
Gp-100 as a Novel Therapeutic Target in Uveal Melanoma


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Discussion
3.1. Glycoprotein 100
3.2. Vaccines Targeting Gp-100 Peptides
3.3. Adoptive Cell Therapy Targeting Gp-100
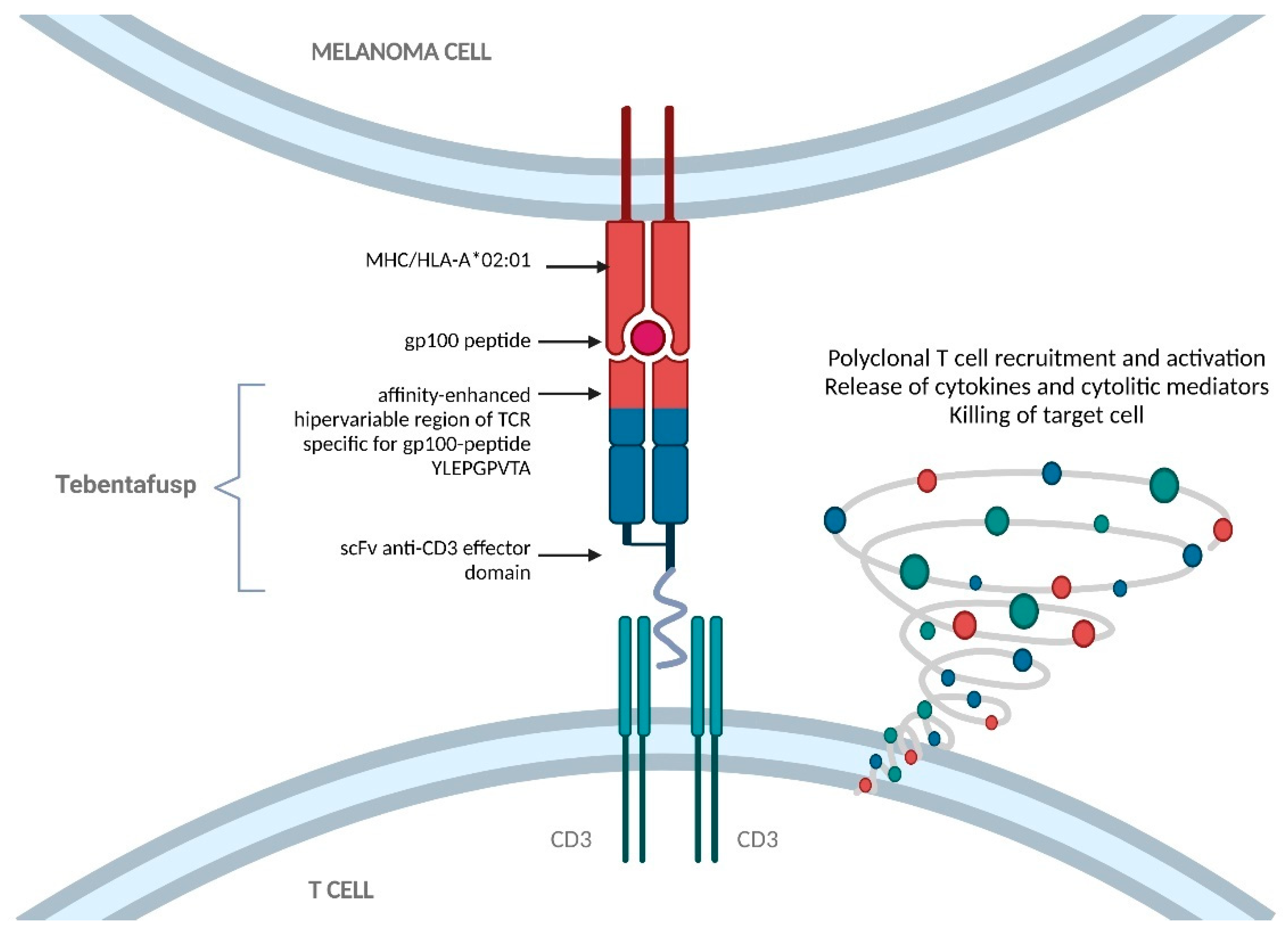
3.4. Tebentafusp: Gp-100 Directed ImmTAC
3.4.1. Preclinical Development of ImmTACs
3.4.2. Preclinical Development of Tebentafusp
3.4.3. Clinical Development of Tebentafusp
3.4.4. Toxicity Profile of Tebentafusp
3.4.5. Possible Mechanisms of Resistance to ImmTACs
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jager, M.J.; Shields, C.L.; Cebulla, C.M.; Abdel-Rahman, M.H.; Grossniklaus, H.E.; Stern, M.-H.; Carvajal, R.D.; Belfort, R.N.; Jia, R.; Shields, J.A.; et al. Uveal melanoma. Nat. Rev. Dis. Primers 2020, 6, 24. [Google Scholar] [CrossRef]
- Virgili, G.; Gatta, G.; Ciccolallo, L.; Capocaccia, R.; Biggeri, A.; Crocetti, E.; Lutz, J.-M.; Paci, E.; EUROCARE Working Group. Incidence of uveal melanoma in Europe. Ophthalmology 2007, 114, 2309–2315.e2. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Kujala, E.; Mäkitie, T.; Kivelä, T. Very long-term prognosis of patients with malignant uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4651–4659. [Google Scholar] [CrossRef] [Green Version]
- Carvajal, R.D.; Piperno-Neumann, S.; Kapiteijn, E.; Chapman, P.B.; Frank, S.; Joshua, A.; Piulats, J.M.; Wolter, P.; Cocquyt, V.; Chmielowski, B.; et al. Selumetinib in Combination with dacarbazine in patients with metastatic uveal melanoma: A phase III, multicenter, randomized trial (SUMIT). J. Clin. Oncol. 2018, 36, 1232–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piulats, J.M.; Espinosa, E.; Merino, L.D.L.C.; Varela, M.; Carrión, L.A.; Martín-Algarra, S.; Castro, R.L.; Curiel, T.; Rodríguez-Abreu, D.; Redrado, M.; et al. Nivolumab plus ipilimumab for treatment-naïve metastatic uveal melanoma: An open-label, multicenter, Phase II trial by the Spanish multidisciplinary melanoma group (GEM-1402). J. Clin. Oncol. 2021, 39, 586–598. [Google Scholar] [CrossRef]
- Pelster, M.S.; Gruschkus, S.K.; Bassett, R.; Gombos, D.S.; Shephard, M.; Posada, L.; Glover, M.S.; Simien, R.; Diab, A.; Hwu, P.; et al. Nivolumab and ipilimumab in metastatic uveal melanoma: Results from a single-arm phase II study. J. Clin. Oncol. 2021, 39, 599–607. [Google Scholar] [CrossRef]
- Nathan, P.; Ascierto, P.A.; Haanen, J.; Espinosa, E.; Demidov, L.; Garbe, C.; Guida, M.; Lorigan, P.; Sileni, V.C.; Gogas, H.; et al. Safety and efficacy of nivolumab in patients with rare melanoma subtypes who progressed on or after ipilimumab treatment: A single-arm, open-label, phase II study (CheckMate 172). Eur. J. Cancer 2019, 119, 168–178. [Google Scholar] [CrossRef]
- Wagner, S.N.; Wagner, C.; Schultewolter, T.; Goos, M. Analysis of Pmel17/gp100 expression in primary human tissue specimens: Implications for melanoma immuno- and gene-therapy. Cancer Immunol. Immunother. 1997, 44, 239–247. [Google Scholar] [CrossRef]
- Nathan, P.; Hassel, J.C.; Rutkowski, P.; Baurain, J.-F.; Butler, M.O.; Schlaak, M.; Sullivan, R.J.; Ochsenreither, S.; Dummer, R.; Kirkwood, J.M.; et al. Overall survival benefit with tebentafusp in metastatic uveal melanoma. N. Engl. J. Med. 2021, 385, 1196–1206. [Google Scholar] [CrossRef]
- De Vries, T.; Trančikova, D.; Ruiter, D.; Van Muijen, G. High expression of immunotherapy candidate proteins gp100, MART-1, tyrosinase and TRP-1 in uveal melanoma. Br. J. Cancer 1998, 78, 1156–1161. [Google Scholar] [CrossRef] [Green Version]
- Ohsie, S.J.; Sarantopoulos, G.P.; Cochran, A.J.; Binder, S.W. Immunohistochemical characteristics of melanoma. J. Cutan. Pathol. 2008, 35, 433–444. [Google Scholar] [CrossRef]
- Kawakami, Y.; Rosenberg, S.A. Immunobiology of human melanoma antigens MART-1 and gp100 and their use for immuno-gene therapy. Int. Rev. Immunol. 1997, 14, 173–192. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.-H.; Seipp, C.A.; et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, Y.; Eliyahu, S.; Jennings, C.; Sakaguchi, K.; Kang, X.; Southwood, S.; Rosenberg, S.A. Recognition of multiple epitopes in the human melanoma antigen gp100 by tumor-infiltrating T lymphocytes associated with in vivo tumor regression. J. Immunol. 1995, 154, 3961–3968. [Google Scholar]
- Barker, A.B.H.; Schreurs, M.W.J.; Tafazzul, G.; De Boer, A.J.; Kawakami, Y.; Adema, G.J.; Figdor, C.G. Identification of a novel peptide derived from the melanocyte-specific gp100 antigen as the dominant epitope recognized by an HLA-A2.1-restricted anti-melanoma CTL line. Int. J. Cancer 1995, 62, 97–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartzentruber, D.J.; Lawson, D.H.; Richards, J.M.; Conry, R.M.; Miller, D.M.; Treisman, J.; Gailani, F.; Riley, L.; Conlon, K.; Pockaj, B.; et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N. Engl. J. Med. 2011, 364, 2119–2127. [Google Scholar] [CrossRef] [Green Version]
- Van Lint, S.; Wilgenhof, S.; Heirman, C.; Corthals, J.; Breckpot, K.; Bonehill, A.; Neyns, B.; Thielemans, K. Optimized dendritic cell-based immunotherapy for melanoma: The trimix-formula. Cancer Immunol. Immunother. 2014, 63, 959–967. [Google Scholar] [CrossRef]
- Jansen, Y.; Kruse, V.; Corthals, J.; Schats, K.; Van Dam, P.-J.; Seremet, T.; Heirman, C.; Brochez, L.; Kockx, M.; Thielemans, K.; et al. A randomized controlled phase II clinical trial on mRNA electroporated autologous monocyte-derived dendritic cells (TriMixDC-MEL) as adjuvant treatment for stage III/IV melanoma patients who are disease-free following the resection of macrometastases. Cancer Immunol. Immunother. 2020, 69, 2589–2598. [Google Scholar] [CrossRef] [PubMed]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; Van Baren, N.; Lucas, S.; Kvistborg, P.; Thielemans, K.; Neyns, B. Phase II study of autologous monocyte-derived mRNA electroporated dendritic cells (TriMixDC-MEL) plus ipilimumab in patients with pretreated advanced melanoma. J. Clin. Oncol. 2016, 34, 1330–1338. [Google Scholar] [CrossRef]
- Targeted Oncology (19 February 2021). Tebentafusp Granted FDA Breakthrough Therapy Designation for Unresectable or Metastatic Uveal Melanoma [Press Release]. Available online: https://www.targetedonc.com/view/tebentafusp-granted-fda-breakthrough-therapy-designation-for-unresectable-or-metastatic-uveal-melanoma (accessed on 30 September 2021).
- Damato, B.E.; Dukes, J.; Goodall, H.; Carvajal, R.D. Tebentafusp: T Cell redirection for the treatment of metastatic uveal melanoma. Cancers 2019, 11, 971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddy, N.; Bossi, G.; Adams, K.J.; Lissina, A.; Mahon, T.M.; Hassan, N.J.; Gavarret, J.; Bianchi, F.C.; Pumphrey, N.J.; Ladell, K.; et al. Monoclonal TCR-redirected tumor cell killing. Nat. Med. 2012, 18, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Oates, J.; Hassan, N.J.; Jakobsen, B.K. ImmTACs for targeted cancer therapy: Why, what, how, and which. Mol. Immunol. 2015, 67 Pt A, 67–74. [Google Scholar] [CrossRef]
- Ellis, J.M.; Henson, V.; Slack, R.; Ng, J.; Hartzman, R.J.; Hurley, C.K. Frequencies of HLA-A2 alleles in five U.S. population groups: Predominance of A∗02011 and identification of HLA-A∗0231. Hum. Immunol. 2000, 61, 334–340. [Google Scholar] [CrossRef]
- Immunocore 2021. Pipeline. Available online: https://www.immunocore.com/our-science/pipeline (accessed on 19 October 2021).
- Boudousquie, C.; Bossi, G.; Hurst, J.M.; Rygiel, K.A.; Jakobsen, B.K.; Hassan, N.J. Polyfunctional response by ImmTAC (IMCgp100) redirected CD8+ and CD4+ T cells. Immunology 2017, 152, 425–438. [Google Scholar] [CrossRef] [Green Version]
- Bossi, G.; Buisson, S.; Oates, J.; Jakobsen, B.K.; Hassan, N.J. ImmTAC-redirected tumour cell killing induces and potentiates antigen cross-presentation by dendritic cells. Cancer Immunol. Immunother. 2014, 63, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Middleton, M.R.; McAlpine, C.; Woodcock, V.K.; Corrie, P.; Infante, J.R.; Steven, N.M.; Evans, T.R.J.; Anthoney, A.; Shoushtari, A.N.; Hamid, O.; et al. Tebentafusp, a TCR/anti-CD3 bispecific fusion protein targeting gp100, potently activated antitumor immune responses in patients with metastatic melanoma. Clin. Cancer Res. 2020, 26, 5869–5878. [Google Scholar] [CrossRef] [PubMed]
- Groom, J.R.; Luster, A.D. CXCR3 in T cell function. Exp. Cell Res. 2011, 317, 620–631. [Google Scholar] [CrossRef]
- Sacco, J.; Carvajal, R.; Butler, M.; Shoushtari, A.; Hassel, J.; Ikeguchi, A.; Hernandez-Aya, L.; Nathan, P.; Hamid, O.; Rodriguez, J.P.; et al. A Phase (ph) II, Multi-Center Study of the Safety and Efficacy of Tebentafusp (tebe) (IMCgp100) in Patients (pts) with Metastatic Uveal Melanoma (mUM); Oxford University Press: Oxford, UK, 2020. [Google Scholar] [CrossRef]
- Shoushtari, A.; Collins, L.; Espinosa, E.; Sethi, H.; Stanhope, S.; Abdullah, S.; Ikeguchi, A.; Ranade, K.; Hamid, O. 1757O Early reduction in ctDNA, regardless of best RECIST response, is associated with overall survival (OS) on tebentafusp in previously treated metastatic uveal melanoma (mUM) patients. Ann. Oncol. 2021, 32 (Suppl. 5), S1211–S1226. [Google Scholar] [CrossRef]
- De la Cruz Merino, L.; Eroglu, Z.; Collins, L.; Greenshields-Watson, A.; Stanhope, S.; Abdullah, S.; Sacco, J. 1770P—Genomic correlates of clinical outcomes in patients with metastatic uveal melanoma treated with tebentafusp. Ann. Oncol. 2021, 32 (Suppl. 5), S829–S866. [Google Scholar] [CrossRef]
- Piulats, J.M.; Sato, T.; Luke, J.J.; Collins, L.; Edukulla, R.; Abdullah, S.E.; Leyvraz, S. 1013P—Similar overall survival in tebentafusp-treated 2L+ metastatic uveal melanoma regardless of prior immunotherapy. Ann. Oncol. 2021, 32 (Suppl. 5), S829–S866. [Google Scholar] [CrossRef]
- Stäger, R.; Stanhope, S.; Greenshields-Watson, A.; Collins, L.; Ramelyte, E.; Kolm, I.; Meier-Schiesser, B. 1772P—Demonstration of T cell redirection and immune activation in skin rash following tebentafusp treatment. Ann. Oncol. 2021, 32 (Suppl. 5), S1211–S1226. [Google Scholar] [CrossRef]
- Chandran, S.S.; Klebanoff, C.A. T cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance. Immunol. Rev. 2019, 290, 127–147. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, K.; Depoil, D.; Gascoyne, D.M.; Page, K.; Curnock, A.; Benlahrech, A. 1016P—ImmTAC redirect exhausted tumor-infiltrating T-cells: An effect enhanced by pembrolizumab against PD-L1+ tumors. Ann. Oncol. 2021, 32 (Suppl. 5), S829–S866. [Google Scholar] [CrossRef]


{kind=link}
| Clinical Trial | Design | Disease and N | DCR and ORR (%) | PFS | OS |
|---|---|---|---|---|---|
| IMCGp-100-01 [27] | Phase I | mUM (n = 19) mCM (n = 61) | 57% and 14% 18% and 6% | - | mOS: 33.4 months 1Y-OS: 65% |
| IMCGp-100-102 [29] | Phase I cohort | heavily pretreated mUM n = 19 | 31% and 10% | 1Y-PFS: 66% | mOS: NR 1Y-OS: 74% |
| Phase II cohort | pretreated (1 line) mUM n = 130 | 50% and 5% | 2.8 months | mOS: 16.8 months 1Y-OS: 62% | |
| IMCGp-100-202 [7] | Controlled phase III | untreated mUM (n = 378) T (n = 252) vs. ICOT (n = 126) | T: 46% and 9% ICOT: 27% and 5% | 3.3 vs. 2.9 months (HR, 0.73; 95% CI, 0.58–0.94) | 21.7 vs. 16.0 months (HR, 0.51; 95% CI, 0.37–0.71) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez-Perez, D.; Viñal, D.; Solares, I.; Espinosa, E.; Feliu, J. Gp-100 as a Novel Therapeutic Target in Uveal Melanoma. Cancers 2021, 13, 5968. https://doi.org/10.3390/cancers13235968
Martinez-Perez D, Viñal D, Solares I, Espinosa E, Feliu J. Gp-100 as a Novel Therapeutic Target in Uveal Melanoma. Cancers. 2021; 13(23):5968. https://doi.org/10.3390/cancers13235968
Chicago/Turabian StyleMartinez-Perez, Daniel, David Viñal, Isabel Solares, Enrique Espinosa, and Jaime Feliu. 2021. "Gp-100 as a Novel Therapeutic Target in Uveal Melanoma" Cancers 13, no. 23: 5968. https://doi.org/10.3390/cancers13235968
APA StyleMartinez-Perez, D., Viñal, D., Solares, I., Espinosa, E., & Feliu, J. (2021). Gp-100 as a Novel Therapeutic Target in Uveal Melanoma. Cancers, 13(23), 5968. https://doi.org/10.3390/cancers13235968