TNM Stage Groupings in the Forthcoming (Eighth) Edition of the TNM Classification for Lung Cancer. J. Thorac. Oncol. 2016, 11, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [Green Version]
- Mendelsohn, J.; Baselga, J. The EGF receptor family as targets for cancer therapy. Oncogene 2000, 19, 6550–6565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef] [Green Version]
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.; Dezube, B.J.; Janne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Sequist, L.V.; Yang, J.C.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.M.; Boyer, M.; et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crino, L.; Ahn, M.J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alamgeer, M.; Ganju, V.; Watkins, D.N. Novel therapeutic targets in non-small cell lung cancer. Curr. Opin. Pharmacol. 2013, 13, 394–401. [Google Scholar] [CrossRef]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piotrowska, Z.; Isozaki, H.; Lennerz, J.K.; Gainor, J.F.; Lennes, I.T.; Zhu, V.W.; Marcoux, N.; Banwait, M.K.; Digumarthy, S.R.; Su, W.; et al. Landscape of Acquired Resistance to Osimertinib in EGFR-Mutant NSCLC and Clinical Validation of Combined EGFR and RET Inhibition with Osimertinib and BLU-667 for Acquired RET Fusion. Cancer Discov. 2018, 8, 1529–1539. [Google Scholar] [CrossRef] [Green Version]
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci. Transl. Med. 2012, 4, 120ra17. [Google Scholar] [CrossRef] [Green Version]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef] [Green Version]
- Sequist, L.V.; Soria, J.C.; Goldman, J.W.; Wakelee, H.A.; Gadgeel, S.M.; Varga, A.; Papadimitrakopoulou, V.; Solomon, B.J.; Oxnard, G.R.; Dziadziuszko, R.; et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1700–1709. [Google Scholar] [CrossRef]
- O’Bryan, J.P.; Frye, R.A.; Cogswell, P.C.; Neubauer, A.; Kitch, B.; Prokop, C.; Espinosa, R., 3rd; Le Beau, M.M.; Earp, H.S.; Liu, E.T. Axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell. Biol. 1991, 11, 5016–5031. [Google Scholar] [PubMed] [Green Version]
- Graham, D.K.; Dawson, T.L.; Mullaney, D.L.; Snodgrass, H.R.; Earp, H.S. Cloning and mRNA expression analysis of a novel human protooncogene, c-mer. Cell Growth Differ. 1994, 5, 647–657. [Google Scholar] [PubMed]
- Caberoy, N.B.; Zhou, Y.; Li, W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 2010, 29, 3898–3910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caberoy, N.B.; Alvarado, G.; Bigcas, J.L.; Li, W. Galectin-3 is a new MerTK-specific eat-me signal. J. Cell. Physiol. 2012, 227, 401–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manfioletti, G.; Brancolini, C.; Avanzi, G.; Schneider, C. The protein encoded by a growth arrest-specific gene (gas6) is a new member of the vitamin K-dependent proteins related to protein S, a negative coregulator in the blood coagulation cascade. Mol. Cell. Biol. 1993, 13, 4976–4985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lew, E.D.; Oh, J.; Burrola, P.G.; Lax, I.; Zagorska, A.; Traves, P.G.; Schlessinger, J.; Lemke, G. Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. eLife 2014, 3. [Google Scholar] [CrossRef]
- Nagata, K.; Ohashi, K.; Nakano, T.; Arita, H.; Zong, C.; Hanafusa, H.; Mizuno, K. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J. Biol. Chem. 1996, 271, 30022–30027. [Google Scholar] [CrossRef] [Green Version]
- Kasikara, C.; Kumar, S.; Kimani, S.; Tsou, W.I.; Geng, K.; Davra, V.; Sriram, G.; Devoe, C.; Nguyen, K.N.; Antes, A.; et al. Phosphatidylserine Sensing by TAM Receptors Regulates AKT-Dependent Chemoresistance and PD-L1 Expression. Mol. Cancer Res. 2017, 15, 753–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsou, W.I.; Nguyen, K.Q.; Calarese, D.A.; Garforth, S.J.; Antes, A.L.; Smirnov, S.V.; Almo, S.C.; Birge, R.B.; Kotenko, S.V. Receptor tyrosine kinases, TYRO3, AXL, and MER, demonstrate distinct patterns and complex regulation of ligand-induced activation. J. Biol. Chem. 2014, 289, 25750–25763. [Google Scholar] [CrossRef] [Green Version]
- Crosier, P.S.; Freeman, S.A.; Orlic, D.; Bodine, D.M.; Crosier, K.E. The Dtk receptor tyrosine kinase, which binds protein S, is expressed during hematopoiesis. Exp. Hematol. 1996, 24, 318–323. [Google Scholar]
- Faust, M.; Ebensperger, C.; Schulz, A.S.; Schleithoff, L.; Hameister, H.; Bartram, C.R.; Janssen, J.W. The murine ufo receptor: Molecular cloning, chromosomal localization and in situ expression analysis. Oncogene 1992, 7, 1287–1293. [Google Scholar] [PubMed]
- Lu, Q.; Lemke, G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science 2001, 293, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.S.; McMahon, E.J.; Pop, S.M.; Reap, E.A.; Caricchio, R.; Cohen, P.L.; Earp, H.S.; Matsushima, G.K. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 2001, 411, 207–211. [Google Scholar] [CrossRef]
- Lu, Q.; Gore, M.; Zhang, Q.; Camenisch, T.; Boast, S.; Casagranda, F.; Lai, C.; Skinner, M.K.; Klein, R.; Matsushima, G.K.; et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature 1999, 398, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Camenisch, T.D.; Koller, B.H.; Earp, H.S.; Matsushima, G.K. A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J. Immunol. 1999, 162, 3498–3503. [Google Scholar] [PubMed]
- Caraux, A.; Lu, Q.; Fernandez, N.; Riou, S.; Di Santo, J.P.; Raulet, D.H.; Lemke, G.; Roth, C. Natural killer cell differentiation driven by Tyro3 receptor tyrosine kinases. Nat. Immunol. 2006, 7, 747–754. [Google Scholar] [CrossRef]
- Rahman, Z.S.; Shao, W.H.; Khan, T.N.; Zhen, Y.; Cohen, P.L. Impaired apoptotic cell clearance in the germinal center by Mer-deficient tingible body macrophages leads to enhanced antibody-forming cell and germinal center responses. J. Immunol. 2010, 185, 5859–5868. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P.L.; Caricchio, R.; Abraham, V.; Camenisch, T.D.; Jennette, J.C.; Roubey, R.A.; Earp, H.S.; Matsushima, G.; Reap, E.A. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J. Exp. Med. 2002, 196, 135–140. [Google Scholar] [CrossRef]
- Angelillo-Scherrer, A.; Burnier, L.; Flores, N.; Savi, P.; DeMol, M.; Schaeffer, P.; Herbert, J.M.; Lemke, G.; Goff, S.P.; Matsushima, G.K.; et al. Role of Gas6 receptors in platelet signaling during thrombus stabilization and implications for antithrombotic therapy. J. Clin. Investig. 2005, 115, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Wallet, M.A.; Sen, P.; Flores, R.R.; Wang, Y.; Yi, Z.; Huang, Y.; Mathews, C.E.; Earp, H.S.; Matsushima, G.; Wang, B.; et al. MerTK is required for apoptotic cell-induced T cell tolerance. J. Exp. Med. 2008, 205, 219–232. [Google Scholar] [CrossRef]
- Linger, R.M.; Cohen, R.A.; Cummings, C.T.; Sather, S.; Migdall-Wilson, J.; Middleton, D.H.; Lu, X.; Baron, A.E.; Franklin, W.A.; Merrick, D.T.; et al. Mer or Axl receptor tyrosine kinase inhibition promotes apoptosis, blocks growth and enhances chemosensitivity of human non-small cell lung cancer. Oncogene 2013, 32, 3420–3431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wimmel, A.; Glitz, D.; Kraus, A.; Roeder, J.; Schuermann, M. Axl receptor tyrosine kinase expression in human lung cancer cell lines correlates with cellular adhesion. Eur. J. Cancer 2001, 37, 2264–2274. [Google Scholar] [CrossRef]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Yamada, T.; Wang, R.; Tanimura, K.; Adachi, Y.; Nishiyama, A.; Tanimoto, A.; Takeuchi, S.; Araujo, L.H.; Boroni, M.; et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat. Commun. 2019, 10, 259. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Li, Y.; Li, X.; Wang, L.; Yang, N.; Wang, Y.; Wei, H. Mer receptor tyrosine kinase is frequently overexpressed in human non-small cell lung cancer, confirming resistance to erlotinib. Oncotarget 2015, 6, 9206–9219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iida, S.; Miki, Y.; Suzuki, T.; Mori, K.; Saito, M.; Niikawa, H.; Kondo, T.; Yamada-Okabe, H.; Sasano, H. Activation of AXL and antitumor effects of a monoclonal antibody to AXL in lung adenocarcinoma. Anticancer Res. 2014, 34, 1821–1827. [Google Scholar] [PubMed]
- Qu, X.H.; Liu, J.L.; Zhong, X.W.; Li, X.I.; Zhang, Q.G. Insights into the roles of hnRNP A2/B1 and AXL in non-small cell lung cancer. Oncol. Lett. 2015, 10, 1677–1685. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Bai, F.; Fan, L.; Pang, W.; Han, R.; Wang, J.; Liu, Y.; Yan, X.; Duan, H.; Xing, L. Coexpression of receptor tyrosine kinase AXL and EGFR in human primary lung adenocarcinomas. Hum. Pathol. 2015, 46, 1935–1944. [Google Scholar] [CrossRef] [PubMed]
- Ramkumar, K.; Stewart, C.A.; Cargill, K.R.; Della Corte, C.M.; Wang, Q.; Shen, L.; Diao, L.; Cardnell, R.J.; Peng, D.H.; Rodriguez, B.L.; et al. AXL Inhibition Induces DNA Damage and Replication Stress in Non-Small Cell Lung Cancer Cells and Promotes Sensitivity to ATR Inhibitors. Mol. Cancer Res. 2021, 19, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Safaric Tepes, P.; Pal, D.; Lindsted, T.; Ibarra, I.; Lujambio, A.; Jimenez Sabinina, V.; Senturk, S.; Miller, M.; Korimerla, N.; Huang, J.; et al. An epigenetic switch regulates the ontogeny of AXL-positive/EGFR-TKi-resistant cells by modulating miR-335 expression. eLife 2021, 10. [Google Scholar] [CrossRef]
- Shieh, Y.S.; Lai, C.Y.; Kao, Y.R.; Shiah, S.G.; Chu, Y.W.; Lee, H.S.; Wu, C.W. Expression of axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia 2005, 7, 1058–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, M.; Sonobe, M.; Nakayama, E.; Kobayashi, M.; Kikuchi, R.; Kitamura, J.; Imamura, N.; Date, H. Higher expression of receptor tyrosine kinase Axl, and differential expression of its ligand, Gas6, predict poor survival in lung adenocarcinoma patients. Ann. Surg. Oncol. 2013, 20 (Suppl. 3), S467–S476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melchionna, R.; Spada, S.; Di Modugno, F.; D’Andrea, D.; Di Carlo, A.; Panetta, M.; Mileo, A.M.; Sperduti, I.; Antoniani, B.; Gallo, E.; et al. The actin modulator hMENA regulates GAS6-AXL axis and pro-tumor cancer/stromal cell cooperation. EMBO Rep. 2020, 21, e50078. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ye, X.; Tan, C.; Hongo, J.A.; Zha, J.; Liu, J.; Kallop, D.; Ludlam, M.J.; Pei, L. Axl as a potential therapeutic target in cancer: Role of Axl in tumor growth, metastasis and angiogenesis. Oncogene 2009, 28, 3442–3455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.; Suda, K.; Shimizu, S.; Sakai, K.; Mizuuchi, H.; Tomizawa, K.; Takemoto, T.; Nishio, K.; Mitsudomi, T. Clinical, Pathological, and Molecular Features of Lung Adenocarcinomas with AXL Expression. PLoS ONE 2016, 11, e0154186. [Google Scholar] [CrossRef]
- Cummings, C.T.; Linger, R.M.; Cohen, R.A.; Sather, S.; Kirkpatrick, G.D.; Davies, K.D.; DeRyckere, D.; Earp, H.S.; Graham, D.K. Mer590, a novel monoclonal antibody targeting MER receptor tyrosine kinase, decreases colony formation and increases chemosensitivity in non-small cell lung cancer. Oncotarget 2014, 5, 10434–10445. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; DeRyckere, D.; Hunter, D.; Liu, J.; Stashko, M.A.; Minson, K.A.; Cummings, C.T.; Lee, M.; Glaros, T.G.; Newton, D.L.; et al. UNC2025, a potent and orally bioavailable MER/FLT3 dual inhibitor. J. Med. Chem. 2014, 57, 7031–7041. [Google Scholar] [CrossRef] [Green Version]
- Cummings, C.T.; Zhang, W.; Davies, K.D.; Kirkpatrick, G.D.; Zhang, D.; DeRyckere, D.; Wang, X.; Frye, S.V.; Earp, H.S.; Graham, D.K. Small Molecule Inhibition of MERTK Is Efficacious in Non-Small Cell Lung Cancer Models Independent of Driver Oncogene Status. Mol. Cancer Ther. 2015, 14, 2014–2022. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Parker, R.E.; Wang, X.; Frye, S.V.; Earp, H.S., 3rd; DeRyckere, D.; Graham, D.K. MERTK Promotes Resistance to Irreversible EGFR Tyrosine Kinase Inhibitors in Non-small Cell Lung Cancers Expressing Wild-type EGFR Family Members. Clin. Cancer Res. 2018, 24, 6523–6535. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Li, Y.; Stawicki, S.; Couto, S.; Eastham-Anderson, J.; Kallop, D.; Weimer, R.; Wu, Y.; Pei, L. An anti-Axl monoclonal antibody attenuates xenograft tumor growth and enhances the effect of multiple anticancer therapies. Oncogene 2010, 29, 5254–5264. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, T.A.; Rafat, M.; Castellini, L.; Shehade, H.; Kariolis, M.S.; Hui, A.B.; Stehr, H.; von Eyben, R.; Jiang, D.; Ellies, L.G.; et al. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat. Commun. 2016, 7, 13898. [Google Scholar] [CrossRef] [Green Version]
- Brand, T.M.; Iida, M.; Stein, A.P.; Corrigan, K.L.; Braverman, C.M.; Luthar, N.; Toulany, M.; Gill, P.S.; Salgia, R.; Kimple, R.J.; et al. AXL mediates resistance to cetuximab therapy. Cancer Res. 2014, 74, 5152–5164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rho, J.K.; Choi, Y.J.; Kim, S.Y.; Kim, T.W.; Choi, E.K.; Yoon, S.J.; Park, B.M.; Park, E.; Bae, J.H.; Choi, C.M.; et al. MET and AXL inhibitor NPS-1034 exerts efficacy against lung cancer cells resistant to EGFR kinase inhibitors because of MET or AXL activation. Cancer Res. 2014, 74, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Li, J.; Jang, C.; Wang, J.; Xiong, J. The role of Axl in drug resistance and epithelial-to-mesenchymal transition of non-small cell lung carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 6653–6661. [Google Scholar] [PubMed]
- Bae, S.Y.; Hong, J.Y.; Lee, H.J.; Park, H.J.; Lee, S.K. Targeting the degradation of AXL receptor tyrosine kinase to overcome resistance in gefitinib-resistant non-small cell lung cancer. Oncotarget 2015, 6, 10146–10160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkabets, M.; Pazarentzos, E.; Juric, D.; Sheng, Q.; Pelossof, R.A.; Brook, S.; Benzaken, A.O.; Rodon, J.; Morse, N.; Yan, J.J.; et al. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell 2015, 27, 533–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, T.; Tong, P.; Diao, L.; Li, L.; Fan, Y.; Hoff, J.; Heymach, J.V.; Wang, J.; Byers, L.A. Targeting AXL and mTOR Pathway Overcomes Primary and Acquired Resistance to WEE1 Inhibition in Small-Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 6239–6253. [Google Scholar] [CrossRef] [Green Version]
- McDaniel, N.K.; Cummings, C.T.; Iida, M.; Hulse, J.; Pearson, H.E.; Vasileiadi, E.; Parker, R.E.; Orbuch, R.A.; Ondracek, O.J.; Welke, N.B.; et al. MERTK Mediates Intrinsic and Adaptive Resistance to AXL-targeting Agents. Mol. Cancer Ther. 2018, 17, 2297–2308. [Google Scholar] [CrossRef] [Green Version]
- Nakamichi, S.; Seike, M.; Miyanaga, A.; Chiba, M.; Zou, F.; Takahashi, A.; Ishikawa, A.; Kunugi, S.; Noro, R.; Kubota, K.; et al. Overcoming drug-tolerant cancer cell subpopulations showing AXL activation and epithelial-mesenchymal transition is critical in conquering ALK-positive lung cancer. Oncotarget 2018, 9, 27242–27255. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Huelse, J.; Parker, R.; Tan, Z.; Wang, X.; Frye, S.V.; Earp, H.S.; DeRyckere, D.; Graham, D.K. MERTK drives residual tumor growth in EGFR-mutated non-small cell lung cancer cells treated with osimertinib. Cancer Res. 2020, 80. [Google Scholar] [CrossRef]
- Yan, D.; Tan, Z.; Wang, X.; Frye, S.V.; Earp, H.S., III; DeRyckere, D.; Graham, D.K. A novel strategy to cope with osimertinib resistance in non-small cell lung cancer by treatment with a PIM kinase inhibitor in combination with a MERTK-selective kinase inhibitor. Cancer Res. 2021, 81. [Google Scholar] [CrossRef]
- Okura, N.; Nishioka, N.; Yamada, T.; Taniguchi, H.; Tanimura, K.; Katayama, Y.; Yoshimura, A.; Watanabe, S.; Kikuchi, T.; Shiotsu, S.; et al. ONO-7475, a Novel AXL Inhibitor, Suppresses the Adaptive Resistance to Initial EGFR-TKI Treatment in EGFR-Mutated Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 2244–2256. [Google Scholar] [CrossRef] [PubMed]
- Rios-Doria, J.; Favata, M.; Lasky, K.; Feldman, P.; Lo, Y.; Yang, G.; Stevens, C.; Wen, X.; Sehra, S.; Katiyar, K.; et al. A Potent and Selective Dual Inhibitor of AXL and MERTK Possesses Both Immunomodulatory and Tumor-Targeted Activity. Front. Oncol. 2020, 10, 598477. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.H.; Wu, C.C.; Huang, K.Y.; Leu, Y.L.; Yang, S.C.; Chen, C.L.; Chen, C.Y. Integrated Omics Analysis of Non-Small-Cell Lung Cancer Cells Harboring the EGFR C797S Mutation Reveals the Potential of AXL as a Novel Therapeutic Target in TKI-Resistant Lung Cancer. Cancers 2020, 13, 111. [Google Scholar] [CrossRef]
- Konen, J.M.; Rodriguez, B.L.; Padhye, A.; Ochieng, J.K.; Gibson, L.; Diao, L.; Fowlkes, N.W.; Fradette, J.J.; Peng, D.H.; Cardnell, R.J.; et al. Dual Inhibition of MEK and AXL Targets Tumor Cell Heterogeneity and Prevents Resistant Outgrowth Mediated by the Epithelial-to-Mesenchymal Transition in NSCLC. Cancer Res. 2021, 81, 1398–1412. [Google Scholar] [CrossRef]
- Tirado-Gonzalez, I.; Descot, A.; Soetopo, D.; Nevmerzhitskaya, A.; Schaffer, A.; Czlonka, E.; Wachtel, C.; Tsoukala, I.; Muller, L. AXL Inhibition in Macrophages Stimulates Host-versus-Leukemia Immunity and Eradicates Naive and Treatment-Resistant Leukemia. Cancer Discov. 2021, 11. [Google Scholar] [CrossRef]
- Wilson, C.; Ye, X.; Pham, T.; Lin, E.; Chan, S.; McNamara, E.; Neve, R.M.; Belmont, L.; Koeppen, H.; Yauch, R.L.; et al. AXL inhibition sensitizes mesenchymal cancer cells to antimitotic drugs. Cancer Res. 2014, 74, 5878–5890. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.C.; Baek, S.H.; Lee, C. Curcumin-induced downregulation of Axl receptor tyrosine kinase inhibits cell proliferation and circumvents chemoresistance in non-small lung cancer cells. Int. J. Oncol. 2015, 47, 2296–2303. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Jin, H.; Wang, N.; Fan, S.; Wang, Y.; Zhang, Y.; Wei, L.; Tao, X.; Gu, D.; Zhao, F.; et al. Gas6/Axl Axis Contributes to Chemoresistance and Metastasis in Breast Cancer through Akt/GSK-3beta/beta-catenin Signaling. Theranostics 2016, 6, 1205–1219. [Google Scholar] [CrossRef]
- Shin, D.H.; Lee, H.J.; Min, H.Y.; Choi, S.P.; Lee, M.S.; Lee, J.W.; Johnson, F.M.; Mehta, K.; Lippman, S.M.; Glisson, B.S.; et al. Combating resistance to anti-IGFR antibody by targeting the integrin beta3-Src pathway. J. Natl. Cancer Inst. 2013, 105, 1558–1570. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Hurlburt, W.; Greer, A.; Reeves, K.A.; Hillerman, S.; Chang, H.; Fargnoli, J.; Graf Finckenstein, F.; Gottardis, M.M.; Carboni, J.M. Differential mechanisms of acquired resistance to insulin-like growth factor-i receptor antibody therapy or to a small-molecule inhibitor, BMS-754807, in a human rhabdomyosarcoma model. Cancer Res. 2010, 70, 7221–7231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boshuizen, J.; Koopman, L.A.; Krijgsman, O.; Shahrabi, A.; van den Heuvel, E.G.; Ligtenberg, M.A.; Vredevoogd, D.W.; Kemper, K.; Kuilman, T.; Song, J.Y.; et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK inhibitors. Nat. Med. 2018, 24, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Taverna, J.A.; Hung, C.N.; DeArmond, D.T.; Chen, M.; Lin, C.L.; Osmulski, P.A.; Gaczynska, M.E.; Wang, C.M.; Lucio, N.D.; Chou, C.W.; et al. Single-Cell Proteomic Profiling Identifies Combined AXL and JAK1 Inhibition as a Novel Therapeutic Strategy for Lung Cancer. Cancer Res. 2020, 80, 1551–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, D.K.; Bowman, G.W.; Dawson, T.L.; Stanford, W.L.; Earp, H.S.; Snodgrass, H.R. Cloning and developmental expression analysis of the murine c-mer tyrosine kinase. Oncogene 1995, 10, 2349–2359. [Google Scholar] [PubMed]
- Quong, R.Y.; Bickford, S.T.; Ing, Y.L.; Terman, B.; Herlyn, M.; Lassam, N.J. Protein kinases in normal and transformed melanocytes. Melanoma Res. 1994, 4, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Lee-Sherick, A.B.; Eisenman, K.M.; Sather, S.; McGranahan, A.; Armistead, P.M.; McGary, C.S.; Hunsucker, S.A.; Schlegel, J.; Martinson, H.; Cannon, C.; et al. Aberrant Mer receptor tyrosine kinase expression contributes to leukemogenesis in acute myeloid leukemia. Oncogene 2016, 35, 6270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huey, M.G.; Minson, K.A.; Earp, H.S.; DeRyckere, D.; Graham, D.K. Targeting the TAM Receptors in Leukemia. Cancers 2016, 8, 101. [Google Scholar] [CrossRef]
- Meric, F.; Lee, W.P.; Sahin, A.; Zhang, H.; Kung, H.J.; Hung, M.C. Expression profile of tyrosine kinases in breast cancer. Clin. Cancer Res. 2002, 8, 361–367. [Google Scholar]
- Craven, R.J.; Xu, L.H.; Weiner, T.M.; Fridell, Y.W.; Dent, G.A.; Srivastava, S.; Varnum, B.; Liu, E.T.; Cance, W.G. Receptor tyrosine kinases expressed in metastatic colon cancer. Int. J. Cancer 1995, 60, 791–797. [Google Scholar] [CrossRef]
- Tsou, A.P.; Wu, K.M.; Tsen, T.Y.; Chi, C.W.; Chiu, J.H.; Lui, W.Y.; Hu, C.P.; Chang, C.; Chou, C.K.; Tsai, S.F. Parallel hybridization analysis of multiple protein kinase genes: Identification of gene expression patterns characteristic of human hepatocellular carcinoma. Genomics 1998, 50, 331–340. [Google Scholar] [CrossRef]
- Wu, C.W.; Li, A.F.; Chi, C.W.; Lai, C.H.; Huang, C.L.; Lo, S.S.; Lui, W.Y.; Lin, W.C. Clinical significance of AXL kinase family in gastric cancer. Anticancer Res. 2002, 22, 1071–1078. [Google Scholar] [PubMed]
- Jacob, A.N.; Kalapurakal, J.; Davidson, W.R.; Kandpal, G.; Dunson, N.; Prashar, Y.; Kandpal, R.P. A receptor tyrosine kinase, UFO/Axl, and other genes isolated by a modified differential display PCR are overexpressed in metastatic prostatic carcinoma cell line DU145. Cancer Detect. Prev. 1999, 23, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.M.; Robinson, D.R.; Kung, H.J. Signal pathways in up-regulation of chemokines by tyrosine kinase MER/NYK in prostate cancer cells. Cancer Res. 2004, 64, 7311–7320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rankin, E.B.; Fuh, K.C.; Taylor, T.E.; Krieg, A.J.; Musser, M.; Yuan, J.; Wei, K.; Kuo, C.J.; Longacre, T.A.; Giaccia, A.J. AXL is an essential factor and therapeutic target for metastatic ovarian cancer. Cancer Res. 2010, 70, 7570–7579. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.O.; Young, A.N.; Brown, M.R.; Brat, D.J.; Parks, J.S.; Neish, A.S.; Oyesiku, N.M. Novel patterns of gene expression in pituitary adenomas identified by complementary deoxyribonucleic acid microarrays and quantitative reverse transcription-polymerase chain reaction. J. Clin. Endocrinol. Metab. 2001, 86, 3097–3107. [Google Scholar] [CrossRef] [Green Version]
- Vajkoczy, P.; Knyazev, P.; Kunkel, A.; Capelle, H.H.; Behrndt, S.; von Tengg-Kobligk, H.; Kiessling, F.; Eichelsbacher, U.; Essig, M.; Read, T.A.; et al. Dominant-negative inhibition of the Axl receptor tyrosine kinase suppresses brain tumor cell growth and invasion and prolongs survival. Proc. Natl. Acad. Sci. USA 2006, 103, 5799–5804. [Google Scholar] [CrossRef] [Green Version]
- Ling, L.; Kung, H.J. Mitogenic signals and transforming potential of Nyk, a newly identified neural cell adhesion molecule-related receptor tyrosine kinase. Mol. Cell. Biol. 1995, 15, 6582–6592. [Google Scholar] [CrossRef] [Green Version]
- Lierman, E.; Van Miegroet, H.; Beullens, E.; Cools, J. Identification of protein tyrosine kinases with oncogenic potential using a retroviral insertion mutagenesis screen. Haematologica 2009, 94, 1440–1444. [Google Scholar] [CrossRef] [Green Version]
- Georgescu, M.M.; Kirsch, K.H.; Shishido, T.; Zong, C.; Hanafusa, H. Biological effects of c-Mer receptor tyrosine kinase in hematopoietic cells depend on the Grb2 binding site in the receptor and activation of NF-kappaB. Mol. Cell. Biol. 1999, 19, 1171–1181. [Google Scholar] [CrossRef] [Green Version]
- Janssen, J.W.; Schulz, A.S.; Steenvoorden, A.C.; Schmidberger, M.; Strehl, S.; Ambros, P.F.; Bartram, C.R. A novel putative tyrosine kinase receptor with oncogenic potential. Oncogene 1991, 6, 2113–2120. [Google Scholar]
- Vuoriluoto, K.; Haugen, H.; Kiviluoto, S.; Mpindi, J.P.; Nevo, J.; Gjerdrum, C.; Tiron, C.; Lorens, J.B.; Ivaska, J. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene 2011, 30, 1436–1448. [Google Scholar] [CrossRef] [Green Version]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaji, K.; Vijayaraghavan, S.; Diao, L.; Tong, P.; Fan, Y.; Carey, J.P.; Bui, T.N.; Warner, S.; Heymach, J.V.; Hunt, K.K.; et al. AXL Inhibition Suppresses the DNA Damage Response and Sensitizes Cells to PARP Inhibition in Multiple Cancers. Mol. Cancer Res. 2017, 15, 45–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gjerdrum, C.; Tiron, C.; Hoiby, T.; Stefansson, I.; Haugen, H.; Sandal, T.; Collett, K.; Li, S.; McCormack, E.; Gjertsen, B.T.; et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc. Natl. Acad. Sci. USA 2010, 107, 1124–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Suda, K.; Tomizawa, K.; Fujii, M.; Murakami, H.; Osada, H.; Maehara, Y.; Yatabe, Y.; Sekido, Y.; Mitsudomi, T. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J. Thorac. Oncol. 2011, 6, 1152–1161. [Google Scholar] [CrossRef] [Green Version]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; You, Y.; Jiang, H.; Wang, Z.Z. Epithelial-mesenchymal transition (EMT): A biological process in the development, stem cell differentiation, and tumorigenesis. J. Cell. Physiol. 2017, 232, 3261–3272. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Bansal, N.; Mishra, P.J.; Stein, M.; DiPaola, R.S.; Bertino, J.R. Axl receptor tyrosine kinase is up-regulated in metformin resistant prostate cancer cells. Oncotarget 2015, 6, 15321–15331. [Google Scholar] [CrossRef] [Green Version]
- Debruyne, D.N.; Bhatnagar, N.; Sharma, B.; Luther, W.; Moore, N.F.; Cheung, N.K.; Gray, N.S.; George, R.E. ALK inhibitor resistance in ALK(F1174L)-driven neuroblastoma is associated with AXL activation and induction of EMT. Oncogene 2016, 35, 3681–3691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cichon, M.A.; Szentpetery, Z.; Caley, M.P.; Papadakis, E.S.; Mackenzie, I.C.; Brennan, C.H.; O’Toole, E.A. The receptor tyrosine kinase Axl regulates cell-cell adhesion and stemness in cutaneous squamous cell carcinoma. Oncogene 2014, 33, 4185–4192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lay, J.D.; Hong, C.C.; Huang, J.S.; Yang, Y.Y.; Pao, C.Y.; Liu, C.H.; Lai, Y.P.; Lai, G.M.; Cheng, A.L.; Su, I.J.; et al. Sulfasalazine suppresses drug resistance and invasiveness of lung adenocarcinoma cells expressing AXL. Cancer Res. 2007, 67, 3878–3887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, K.Y.; Shieh, Y.S.; Lee, C.S.; Shiah, S.G.; Wu, C.W. Axl promotes cell invasion by inducing MMP-9 activity through activation of NF-kappaB and Brg-1. Oncogene 2008, 27, 4044–4055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 574. [Google Scholar] [CrossRef] [Green Version]
- Lawson, D.A.; Bhakta, N.R.; Kessenbrock, K.; Prummel, K.D.; Yu, Y.; Takai, K.; Zhou, A.; Eyob, H.; Balakrishnan, S.; Wang, C.Y.; et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015, 526, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Eom, H.; Kaushik, N.; Yoo, K.C.; Shim, J.K.; Kwon, M.; Choi, M.Y.; Yoon, T.; Kang, S.G.; Lee, S.J. MerTK mediates STAT3-KRAS/SRC-signaling axis for glioma stem cell maintenance. Artif. Cells Nanomed. Biotechnol. 2018, 46, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asiedu, M.K.; Beauchamp-Perez, F.D.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. AXL induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene 2014, 33, 1316–1324. [Google Scholar] [CrossRef] [Green Version]
- Cackowski, F.C.; Eber, M.R.; Rhee, J.; Decker, A.M.; Yumoto, K.; Berry, J.E.; Lee, E.; Shiozawa, Y.; Jung, Y.; Aguirre-Ghiso, J.A.; et al. Mer Tyrosine Kinase Regulates Disseminated Prostate Cancer Cellular Dormancy. J. Cell. Biochem. 2017, 118, 891–902. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.; Decker, A.M.; Wang, J.; Lee, E.; Kana, L.A.; Yumoto, K.; Cackowski, F.C.; Rhee, J.; Carmeliet, P.; Buttitta, L.; et al. Endogenous GAS6 and Mer receptor signaling regulate prostate cancer stem cells in bone marrow. Oncotarget 2016, 7, 25698–25711. [Google Scholar] [CrossRef] [Green Version]
- Ling, L.; Templeton, D.; Kung, H.J. Identification of the major autophosphorylation sites of Nyk/Mer, an NCAM-related receptor tyrosine kinase. J. Biol. Chem. 1996, 271, 18355–18362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinger, J.G.; Gohari, P.; Yan, Y.; Backer, J.M.; Varnum, B.; Shafit-Zagardo, B. In brain, Axl recruits Grb2 and the p85 regulatory subunit of PI3 kinase; in vitro mutagenesis defines the requisite binding sites for downstream Akt activation. J. Neurochem. 2008, 106, 134–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunger, J.; Schleithoff, L.; Schulz, A.S.; Kessler, H.; Lammers, R.; Ullrich, A.; Bartram, C.R.; Janssen, J.W. Intracellular signaling of the Ufo/Axl receptor tyrosine kinase is mediated mainly by a multi-substrate docking-site. Oncogene 1997, 14, 2619–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridell, Y.W.; Jin, Y.; Quilliam, L.A.; Burchert, A.; McCloskey, P.; Spizz, G.; Varnum, B.; Der, C.; Liu, E.T. Differential activation of the Ras/extracellular-signal-regulated protein kinase pathway is responsible for the biological consequences induced by the Axl receptor tyrosine kinase. Mol. Cell. Biol. 1996, 16, 135–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goruppi, S.; Ruaro, E.; Varnum, B.; Schneider, C. Requirement of phosphatidylinositol 3-kinase-dependent pathway and Src for Gas6-Axl mitogenic and survival activities in NIH 3T3 fibroblasts. Mol. Cell. Biol. 1997, 17, 4442–4453. [Google Scholar] [CrossRef] [Green Version]
- Rogers, A.E.; Le, J.P.; Sather, S.; Pernu, B.M.; Graham, D.K.; Pierce, A.M.; Keating, A.K. Mer receptor tyrosine kinase inhibition impedes glioblastoma multiforme migration and alters cellular morphology. Oncogene 2012, 31, 4171–4181. [Google Scholar] [CrossRef] [Green Version]
- Besser, D.; Bromberg, J.F.; Darnell, J.E., Jr.; Hanafusa, H. A single amino acid substitution in the v-Eyk intracellular domain results in activation of Stat3 and enhances cellular transformation. Mol. Cell. Biol. 1999, 19, 1401–1409. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, N.P.; Whang, Y.E.; Mohler, J.L.; Earp, H.S. Activated tyrosine kinase Ack1 promotes prostate tumorigenesis: Role of Ack1 in polyubiquitination of tumor suppressor Wwox. Cancer Res. 2005, 65, 10514–10523. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.K.; Secreto, C.; Boysen, J.; Sassoon, T.; Shanafelt, T.D.; Mukhopadhyay, D.; Kay, N.E. The novel receptor tyrosine kinase Axl is constitutively active in B-cell chronic lymphocytic leukemia and acts as a docking site of nonreceptor kinases: Implications for therapy. Blood 2011, 117, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Komurov, K.; Padron, D.; Cheng, T.; Roth, M.; Rosenblatt, K.P.; White, M.A. Comprehensive mapping of the human kinome to epidermal growth factor receptor signaling. J. Biol. Chem. 2010, 285, 21134–21142. [Google Scholar] [CrossRef] [Green Version]
- Zahuczky, G.; Kristof, E.; Majai, G.; Fesus, L. Differentiation and glucocorticoid regulated apopto-phagocytic gene expression patterns in human macrophages. Role of Mertk in enhanced phagocytosis. PLoS ONE 2011, 6, e21349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrens, E.M.; Gadue, P.; Gong, S.Y.; Garrett, S.; Stein, P.L.; Cohen, P.L. The mer receptor tyrosine kinase: Expression and function suggest a role in innate immunity. Eur. J. Immunol. 2003, 33, 2160–2167. [Google Scholar] [CrossRef]
- Zizzo, G.; Hilliard, B.A.; Monestier, M.; Cohen, P.L. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J. Immunol. 2012, 189, 3508–3520. [Google Scholar] [CrossRef] [Green Version]
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, K.V.; Amend, S.R.; Pienta, K.J. Targeting Tyro3, Axl and MerTK (TAM receptors): Implications for macrophages in the tumor microenvironment. Mol. Cancer 2019, 18, 94. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, N.P.; Earp, H.S. An SH2 domain-dependent, phosphotyrosine-independent interaction between Vav1 and the Mer receptor tyrosine kinase: A mechanism for localizing guanine nucleotide-exchange factor action. J. Biol. Chem. 2003, 278, 42596–42603. [Google Scholar] [CrossRef] [Green Version]
- Crittenden, M.R.; Baird, J.; Friedman, D.; Savage, T.; Uhde, L.; Alice, A.; Cottam, B.; Young, K.; Newell, P.; Nguyen, C.; et al. Mertk on tumor macrophages is a therapeutic target to prevent tumor recurrence following radiation therapy. Oncotarget 2016, 7, 78653–78666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodelja, V.; Muller, C.; Tenorio, S.; Schebesch, C.; Orfanos, C.E.; Goerdt, S. Differences in angiogenic potential of classically vs alternatively activated macrophages. Immunobiology 1997, 197, 478–493. [Google Scholar] [CrossRef]
- Jetten, N.; Verbruggen, S.; Gijbels, M.J.; Post, M.J.; De Winther, M.P.; Donners, M.M. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 2014, 17, 109–118. [Google Scholar] [CrossRef]
- Alciato, F.; Sainaghi, P.P.; Sola, D.; Castello, L.; Avanzi, G.C. TNF-alpha, IL-6, and IL-1 expression is inhibited by GAS6 in monocytes/macrophages. J. Leukoc. Biol. 2010, 87, 869–875. [Google Scholar] [CrossRef]
- Kim, S.Y.; Lim, E.J.; Yoon, Y.S.; Ahn, Y.H.; Park, E.M.; Kim, H.S.; Kang, J.L. Liver X receptor and STAT1 cooperate downstream of Gas6/Mer to induce anti-inflammatory arginase 2 expression in macrophages. Sci. Rep. 2016, 6, 29673. [Google Scholar] [CrossRef] [Green Version]
- Ito, K. Impact of post-translational modifications of proteins on the inflammatory process. Biochem. Soc. Trans. 2007, 35, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Covert, M.W.; Leung, T.H.; Gaston, J.E.; Baltimore, D. Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science 2005, 309, 1854–1857. [Google Scholar] [CrossRef]
- Cook, R.S.; Jacobsen, K.M.; Wofford, A.M.; DeRyckere, D.; Stanford, J.; Prieto, A.L.; Redente, E.; Sandahl, M.; Hunter, D.M.; Strunk, K.E.; et al. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J. Clin. Investig. 2013, 123, 3231–3242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [Green Version]
- Cabezon, R.; Carrera-Silva, E.A.; Florez-Grau, G.; Errasti, A.E.; Calderon-Gomez, E.; Lozano, J.J.; Espana, C.; Ricart, E.; Panes, J.; Rothlin, C.V.; et al. MERTK as negative regulator of human T cell activation. J. Leukoc. Biol. 2015, 97, 751–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, P.; Wallet, M.A.; Yi, Z.; Huang, Y.; Henderson, M.; Mathews, C.E.; Earp, H.S.; Matsushima, G.; Baldwin, A.S., Jr.; Tisch, R.M. Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-kappaB activation in dendritic cells. Blood 2007, 109, 653–660. [Google Scholar] [CrossRef] [Green Version]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, R.; Naka, T.; Tsutsui, H.; Fujimoto, M.; Kimura, A.; Abe, T.; Seki, E.; Sato, S.; Takeuchi, O.; Takeda, K.; et al. SOCS-1 participates in negative regulation of LPS responses. Immunity 2002, 17, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076. [Google Scholar] [CrossRef]
- Lumbroso, D.; Soboh, S.; Maimon, A.; Schif-Zuck, S.; Ariel, A.; Burstyn-Cohen, T. Macrophage-Derived Protein S Facilitates Apoptotic Polymorphonuclear Cell Clearance by Resolution Phase Macrophages and Supports Their Reprogramming. Front. Immunol. 2018, 9, 358. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fei, M.; Zhang, G.; Liang, W.C.; Lin, W.; Wu, Y.; Piskol, R.; Ridgway, J.; McNamara, E.; Huang, H.; et al. Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2X7R-Dependent STING Activation by Tumor-Derived cGAMP. Immunity 2020, 52, 357–373.e359. [Google Scholar] [CrossRef]
- Scutera, S.; Fraone, T.; Musso, T.; Cappello, P.; Rossi, S.; Pierobon, D.; Orinska, Z.; Paus, R.; Bulfone-Paus, S.; Giovarelli, M. Survival and migration of human dendritic cells are regulated by an IFN-alpha-inducible Axl/Gas6 pathway. J. Immunol. 2009, 183, 3004–3013. [Google Scholar] [CrossRef] [Green Version]
- Sharif, M.N.; Sosic, D.; Rothlin, C.V.; Kelly, E.; Lemke, G.; Olson, E.N.; Ivashkiv, L.B. Twist mediates suppression of inflammation by type I IFNs and Axl. J. Exp. Med. 2006, 203, 1891–1901. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, A.; Nishinakamura, H.; Matsumura, Y.; Hanada, T. Negative regulation of cytokine signaling and immune responses by SOCS proteins. Arthritis Res. Ther. 2005, 7, 100–110. [Google Scholar] [CrossRef] [Green Version]
- Mansell, A.; Smith, R.; Doyle, S.L.; Gray, P.; Fenner, J.E.; Crack, P.J.; Nicholson, S.E.; Hilton, D.J.; O’Neill, L.A.; Hertzog, P.J. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat. Immunol. 2006, 7, 148–155. [Google Scholar] [CrossRef]
- Zagorska, A.; Traves, P.G.; Lew, E.D.; Dransfield, I.; Lemke, G. Diversification of TAM receptor tyrosine kinase function. Nat. Immunol. 2014, 15, 920–928. [Google Scholar] [CrossRef] [Green Version]
- Du, W.; Huang, H.; Sorrelle, N.; Brekken, R.A. Sitravatinib potentiates immune checkpoint blockade in refractory cancer models. JCI Insight 2018, 3, e124184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolino, M.; Penninger, J.M. The Role of TAM Family Receptors in Immune Cell Function: Implications for Cancer Therapy. Cancers 2016, 8, 97. [Google Scholar] [CrossRef] [Green Version]
- Lee-Sherick, A.B.; Jacobsen, K.M.; Henry, C.J.; Huey, M.G.; Parker, R.E.; Page, L.S.; Hill, A.A.; Wang, X.; Frye, S.V.; Earp, H.S.; et al. MERTK inhibition alters the PD-1 axis and promotes anti-leukemia immunity. JCI Insight 2018, 3, e97941. [Google Scholar] [CrossRef] [PubMed]
- Kasikara, C.; Davra, V.; Calianese, D.; Geng, K.; Spires, T.E.; Quigley, M.; Wichroski, M.; Sriram, G.; Suarez-Lopez, L.; Yaffe, M.B.; et al. Pan-TAM tyrosine kinase inhibitor BMS-777607 enhances anti-PD-1 mAb efficacy in a murine model of triple-negative breast cancer. Cancer Res. 2019, 79, 2669–2683. [Google Scholar] [CrossRef] [Green Version]
- Tsukita, Y.; Fujino, N.; Miyauchi, E.; Saito, R.; Fujishima, F.; Itakura, K.; Kyogoku, Y.; Okutomo, K.; Yamada, M.; Okazaki, T.; et al. Axl kinase drives immune checkpoint and chemokine signalling pathways in lung adenocarcinomas. Mol. Cancer 2019, 18, 24. [Google Scholar] [CrossRef] [Green Version]
- Terry, S.; Dalban, C.; Rioux Leclercq, N.; Adam, J.; Meylan, M.; Buart, S.; Bougouin, A.; Lespagnol, A.; Dugay, F.; Colina Moreno, I.; et al. Association of AXL and PD-L1 expression with clinical outcomes in patients with advanced renal cell carcinoma treated with PD-1 blockade. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Terry, S.; Abdou, A.; Engelsen, A.S.T.; Buart, S.; Dessen, P.; Corgnac, S.; Collares, D.; Meurice, G.; Gausdal, G.; Baud, V.; et al. AXL Targeting Overcomes Human Lung Cancer Cell Resistance to NK- and CTL-Mediated Cytotoxicity. Cancer Immunol. Res. 2019, 7, 1789–1802. [Google Scholar] [CrossRef]
- Grabiec, A.M.; Goenka, A.; Fife, M.E.; Fujimori, T.; Hussell, T. Axl and MerTK receptor tyrosine kinases maintain human macrophage efferocytic capacity in the presence of viral triggers. Eur. J. Immunol. 2018, 48, 855–860. [Google Scholar] [CrossRef]
- Seitz, H.M.; Camenisch, T.D.; Lemke, G.; Earp, H.S.; Matsushima, G.K. Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J. Immunol. 2007, 178, 5635–5642. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Pellegatti, P.; Raffaghello, L.; Bianchi, G.; Piccardi, F.; Pistoia, V.; Di Virgilio, F. Increased level of extracellular ATP at tumor sites: In vivo imaging with plasma membrane luciferase. PLoS ONE 2008, 3, e2599. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surprenant, A.; Rassendren, F.; Kawashima, E.; North, R.A.; Buell, G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 1996, 272, 735–738. [Google Scholar] [CrossRef]
- Savio, L.E.B.; de Andrade Mello, P.; da Silva, C.G.; Coutinho-Silva, R. The P2X7 Receptor in Inflammatory Diseases: Angel or Demon? Front. Pharmacol. 2018, 9, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, B.R.; Lynch, K.J.; Touma, E.; Niforatos, W.; Burgard, E.C.; Alexander, K.M.; Park, H.S.; Yu, H.; Metzger, R.; Kowaluk, E.; et al. Pharmacological characterization of recombinant human and rat P2X receptor subtypes. Eur. J. Pharmacol. 1999, 376, 127–138. [Google Scholar] [CrossRef]
- Wang, H.; Hu, S.; Chen, X.; Shi, H.; Chen, C.; Sun, L.; Chen, Z.J. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc. Natl. Acad. Sci. USA 2017, 114, 1637–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanvetyanon, T.; Gray, J.E.; Antonia, S.J. PD-1 checkpoint blockade alone or combined PD-1 and CTLA-4 blockade as immunotherapy for lung cancer? Expert Opin. Biol. Ther. 2017, 17, 305–312. [Google Scholar] [CrossRef]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [Green Version]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [Green Version]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef] [Green Version]
- Riely, G.J.; Pao, W.; Pham, D.; Li, A.R.; Rizvi, N.; Venkatraman, E.S.; Zakowski, M.F.; Kris, M.G.; Ladanyi, M.; Miller, V.A. Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin. Cancer Res. 2006, 12, 839–844. [Google Scholar] [CrossRef] [Green Version]
- Jackman, D.M.; Yeap, B.Y.; Sequist, L.V.; Lindeman, N.; Holmes, A.J.; Joshi, V.A.; Bell, D.W.; Huberman, M.S.; Halmos, B.; Rabin, M.S.; et al. Exon 19 deletion mutations of epidermal growth factor receptor are associated with prolonged survival in non-small cell lung cancer patients treated with gefitinib or erlotinib. Clin. Cancer Res. 2006, 12, 3908–3914. [Google Scholar] [CrossRef] [Green Version]
- Ogino, A.; Kitao, H.; Hirano, S.; Uchida, A.; Ishiai, M.; Kozuki, T.; Takigawa, N.; Takata, M.; Kiura, K.; Tanimoto, M. Emergence of epidermal growth factor receptor T790M mutation during chronic exposure to gefitinib in a non small cell lung cancer cell line. Cancer Res. 2007, 67, 7807–7814. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balak, M.N.; Gong, Y.; Riely, G.J.; Somwar, R.; Li, A.R.; Zakowski, M.F.; Chiang, A.; Yang, G.; Ouerfelli, O.; Kris, M.G.; et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin. Cancer Res. 2006, 12, 6494–6501. [Google Scholar] [CrossRef] [Green Version]
- Janne, P.A.; Yang, J.C.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Ma, Y.; Yu, M.; Li, X.; Chen, X.; Gao, Y.; Cheng, P.; Zhang, G.; Wang, X. Identification of Hub Genes and Small Molecule Drugs Associated with Acquired Resistance to Gefitinib in Non-Small Cell Lung Cancer. J. Cancer 2021, 12, 5286–5295. [Google Scholar] [CrossRef]
- Namba, K.; Shien, K.; Takahashi, Y.; Torigoe, H.; Sato, H.; Yoshioka, T.; Takeda, T.; Kurihara, E.; Ogoshi, Y.; Yamamoto, H.; et al. Activation of AXL as a Preclinical Acquired Resistance Mechanism Against Osimertinib Treatment in EGFR-Mutant Non-Small Cell Lung Cancer Cells. Mol. Cancer Res. 2019, 17, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Bach, D.H.; Fan, Y.H.; Luu, T.T.; Hong, J.Y.; Park, H.J.; Lee, S.K. AXL degradation in combination with EGFR-TKI can delay and overcome acquired resistance in human non-small cell lung cancer cells. Cell Death Dis. 2019, 10, 361. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Liu, X.; Boris, A. Bartholdy, Haiying Cheng, Balazs Halmos. Blockade of AXL activation overcomes acquired resistance to EGFR tyrosine kinase inhibition in non-small cell lung cancer. Transl. Cancer Res. 2019, 8, 4. [Google Scholar] [CrossRef]
- Jimbo, T.; Hatanaka, M.; Komatsu, T.; Taira, T.; Kumazawa, K.; Maeda, N.; Suzuki, T.; Ota, M.; Haginoya, N.; Isoyama, T.; et al. DS-1205b, a novel selective inhibitor of AXL kinase, blocks resistance to EGFR-tyrosine kinase inhibitors in a non-small cell lung cancer xenograft model. Oncotarget 2019, 10, 5152–5167. [Google Scholar] [CrossRef] [Green Version]
- Giles, K.M.; Kalinowski, F.C.; Candy, P.A.; Epis, M.R.; Zhang, P.M.; Redfern, A.D.; Stuart, L.M.; Goodall, G.J.; Leedman, P.J. Axl mediates acquired resistance of head and neck cancer cells to the epidermal growth factor receptor inhibitor erlotinib. Mol. Cancer Ther. 2013, 12, 2541–2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.; Takahashi, M.; Chirieac, L.R.; Padera, R.F.; Shapiro, G.I.; Baum, A.; Himmelsbach, F.; et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008, 27, 4702–4711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelman, J.A.; Zejnullahu, K.; Gale, C.M.; Lifshits, E.; Gonzales, A.J.; Shimamura, T.; Zhao, F.; Vincent, P.W.; Naumov, G.N.; Bradner, J.E.; et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007, 67, 11924–11932. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Shimamura, T.; Ji, H.; Chen, L.; Haringsma, H.J.; McNamara, K.; Liang, M.C.; Perera, S.A.; Zaghlul, S.; Borgman, C.L.; et al. Bronchial and peripheral murine lung carcinomas induced by T790M-L858R mutant EGFR respond to HKI-272 and rapamycin combination therapy. Cancer Cell 2007, 12, 81–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.S.; Jang, W.J.; Chun, K.S.; Jeong, C.H. Hsp90 inhibition by WK88-1 potently suppresses the growth of gefitinib-resistant H1975 cells harboring the T790M mutation in EGFR. Oncol. Rep. 2014, 31, 2619–2624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, A.O.; Sjin, R.T.; Haringsma, H.J.; Ohashi, K.; Sun, J.; Lee, K.; Dubrovskiy, A.; Labenski, M.; Zhu, Z.; Wang, Z.; et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013, 3, 1404–1415. [Google Scholar] [CrossRef] [Green Version]
- Bean, J.; Riely, G.J.; Balak, M.; Marks, J.L.; Ladanyi, M.; Miller, V.A.; Pao, W. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin. Cancer Res. 2008, 14, 7519–7525. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.A.; Mirshahidi, S.; Mirshahidi, H.R. A novel insertion mutation on exon 20 of epidermal growth factor receptor, conferring resistance to erlotinib. Case Rep. Oncol. 2014, 7, 491–496. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef] [Green Version]
- Kubo, T.; Yamamoto, H.; Lockwood, W.W.; Valencia, I.; Soh, J.; Peyton, M.; Jida, M.; Otani, H.; Fujii, T.; Ouchida, M.; et al. MET gene amplification or EGFR mutation activate MET in lung cancers untreated with EGFR tyrosine kinase inhibitors. J. Int. Cancer 2009, 124, 1778–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, S.; Wang, W.; Li, Q.; Matsumoto, K.; Sakurama, H.; Nakamura, T.; Ogino, H.; Kakiuchi, S.; Hanibuchi, M.; Nishioka, Y.; et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008, 68, 9479–9487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guix, M.; Faber, A.C.; Wang, S.E.; Olivares, M.G.; Song, Y.; Qu, S.; Rinehart, C.; Seidel, B.; Yee, D.; Arteaga, C.L.; et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J. Clin. Investig. 2008, 118, 2609–2619. [Google Scholar] [CrossRef] [PubMed]
- Azuma, K.; Kawahara, A.; Sonoda, K.; Nakashima, K.; Tashiro, K.; Watari, K.; Izumi, H.; Kage, M.; Kuwano, M.; Ono, M.; et al. FGFR1 activation is an escape mechanism in human lung cancer cells resistant to afatinib, a pan-EGFR family kinase inhibitor. Oncotarget 2014, 5, 5908–5919. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.K.; Kim, S.; Lee, J.S.; Lee, J.E.; Kim, S.M.; Yang, I.S.; Kim, H.R.; Lee, J.H.; Kim, S.; Cho, B.C. Next-generation sequencing reveals novel resistance mechanisms and molecular heterogeneity in EGFR-mutant non-small cell lung cancer with acquired resistance to EGFR-TKIs. Lung Cancer 2017, 113, 106–114. [Google Scholar] [CrossRef]
- Ohashi, K.; Sequist, L.V.; Arcila, M.E.; Moran, T.; Chmielecki, J.; Lin, Y.L.; Pan, Y.; Wang, L.; de Stanchina, E.; Shien, K.; et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc. Natl. Acad. Sci. USA 2012, 109, E2127–E2133. [Google Scholar] [CrossRef] [Green Version]
- Schrock, A.B.; Zhu, V.W.; Hsieh, W.S.; Madison, R.; Creelan, B.; Silberberg, J.; Costin, D.; Bharne, A.; Bonta, I.; Bosemani, T.; et al. Receptor Tyrosine Kinase Fusions and BRAF Kinase Fusions are Rare but Actionable Resistance Mechanisms to EGFR Tyrosine Kinase Inhibitors. J. Thorac. Oncol. 2018, 13, 1312–1323. [Google Scholar] [CrossRef] [Green Version]
- de Bruin, E.C.; Cowell, C.; Warne, P.H.; Jiang, M.; Saunders, R.E.; Melnick, M.A.; Gettinger, S.; Walther, Z.; Wurtz, A.; Heynen, G.J.; et al. Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer Discov. 2014, 4, 606–619. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Yun, M.R.; Hong, Y.K.; Solca, F.; Kim, J.H.; Kim, H.J.; Cho, B.C. Glycolysis inhibition sensitizes non-small cell lung cancer with T790M mutation to irreversible EGFR inhibitors via translational suppression of Mcl-1 by AMPK activation. Mol. Cancer Ther. 2013, 12, 2145–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ercan, D.; Zhou, W.; Yanagita, M.; Capelletti, M.; Rogers, A.; Xiao, Y.; Gray, N.S.; Janne, P.A. Amplification of ERK2 mediates resistance to the novel irreversible EGFR inhibitor WZ4002. Cancer Res. 2011, 71, 4736. [Google Scholar] [CrossRef]
- Kim, S.M.; Kwon, O.J.; Hong, Y.K.; Kim, J.H.; Solca, F.; Ha, S.J.; Soo, R.A.; Christensen, J.G.; Lee, J.H.; Cho, B.C. Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol. Cancer Ther. 2012, 11, 2254–2264. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.; Fenoglio, S.; Gao, D.C.; Camiolo, M.; Stiles, B.; Lindsted, T.; Schlederer, M.; Johns, C.; Altorki, N.; Mittal, V.; et al. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 15535–15540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Smith, M.A.; Doshi, P.; Sasser, K.; Fulp, W.; Altiok, S.; Haura, E.B. Antitumor efficacy of the anti-interleukin-6 (IL-6) antibody siltuximab in mouse xenograft models of lung cancer. J. Thorac. Oncol. 2014, 9, 974–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piotrowska, Z.; Thress, K.S.; Mooradian, M.; Heist, R.S.; Azzoli, C.G.; Temel, J.S.; Rizzo, C.; Nagy, R.J.; Lanman, R.B.; Gettinger, S.N.; et al. MET amplification (amp) as a resistance mechanism to osimertinib. J. Clin. Oncol. 2017, 35, 9020. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, A.; Wu, Y.-L.; Han, J.-Y.; Ahn, M.-J.; Ramalingam, S.S.; John, T.; Okamoto, I.; Yang, J.C.-H.; Bulusu, K.C.; Laus, G.; et al. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann. Oncol. 2018, 29, VIII741. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Cheng, Y.; Zhou, C.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.-C.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann. Oncol. 2018, 29, VIII740. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Hu, Y.; Mileham, K.F.; Husain, H.; Costa, D.B.; Tracy, P.; Feeney, N.; Sholl, L.M.; Dahlberg, S.E.; Redig, A.J.; et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018, 4, 1527–1534. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, S.S.; Yang, J.C.; Lee, C.K.; Kurata, T.; Kim, D.W.; John, T.; Nogami, N.; Ohe, Y.; Mann, H.; Rukazenkov, Y.; et al. Osimertinib As First-Line Treatment of EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 841–849. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Yang, N.; Ou, Q.; Xiang, Y.; Jiang, T.; Wu, X.; Bao, H.; Tong, X.; Wang, X.; Shao, Y.W.; et al. Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non-Small Cell Lung Cancer Patients. Clin. Cancer Res. 2018, 24, 3097–3107. [Google Scholar] [CrossRef] [Green Version]
- Oztan, A.; Fischer, S.; Schrock, A.B.; Erlich, R.L.; Lovly, C.M.; Stephens, P.J.; Ross, J.S.; Miller, V.; Ali, S.M.; Ou, S.I.; et al. Emergence of EGFR G724S mutation in EGFR-mutant lung adenocarcinoma post progression on osimertinib. Lung Cancer 2017, 111, 84–87. [Google Scholar] [CrossRef]
- Fassunke, J.; Muller, F.; Keul, M.; Michels, S.; Dammert, M.A.; Schmitt, A.; Plenker, D.; Lategahn, J.; Heydt, C.; Bragelmann, J.; et al. Overcoming EGFR(G724S)-mediated osimertinib resistance through unique binding characteristics of second-generation EGFR inhibitors. Nat. Commun. 2018, 9, 4655. [Google Scholar] [CrossRef] [PubMed]
- Offin, M.; Somwar, R.; Rekhtman, N.; Benayed, R.; Chang, J.C.; Plodkowski, A.; Lui, A.J.W.; Eng, J.; Rosenblum, M.; Li, B.T.; et al. Acquired ALK and RET Gene Fusions as Mechanisms of Resistance to Osimertinib in EGFR-Mutant Lung Cancers. JCO Precis. Oncol. 2018, 2. [Google Scholar] [CrossRef] [PubMed]
- Klempner, S.J.; Bazhenova, L.A.; Braiteh, F.S.; Nikolinakos, P.G.; Gowen, K.; Cervantes, C.M.; Chmielecki, J.; Greenbowe, J.R.; Ross, J.S.; Stephens, P.J.; et al. Emergence of RET rearrangement co-existing with activated EGFR mutation in EGFR-mutated NSCLC patients who had progressed on first- or second-generation EGFR TKI. Lung Cancer 2015, 89, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef]
- Rosell, R.; Moran, T.; Queralt, C.; Porta, R.; Cardenal, F.; Camps, C.; Majem, M.; Lopez-Vivanco, G.; Isla, D.; Provencio, M.; et al. Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med. 2009, 361, 958–967. [Google Scholar] [CrossRef] [Green Version]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [Green Version]
- Schoenfeld, A.J.; Chan, J.M.; Kubota, D.; Sato, H.; Rizvi, H.; Daneshbod, Y.; Chang, J.C.; Paik, P.K.; Offin, M.; Arcila, M.E.; et al. Tumor Analyses Reveal Squamous Transformation and Off-Target Alterations As Early Resistance Mechanisms to First-line Osimertinib in EGFR-Mutant Lung Cancer. Clin. Cancer Res. 2020, 26. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Yamada, T.; Kita, K.; Taniguchi, H.; Arai, S.; Fukuda, K.; Terashima, M.; Ishimura, A.; Nishiyama, A.; Tanimoto, A.; et al. Transient IGF-1R inhibition combined with osimertinib eradicates AXL-low expressing EGFR mutated lung cancer. Nat. Commun. 2020, 11, 4607. [Google Scholar] [CrossRef]
- Minson, K.A.; Smith, C.C.; DeRyckere, D.; Libbrecht, C.; Lee-Sherick, A.B.; Huey, M.G.; Lasater, E.A.; Kirkpatrick, G.D.; Stashko, M.A.; Zhang, W.; et al. The MERTK/FLT3 inhibitor MRX-2843 overcomes resistance-conferring FLT3 mutations in acute myeloid leukemia. JCI Insight 2016, 1, e85630. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Pham, P.C.; Pei, H.; Lim, B.; Hyun, S.Y.; Baek, B.; Kim, B.; Kim, Y.; Kim, M.H.; Kang, N.W.; et al. Development of the phenylpyrazolo[3,4-d]pyrimidine-based, insulin-like growth factor receptor/Src/AXL-targeting small molecule kinase inhibitor. Theranostics 2021, 11, 1918–1936. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.R.; Dong, Y.J.; Wu, H.B.; Yu, D.P.; Zhou, L.J.; Su, D.; Zhang, L.; Chen, X.J. Expression level of CRKL and AXL combined with exon 19 deletion in EGFR and ALK status confer differential prognosis of lung adenocarcinoma subtypes. Oncol. Lett. 2016, 12, 3312–3322. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Yang, S.; Wang, K.; Sun, S.Y. MET inhibitors for targeted therapy of EGFR TKI-resistant lung cancer. J. Hematol. Oncol. 2019, 12, 63. [Google Scholar] [CrossRef]
- Rotow, J.; Bivona, T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef]
- Molina-Arcas, M.; Moore, C.; Rana, S.; van Maldegem, F.; Mugarza, E.; Romero-Clavijo, P.; Herbert, E.; Horswell, S.; Li, L.S.; Janes, M.R.; et al. Development of combination therapies to maximize the impact of KRAS-G12C inhibitors in lung cancer. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Lew, E.D.; Seelige, R.; Tindall, E.A.; Walsh, C.; Fagan, P.C.; Lee, J.Y.; Nevarez, R.; Oh, J.; Tucker, K.D.; et al. Immuno-oncological Efficacy of RXDX-106, a Novel TAM (TYRO3, AXL, MER) Family Small-Molecule Kinase Inhibitor. Cancer Res. 2019, 79, 1996–2008. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Liu, R.; Ma, B.; Li, X.; Yen, H.Y.; Zhou, Y.; Krasnoperov, V.; Xia, Z.; Zhang, X.; Bove, A.M.; et al. Axl receptor tyrosine kinase is a potential therapeutic target in renal cell carcinoma. Br. J. Cancer 2015, 113, 616–625. [Google Scholar] [CrossRef] [Green Version]
- Leconet, W.; Larbouret, C.; Chardes, T.; Thomas, G.; Neiveyans, M.; Busson, M.; Jarlier, M.; Radosevic-Robin, N.; Pugniere, M.; Bernex, F.; et al. Preclinical validation of AXL receptor as a target for antibody-based pancreatic cancer immunotherapy. Oncogene 2014, 33, 5405–5414. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.X.; Knyazev, P.G.; Cheburkin, Y.V.; Sharma, K.; Knyazev, Y.P.; Orfi, L.; Szabadkai, I.; Daub, H.; Keri, G.; Ullrich, A. AXL is a potential target for therapeutic intervention in breast cancer progression. Cancer Res. 2008, 68, 1905–1915. [Google Scholar] [CrossRef] [Green Version]
- Park, I.K.; Mishra, A.; Chandler, J.; Whitman, S.P.; Marcucci, G.; Caligiuri, M.A. Inhibition of the receptor tyrosine kinase Axl impedes activation of the FLT3 internal tandem duplication in human acute myeloid leukemia: Implications for Axl as a potential therapeutic target. Blood 2013, 121, 2064–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kariolis, M.S.; Miao, Y.R.; Jones, D.S., 2nd; Kapur, S.; Mathews, I.I.; Giaccia, A.J.; Cochran, J.R. An engineered Axl ‘decoy receptor’ effectively silences the Gas6-Axl signaling axis. Nat. Chem. Biol. 2014, 10, 977–983. [Google Scholar] [CrossRef]
- Cerchia, L.; Esposito, C.L.; Camorani, S.; Rienzo, A.; Stasio, L.; Insabato, L.; Affuso, A.; de Franciscis, V. Targeting Axl with an high-affinity inhibitory aptamer. Mol. Ther. 2012, 20, 2291–2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahnert, J.R.; Taylor, M.H.; O’Reilly, E.M.; Zhang, J.; Doebele, R.C.; Ben, Y.; Sharp, L.L.; Boyle, W.J.; Chang, C.; Frey, G.; et al. A phase 1/2 dose-escalation and expansion study of a conditionally active anti-AXL humanized monoclonal antibody (BA3011) in patients with advanced solid tumors. J. Clin. Oncol. 2018, 36, TPS12126. [Google Scholar] [CrossRef]
- Sharp, L.L.; Chang, C.; Frey, G.; Wang, J.; Liu, H.; Xing, C.; Yalcin, S.; Walls, M.; Ben, Y.; Boyle, W.J.; et al. Abstract 827: Anti-tumor efficacy of BA3011, a novel Conditionally Active Biologic (CAB) anti-AXL-ADC. Cancer Res. 2018, 78. [Google Scholar] [CrossRef]
- Bonifacio, L.; Dodds, M.; Prohaska, D.; Moss, A.; Giaccia, A.; Tabibiazar, R.; McIntyre, G. Target-Mediated Drug Disposition Pharmacokinetic/Pharmacodynamic Model-Informed Dose Selection for the First-in-Human Study of AVB-S6-500. Clin. Transl. Sci. 2020, 13, 204–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schepisi, G.; Conteduca, V.; Casadei, C.; Gurioli, G.; Rossi, L.; Galla, V.; Cursano, M.C.; Brighi, N.; Lolli, C.; Menna, C.; et al. Potential Application of Chimeric Antigen Receptor (CAR)-T Cell Therapy in Renal Cell Tumors. Front. Oncol. 2020, 10, 565857. [Google Scholar] [CrossRef]
- Burbridge, M.F.; Bossard, C.J.; Saunier, C.; Fejes, I.; Bruno, A.; Leonce, S.; Ferry, G.; Da Violante, G.; Bouzom, F.; Cattan, V.; et al. S49076 is a novel kinase inhibitor of MET, AXL, and FGFR with strong preclinical activity alone and in association with bevacizumab. Mol. Cancer Ther. 2013, 12, 1749–1762. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, G.M.; An, Y.; Cai, Z.W.; Chen, X.T.; Clark, C.; Cornelius, L.A.; Dai, J.; Gullo-Brown, J.; Gupta, A.; Henley, B.; et al. Discovery of N-(4-(2-amino-3-chloropyridin-4-yloxy)-3-fluorophenyl)-4-ethoxy-1-(4-fluorophenyl)-2-oxo-1,2-dihydropyridine-3-carboxamide (BMS-777607), a selective and orally efficacious inhibitor of the Met kinase superfamily. J. Med. Chem. 2009, 52, 1251–1254. [Google Scholar] [CrossRef]
- Yan, S.B.; Peek, V.L.; Ajamie, R.; Buchanan, S.G.; Graff, J.R.; Heidler, S.A.; Hui, Y.H.; Huss, K.L.; Konicek, B.W.; Manro, J.R.; et al. LY2801653 is an orally bioavailable multi-kinase inhibitor with potent activity against MET, MST1R, and other oncoproteins, and displays anti-tumor activities in mouse xenograft models. Investig. New Drugs 2013, 31, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaulieu, N.; Sainte-Croix, H.; Bonfils, C.; Mannion, M.; Raeppel, S.; Isakovic, L.; Claridge, S.; Saavedra, O.; Raeppel, F.; Vaisburg, A.; et al. Abstract 930: Preclinical charaterization of MG516, a novel inhibitor of receptor tyrosine kinases involved in resistance to targeted therapies. Cancer Res. 2013, 73, 930. [Google Scholar]
- Patyna, S.; Laird, A.D.; Mendel, D.B.; O’Farrell, A.M.; Liang, C.; Guan, H.; Vojkovsky, T.; Vasile, S.; Wang, X.; Chen, J.; et al. SU14813: A novel multiple receptor tyrosine kinase inhibitor with potent antiangiogenic and antitumor activity. Mol. Cancer Ther. 2006, 5, 1774–1782. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Jeng, Y.M.; Chen, Y.L.; Chung, L.; Yuan, R.H. Gas6/Axl pathway promotes tumor invasion through the transcriptional activation of Slug in hepatocellular carcinoma. Carcinogenesis 2014, 35, 769–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mita, M.; Gordon, M.; Rosen, L.; Kapoor, N.; Choy, G.; Redkar, S.; Taverna, P.; Oganesian, A.; Sahai, A.; Azab, M.; et al. Phase 1B study of amuvatinib in combination with five standard cancer therapies in adults with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 74, 195–204. [Google Scholar] [CrossRef]
- Dhillon, S. Gilteritinib: First Global Approval. Drugs 2019, 79, 331–339. [Google Scholar] [CrossRef]
- Padda, S.; Neal, J.W.; Wakelee, H.A. MET inhibitors in combination with other therapies in non-small cell lung cancer. Transl. Lung Cancer Res. 2012, 1, 238–253. [Google Scholar] [CrossRef]
- Liu, L.; Wang, Q.; Xie, C.; Xi, N.; Guo, Z.; Li, M.; Hou, X.; Xie, N.; Sun, M.; Li, J.; et al. Drug interaction of ningetinib and gefitinib involving CYP1A1 and efflux transporters in non-small cell lung cancer patients. Br. J. Clin. Pharmacol. 2021, 87, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Wunk-Lipinska, K.; Tiron, C.; Gausdal, G.; Sandal, T.; Frink, R.; Hinz, S.; Hellesøy, M.; Ahmed, L.; Haugen, H.; Liang, X.; et al. Abstract 1747: BGB324, a selective small molecule Axl kinase inhibitor to overcome EMT-associated drug resistance in carcinomas: Therapeutic rationale and early clinical studies. Cancer Res. 2014, 74. [Google Scholar] [CrossRef]
- Wang, X.; Saso, H.; Iwamoto, T.; Xia, W.; Gong, Y.; Pusztai, L.; Woodward, W.A.; Reuben, J.M.; Warner, S.L.; Bearss, D.J.; et al. TIG1 promotes the development and progression of inflammatory breast cancer through activation of Axl kinase. Cancer Res. 2013, 73, 6516–6525. [Google Scholar] [CrossRef] [Green Version]
- Novello, S.; Camps, C.; Grossi, F.; Mazieres, J.; Abrey, L.; Vernejoux, J.M.; Thall, A.; Patyna, S.; Usari, T.; Wang, Z.; et al. Phase II study of sunitinib in patients with non-small cell lung cancer and irradiated brain metastases. J. Thorac. Oncol. 2011, 6, 1260–1266. [Google Scholar] [CrossRef] [Green Version]
- Qian, F.; Engst, S.; Yamaguchi, K.; Yu, P.; Won, K.A.; Mock, L.; Lou, T.; Tan, J.; Li, C.; Tam, D.; et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 2009, 69, 8009–8016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, C.L.; Cerchia, L.; Catuogno, S.; De Vita, G.; Dassie, J.P.; Santamaria, G.; Swiderski, P.; Condorelli, G.; Giangrande, P.H.; de Franciscis, V. Multifunctional aptamer-miRNA conjugates for targeted cancer therapy. Mol. Ther. 2014, 22, 1151–1163. [Google Scholar] [CrossRef] [Green Version]
- Rankin, E.B.; Fuh, K.C.; Castellini, L.; Viswanathan, K.; Finger, E.C.; Diep, A.N.; LaGory, E.L.; Kariolis, M.S.; Chan, A.; Lindgren, D.; et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc. Natl. Acad. Sci. USA 2014, 111, 13373–13378. [Google Scholar] [CrossRef] [Green Version]
- Kariolis, M.S.; Miao, Y.R.; Diep, A.; Nash, S.E.; Olcina, M.M.; Jiang, D.; Jones, D.S., 2nd; Kapur, S.; Mathews, I.I.; Koong, A.C.; et al. Inhibition of the GAS6/AXL pathway augments the efficacy of chemotherapies. J. Clin. Investig. 2017, 127, 183–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.J.; Mok, T.S.; Chen, Z.H.; Guo, A.L.; Zhang, X.C.; Su, J.; Wu, Y.L. Clinicopathologic and molecular features of epidermal growth factor receptor T790M mutation and c-MET amplification in tyrosine kinase inhibitor-resistant Chinese non-small cell lung cancer. Pathol. Oncol. Res. 2009, 15, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.M.; Jang, Y.; Lee, S.H.; Kang, B.; Lim, S.M. AXL/MET dual inhibitor, CB469, has activity in non-small cell lung cancer with acquired resistance to EGFR TKI with AXL or MET activation. Lung Cancer 2020, 146, 70–77. [Google Scholar] [CrossRef]
- Feneyrolles, C.; Spenlinhauer, A.; Guiet, L.; Fauvel, B.; Dayde-Cazals, B.; Warnault, P.; Cheve, G.; Yasri, A. Axl kinase as a key target for oncology: Focus on small molecule inhibitors. Mol. Cancer Ther. 2014, 13, 2141–2148. [Google Scholar] [CrossRef] [Green Version]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef]
- Wu, J.; Frady, L.N.; Bash, R.E.; Cohen, S.M.; Schorzman, A.N.; Su, Y.T.; Irvin, D.M.; Zamboni, W.C.; Wang, X.; Frye, S.V.; et al. MerTK as a therapeutic target in glioblastoma. Neuro-Oncology 2018, 20, 92–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Cruz, P.M.; Yasumura, D.; Weir, J.; Matthes, M.T.; Abderrahim, H.; LaVail, M.M.; Vollrath, D. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum. Mol. Genet. 2000, 9, 645–651. [Google Scholar] [CrossRef] [Green Version]
- Angelillo-Scherrer, A.; de Frutos, P.; Aparicio, C.; Melis, E.; Savi, P.; Lupu, F.; Arnout, J.; Dewerchin, M.; Hoylaerts, M.; Herbert, J.; et al. Deficiency or inhibition of Gas6 causes platelet dysfunction and protects mice against thrombosis. Nat. Med. 2001, 7, 215–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sather, S.; Kenyon, K.D.; Lefkowitz, J.B.; Liang, X.; Varnum, B.C.; Henson, P.M.; Graham, D.K. A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood 2007, 109, 1026–1033. [Google Scholar] [CrossRef]
- Branchford, B.R.; Stalker, T.J.; Law, L.; Acevedo, G.; Sather, S.; Brzezinski, C.; Wilson, K.M.; Minson, K.; Lee-Sherick, A.B.; Davizon-Castillo, P.; et al. The small-molecule MERTK inhibitor UNC2025 decreases platelet activation and prevents thrombosis. J. Thromb. Haemost. 2018, 16, 352–363. [Google Scholar] [CrossRef]
- DeRyckere, D.; Lee-Sherick, A.B.; Huey, M.G.; Hill, A.A.; Tyner, J.W.; Jacobsen, K.M.; Page, L.S.; Kirkpatrick, G.G.; Eryildiz, F.; Montgomery, S.A.; et al. UNC2025, a MERTK Small-Molecule Inhibitor, Is Therapeutically Effective Alone and in Combination with Methotrexate in Leukemia Models. Clin. Cancer Res. 2017, 23, 1481–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
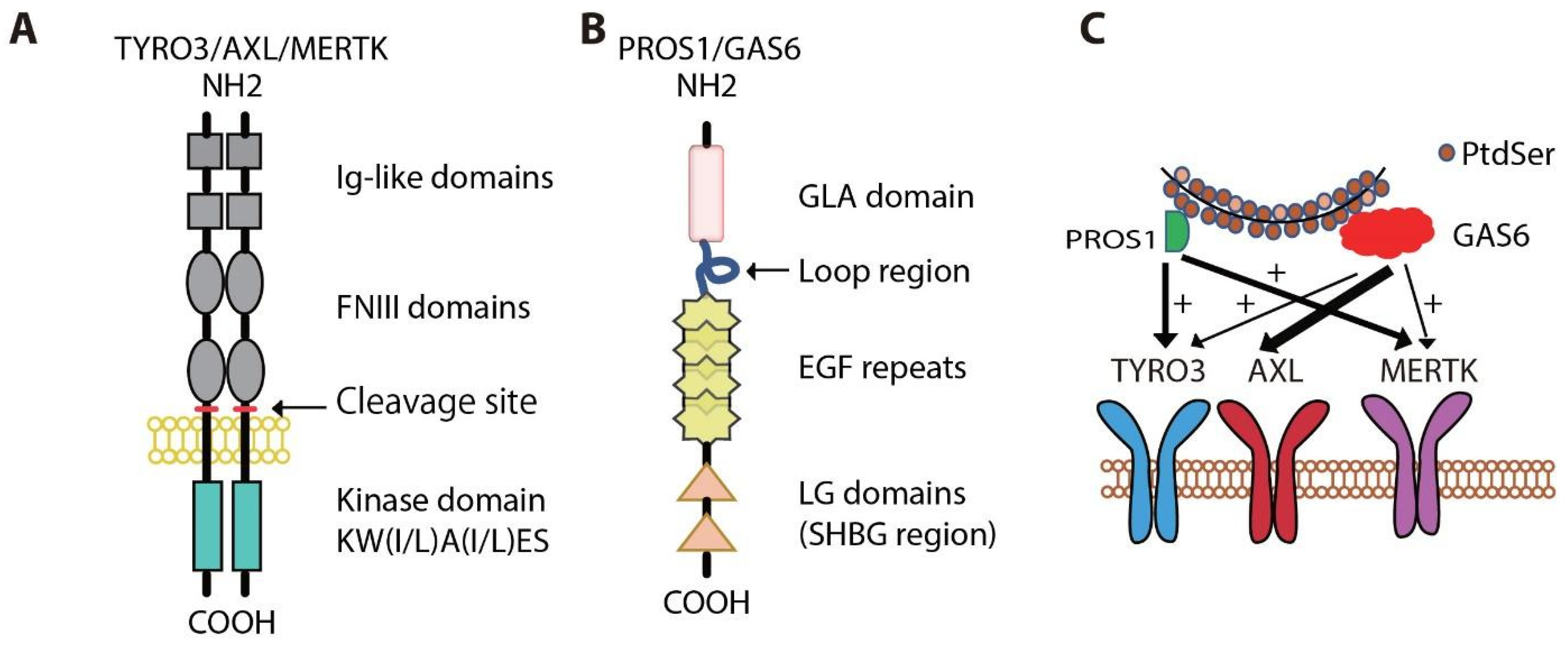



{kind=link}
{kind=link}
{kind=link}
| EGFR TKI | Mechanism of Resistance | Treatment | Reference |
|---|---|---|---|
| Gefitinib/erlotinib | EGFR T790M mutation | afatinib, afatinib + rapamycin, CI-387,785, dacomitinib, HKI-272, CO-1686, osimertinib, WK88-1, afatinib + MET inhibitor ARQ 197, PF00299804 | [192,195,202,203,204,205,206,207,208] |
| EGFR T854A mutation | BIBW2992 | [209] | |
| EGFR exon 20 mutation | [210] | ||
| EMT | MS-275 + erlotinib | [116] | |
| MET amp/hepatocyte growth factor (HGF) overexpression | PHA-665752 + gefitinib, XL880, crizotinib + afatinib or WZ4002, NPS-1034 + gefitinib or erlotinib | [73,205,211,212,213,214] | |
| IGF-1R overexpression | AEW541 + gefitinib | [215] | |
| FGFR1 overexpression/FGFR3 fusion | PD173074 + afatinib | [216] | |
| Small cell lung cancer (SCLC) transformation | Standard SCLC treatments | [117,205] | |
| HER2 amp/mutation | [205] | ||
| EGFR T263P/G719A | afatinib | [217] | |
| NRAS mutation/BRAF mutation/BRAF fusion | MEK inhibitor AZD6244 + erlotinib or BRAF inhibitor vemurafenib + erlotinib | [218,219] | |
| Reduced neurofibromin | MEK inhibitor AZD6244 + erlotinib | [220] | |
| CCDC6-RET fusion | [219] | ||
| Glucose metabolism | 2-deoxy-D-glucose + afatinib | [221] | |
| WZ4002 | ERK2 amp | MEK inhibitor CI-1040 + WZ4002 | [222] |
| Afatinib | IL-6R/JAK1/STAT3 activation or TGF-β- IL-6 axis activation | Pyridone 6 + afatinib | [223,224,225] |
| FGFR3 fusion | [219] | ||
| RET fusion | [219] | ||
| ALK fusion | [219] | ||
| Osimertinib | MET amp/HGF overexpression | [27,226,227,228,229] | |
| EGFR C797S mutation | [27,29,226,227,228,229,230,231] | ||
| EGFR C724S mutation | afatinib | [232,233] | |
| RET fusion | RET inhibitor BLU-667 + osimertinib | [27,229,234,235] | |
| PCBP2-BRAF fusion | MEK inhibitor trametinib | [27] | |
| SCLC transformation | [226,229] | ||
| FGFR1 mutation/FGFR1 amp/FGFR3-TACC3 fusion | [226,229] | ||
| KRAS mutation | [229,230] | ||
| PIK3CA mutation/PIK3CA amp | [226,227,228,229,230] | ||
| HER2 amp/HER2 insertion | [227,228,230] | ||
| BRAF mutation/BRAF fusion | [219,229] | ||
| EGFR T790M loss | [229,230] | ||
| ALK fusion | [219,234] | ||
| JAK2 mutation | [230] |
| Compound | Known Targets | Phase | AXL IC50 | Reference |
|---|---|---|---|---|
| Monoclonal antibody | ||||
| YW327.6S2 | AXL-specific | Preclinical | 340 ng/mL | [70] |
| 12A11 | AXL-specific | Preclinical | ~100 ng/mL | [64] |
| MA b173 | AXL-specific | Preclinical | Unk | [250] |
| D9 and E8 | AXL-specific | Preclinical | Unk | [251] |
| AXL polyclonal antibody | AXL-specific | Preclinical | Unk | [252] |
| Mer590 | MERTK-specific | Preclinical | 6.25 ng/mL | [66,136] |
| Recombinant Protein | ||||
| AXL-Fc | AXL, MERTK, TYRO3 | preclinical | Unk | [37,253] |
| MERTK-Fc | AXL, MERTK, TYRO3 | Preclinical | Unk | [37] |
| Decoy receptor | ||||
| AXL “decoy receptor” | GAS6 | Preclinical | 0.5 mg/kg | [254] |
| Aptamer GL21.T | AXL-specific | Preclinical | 13 nM (Kd) | [255] |
| Antibody-drug conjugate | AXL | I/II | [256] | |
| BA3011/CAB-AXL-ADC | Unk | |||
| Antibody-drug conjugate | AXL | I/II | 0.02–2 µg/mL | [92] |
| HuMax-AXL-ADC | (in vitro) | |||
| Antibody-drug conjugate | AXL | I/II | [257] | |
| CAB-AXL-ADC | Unk | |||
| AXL-Fc | GAS6 | I/II/III | [258] | |
| AVB-S6-500 | Unk | |||
| CAR-T | AXL | I/II | Unk | [259] |
| CCT301-38 |
| Compound | Known Targets | Phase | AXL IC50 | MERTK IC50 | NCT Number | References |
|---|---|---|---|---|---|---|
| MRX-2843 | MERTK, FLT3 | I/Ib | 15 nM (in vitro) | 1.3 nM (in vitro) | NCT03510104 NCT04762199 | [67,69,242] |
| DS-1205c | I | 1.3 nM (in vitro) | 63 nM (in vitro) | NCT03255083 NCT03599518 | [200] | |
| S49076 | AXL, MET, EGFR, ISRC, FGFR1/2/3 | I/II | 7 nM (in vitro) | 2 nM (in vitro) | ISRCTN00759419 | [260] |
| ASLAN002 (BMS-777607) | AXL, MERTK, and MET | I/II | 1.1 nM (in vitro) | 16 nM (in vitro) | NCT01721148 NCT00605618 | [261] |
| LY2801653 | AXL, MET, MST1R | I | 2 nM (in vitro) | 10 nM (in vitro) | NCT01285037 | [262] |
| INCB081776 | AXL, MERTK | I | 0.61 nM (in vitro) | 3.17 nM (in vitro) | NCT03522142 | [83] |
| Sitravatinib (MGCD516) | AXL, MET, RET, TRK, DDR2, KDR, PDGFRA, Kit | I | 1.5 nM (in vitro) | 2 nM (in vitro) | NCT02219711 | [263] |
| SU14813 | FLT3, VEGFR, PDGFR, Kit | I | 84 nM (in vitro) | 66 nM (in vitro) | NCT00982267 | [264] |
| RXDX106 | AXL, MERTK, TYRO3, MET | I | 0.69 nM (in vitro) | 1.89 nM (in vitro) | NCT03454243 | [249] |
| Bosutinib (SKI-606/PF-5208763) | AXL, Src, AbI, TGFB, BMP | I/II | 0.56 µM (in vitro) | Unk | NCT00195260 NCT00319254 | [265] |
| Amuvatinib (MP470) | AXL, c-KIT, PDGFR, FLT3, RAD51, RET | I/Ib/II | <1 µM (in cells) | Unk | NCT00894894 NCT00881166 NCT01357395 | [266] |
| Gilteritinib (ASP2215) | AXL, FLT3 | I/II/III | <1 nM (in vitro) | Unk | NCT02014558 NCT02421939 NCT02752035 NCT02927262 NCT02997202 NCT03182244 NCT02561455 NCT02456883 | [253,267] |
| Glesatinib (MGCD265) | AXL, MET, VEGFR | I/II | Unk | Unk | NCT00697632 NCT00975767 | [268] |
| Ningetinib | VEGFR2, MET, AXL, MERTK, FLT3, RON | I/II | <1 nM (in vitro) | Unk | NCT03758287 NCT04577703 | [269] |
| Merestinib (LY2801653) | MET, RON, FLT3, AXL | I/II | 2 nM (in vitro) | 10 nM (in vitro) | NCT01285037 NCT03027284 NCT02711553 | [262] |
| BGB324 (R428) | AXL | I/II | 14 nM (in vitro) <30 nM (in cells) | 224 nM (in vitro) | NCT024 24617 NCT02488408 NCT02872259 NCT02922777 NCT03184558 NCT03184571 NCT03649321 NCT03654833 | [270] |
| Crizotinib (PF-02341066) | ALK, MET, ROS1, AXL | Ib/II | 0.3 µM | Unk | NCT02034981 NCT02511184 | [24] |
| TP-0903 | AXL | I/II | 27 nM (in vitro) 222 nM (in cells) | Unk | NCT02729298 NCT03572634 | [113] |
| ONO-7475 | AXL, MERTK | I/II | 0.7 nM (in vitro) | 1 nM (in vitro) | NCT03176277 NCT03730337 | [82] |
| SGI-7079 | AXL | II | 58 nM (in vitro) | Unk | NCT00409968 | [112,271] |
| Sunitinib (SU11248) | KIT, FLT3, PDGFR, VEGFR2, AXL | II | 9 nM (in vitro) | Unk | NCT01499121 NCT01034878 NCT00864721 | [272] |
| Foretinib (GSK1363089/XL880) | MET, AXL, VEGFR2, RON, Tie-2 | II | 11 nM (in vitro) | Unk | NCT01068587 | [273] |
| Cabozantinib (XL184) | AXL, MET, VEGFR2, RET, Kit, Flt-1/3/4, Tie2 | II/III | 7 nM (in vitro) 42 nM (in cells) | Unk | NCT01639508 NCT01708954 NCT01866410 | [274] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, D.; Earp, H.S.; DeRyckere, D.; Graham, D.K. Targeting MERTK and AXL in EGFR Mutant Non-Small Cell Lung Cancer. Cancers 2021, 13, 5639. https://doi.org/10.3390/cancers13225639
Yan D, Earp HS, DeRyckere D, Graham DK. Targeting MERTK and AXL in EGFR Mutant Non-Small Cell Lung Cancer. Cancers. 2021; 13(22):5639. https://doi.org/10.3390/cancers13225639
Chicago/Turabian StyleYan, Dan, H. Shelton Earp, Deborah DeRyckere, and Douglas K. Graham. 2021. "Targeting MERTK and AXL in EGFR Mutant Non-Small Cell Lung Cancer" Cancers 13, no. 22: 5639. https://doi.org/10.3390/cancers13225639
APA StyleYan, D., Earp, H. S., DeRyckere, D., & Graham, D. K. (2021). Targeting MERTK and AXL in EGFR Mutant Non-Small Cell Lung Cancer. Cancers, 13(22), 5639. https://doi.org/10.3390/cancers13225639