
The Immune Landscape of Breast Cancer: Strategies for Overcoming Immunotherapy Resistance


Abstract
:Simple Summary
Abstract
1. Introduction
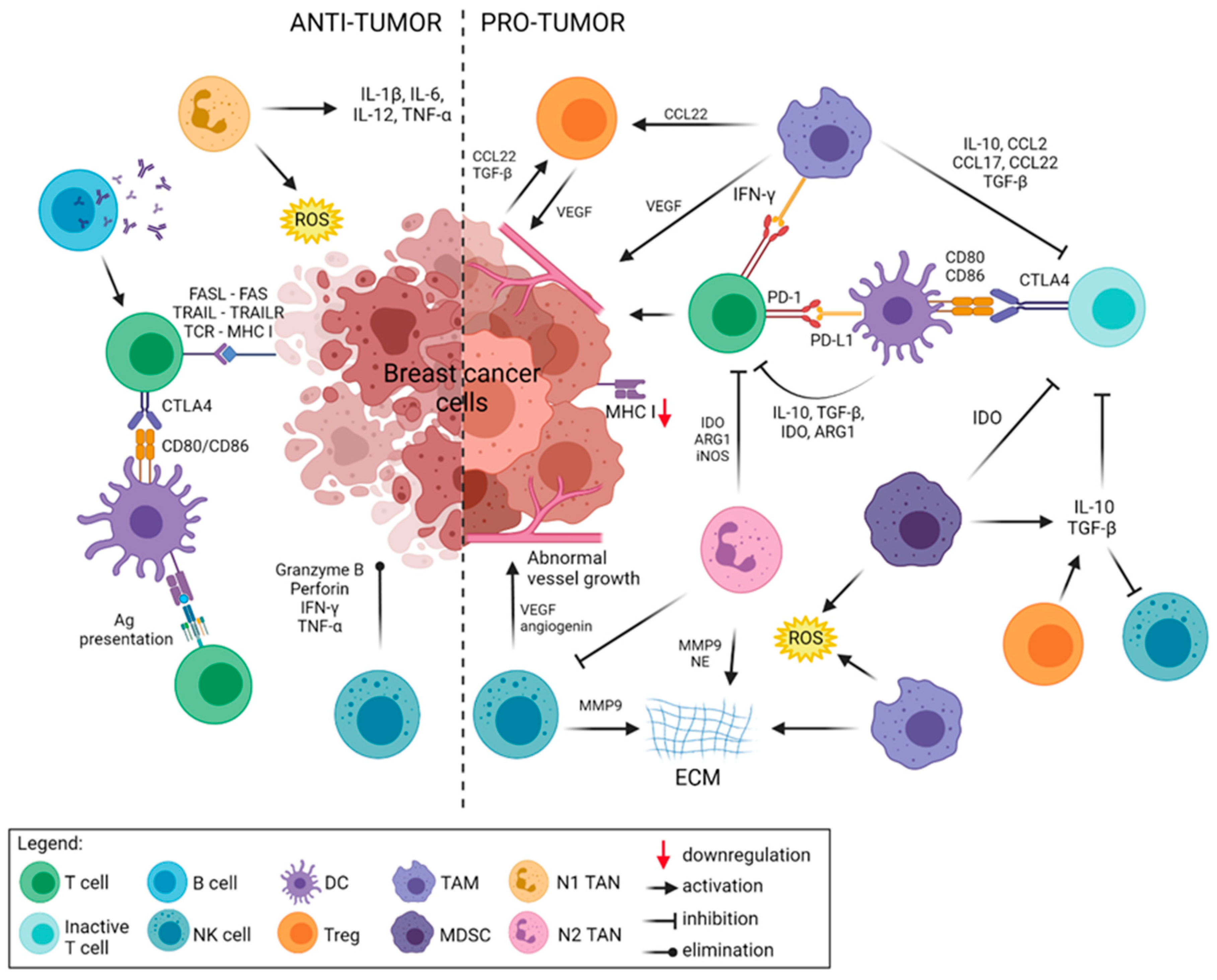
2. Immune Landscape of Breast Cancers
2.1. Components of the Antitumor Immunity
2.2. Tumor Promoting Mechanisms in Breast Cancer
2.2.1. Intrinsic Resistance Mechanisms
2.2.2. Tumor-Extrinsic Factors and Mechanisms of Immune Resistance
2.2.3. Environmental Factors Attributed to Cancer Development
3. Immunotherapeutic Strategies in Breast Cancer
3.1. Passive Immunotherapy: Monoclonal Antibodies for Breast Cancer Treatment
3.2. Active Immunotherapies in Breast Cancer
3.2.1. Combination Therapies in Breast Cancer
3.2.2. Prognostic Significance of Immune Cells in Breast Cancer Immunotherapy
3.3. Adoptive Cell Therapies in Breast Cancer
3.3.1. CAR-Based Therapies in Breast Cancer: Successes and Challenges
3.3.2. Limitations of Adoptive Cell Therapies
3.4. Alternative Treatment Approaches in Breast Cancer
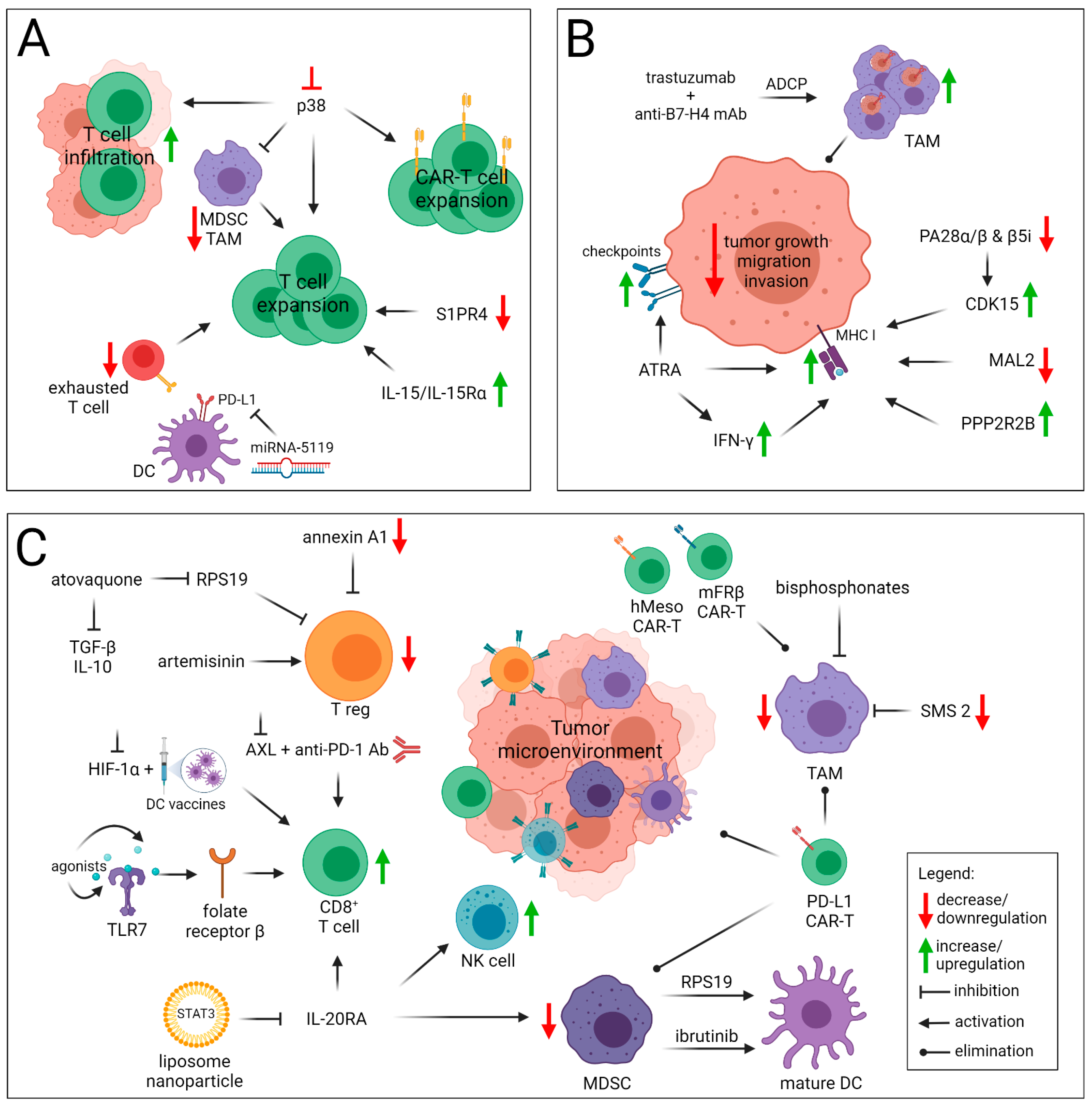
4. Strategies to Overcome Immunotherapy Resistance of Breast Cancer
4.1. Enhancing T Cells Priming and Trafficking within the Tumor
4.2. Improving Antigen Presentation
4.3. Overcoming the Immunosuppressive TME
4.4. Strategies to Overcome CAR-T Resistance
5. Conclusions
6. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Clusan, L.; Le Goff, P.; Flouriot, G.; Pakdel, F. A Closer Look at Estrogen Receptor Mutations in Breast Cancer and Their Implications for Estrogen and Antiestrogen Responses. Int. J. Mol. Sci. 2021, 22, 756. [Google Scholar] [CrossRef]
- Jensen, E.V.; Jordan, V.C. The estrogen receptor: A model for molecular medicine. Clin. Cancer Res. 2003, 9, 1980–1989. [Google Scholar] [PubMed]
- Rimawi, M.F.; Schiff, R.; Osborne, C.K. Targeting HER2 for the treatment of breast cancer. Annu. Rev. Med. 2015, 66, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Saura, C.; Bellet, M.; Munoz-Couselo, E.; Ramirez-Merino, N.; Calvo, V.; Perez, J.; Vidal, M. HER2 and hormone receptor-positive breast cancer--blocking the right target. Nat. Rev. Clin. Oncol. 2011, 8, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salpeter, S.R.; Malter, D.S.; Luo, E.J.; Lin, A.Y.; Stuart, B. Systematic review of cancer presentations with a median survival of six months or less. J. Palliat. Med. 2012, 15, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planes-Laine, G.; Rochigneux, P.; Bertucci, F.; Chretien, A.S.; Viens, P.; Sabatier, R.; Goncalves, A. PD-1/PD-L1 Targeting in Breast Cancer: The First Clinical Evidences Are Emerging. A Literature Review. Cancers 2019, 11, 1033. [Google Scholar] [CrossRef] [Green Version]
- Hartkopf, A.D.; Taran, F.A.; Wallwiener, M.; Walter, C.B.; Kramer, B.; Grischke, E.M.; Brucker, S.Y. PD-1 and PD-L1 Immune Checkpoint Blockade to Treat Breast Cancer. Breast Care 2016, 11, 385–390. [Google Scholar] [CrossRef] [Green Version]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Miles, D.; Kim, S.B.; Im, Y.H.; Im, S.A.; Semiglazov, V.; Ciruelos, E.; Schneeweiss, A.; Loi, S.; Monturus, E.; et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA): End-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 519–530. [Google Scholar] [CrossRef]
- Sadelain, M. Chimeric Antigen Receptors: A Paradigm Shift in Immunotherapy. Annu. Rev. Cancer Biol. 2017, 1, 447–466. [Google Scholar] [CrossRef]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef] [PubMed]
- Soysal, S.D.; Tzankov, A.; Muenst, S.E. Role of the Tumor Microenvironment in Breast Cancer. Pathobiology 2015, 82, 142–152. [Google Scholar] [CrossRef]
- Luen, S.J.; Savas, P.; Fox, S.B.; Salgado, R.; Loi, S. Tumour-infiltrating lymphocytes and the emerging role of immunotherapy in breast cancer. Pathology 2017, 49, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Brown, N.J.; Holen, I. The breast tumor microenvironment: Role in cancer development, progression and response to therapy. Expert Rev. Mol. Diagn. 2018, 18, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Stanton, S.E.; Disis, M.L. Clinical significance of tumor-infiltrating lymphocytes in breast cancer. J. Immunother. Cancer 2016, 4, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, N.R.; Milne, K.; Truong, P.T.; Macpherson, N.; Nelson, B.H.; Watson, P.H. Tumor-infiltrating lymphocytes predict response to anthracycline-based chemotherapy in estrogen receptor-negative breast cancer. Breast Cancer Res. 2011, 13, R126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, W.; Lakkis, F.G.; Chalasani, G. B Cells, Antibodies, and More. Clin. J. Am. Soc. Nephrol. 2016, 11, 137–154. [Google Scholar] [CrossRef] [PubMed]
- Garaud, S.; Buisseret, L.; Solinas, C.; Gu-Trantien, C.; de Wind, A.; Van den Eynden, G.; Naveaux, C.; Lodewyckx, J.N.; Boisson, A.; Duvillier, H.; et al. Tumor infiltrating B-cells signal functional humoral immune responses in breast cancer. JCI Insight 2019, 4, e129641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiLillo, D.J.; Yanaba, K.; Tedder, T.F. B cells are required for optimal CD4+ and CD8+ T cell tumor immunity: Therapeutic B cell depletion enhances B16 melanoma growth in mice. J. Immunol. 2010, 184, 4006–4016. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.Y.; Fu, T.; Jiang, Y.Z.; Shao, Z.M. Natural killer cells in cancer biology and therapy. Mol. Cancer 2020, 19, 120. [Google Scholar] [CrossRef] [PubMed]
- Salemme, V.; Centonze, G.; Cavallo, F.; Defilippi, P.; Conti, L. The Crosstalk between Tumor Cells and the Immune Microenvironment in Breast Cancer: Implications for Immunotherapy. Front. Oncol. 2021, 11, 610303. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Bassani, B.; D’Urso, D.G.; Pitaku, I.; Cassinotti, E.; Pelosi, G.; Boni, L.; Dominioni, L.; Noonan, D.M.; Mortara, L.; et al. Angiogenin and the MMP9-TIMP2 axis are up-regulated in proangiogenic, decidual NK-like cells from patients with colorectal cancer. FASEB J. 2018, 32, 5365–5377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levi, I.; Amsalem, H.; Nissan, A.; Darash-Yahana, M.; Peretz, T.; Mandelboim, O.; Rachmilewitz, J. Characterization of tumor infiltrating natural killer cell subset. Oncotarget 2015, 6, 13835–13843. [Google Scholar] [CrossRef] [Green Version]
- Lapeyre-Prost, A.; Terme, M.; Pernot, S.; Pointet, A.L.; Voron, T.; Tartour, E.; Taieb, J. Immunomodulatory Activity of VEGF in Cancer. Int. Rev. Cell Mol. Biol. 2017, 330, 295–342. [Google Scholar] [CrossRef]
- Segovia-Mendoza, M.; Morales-Montor, J. Immune Tumor Microenvironment in Breast Cancer and the Participation of Estrogen and Its Receptors in Cancer Physiopathology. Front. Immunol. 2019, 10, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fainaru, O.; Almog, N.; Yung, C.W.; Nakai, K.; Montoya-Zavala, M.; Abdollahi, A.; D’Amato, R.; Ingber, D.E. Tumor growth and angiogenesis are dependent on the presence of immature dendritic cells. FASEB J. 2010, 24, 1411–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sisirak, V.; Faget, J.; Gobert, M.; Goutagny, N.; Vey, N.; Treilleux, I.; Renaudineau, S.; Poyet, G.; Labidi-Galy, S.I.; Goddard-Leon, S.; et al. Impaired IFN-alpha production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res. 2012, 72, 5188–5197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, J.P.; Derakhshandeh, R.; Jones, L.; Webb, T.J. Mechanisms of immune evasion in breast cancer. BMC Cancer 2018, 18, 556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haen, S.P.; Loffler, M.W.; Rammensee, H.G.; Brossart, P. Towards new horizons: Characterization, classification and implications of the tumour antigenic repertoire. Nat. Rev. Clin. Oncol. 2020, 17, 595–610. [Google Scholar] [CrossRef]
- Criscitiello, C. Tumor-associated antigens in breast cancer. Breast Care 2012, 7, 262–266. [Google Scholar] [CrossRef] [Green Version]
- Viborg, N.; Ramskov, S.; Andersen, R.S.; Sturm, T.; Fugmann, T.; Bentzen, A.K.; Rafa, V.M.; Straten, P.T.; Svane, I.M.; Met, O.; et al. T cell recognition of novel shared breast cancer antigens is frequently observed in peripheral blood of breast cancer patients. Oncoimmunology 2019, 8, e1663107. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tang, X.; Lu, M.; Tang, Q.; Zhang, H.; Zhu, H.; Xu, N.; Zhang, D.; Xiong, L.; Mao, Y.; et al. Overexpression of MAGE-A9 predicts unfavorable outcome in breast cancer. Exp. Mol. Pathol. 2014, 97, 579–584. [Google Scholar] [CrossRef]
- Rodriguez, J.A. HLA-mediated tumor escape mechanisms that may impair immunotherapy clinical outcomes via T-cell activation. Oncol. Lett. 2017, 14, 4415–4427. [Google Scholar] [CrossRef] [Green Version]
- Vitale, M.; Rezzani, R.; Rodella, L.; Zauli, G.; Grigolato, P.; Cadei, M.; Hicklin, D.J.; Ferrone, S. HLA class I antigen and transporter associated with antigen processing (TAP1 and TAP2) down-regulation in high-grade primary breast carcinoma lesions. Cancer Res. 1998, 58, 737–742. [Google Scholar] [PubMed]
- Kim, H.M.; Lee, J.; Koo, J.S. Clinicopathological and prognostic significance of programmed death ligand-1 expression in breast cancer: A meta-analysis. BMC Cancer 2017, 17, 690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Yang, J.; Jiao, S.; Li, Y.; Zhang, W.; Wang, J. Cytotoxic T lymphocyte antigen 4 expression in human breast cancer: Implications for prognosis. Cancer Immunol. Immunother. 2015, 64, 853–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Zhang, C.; Song, Y.; Wang, Z.; Wang, Y.; Luo, F.; Xu, Y.; Zhao, Y.; Wu, Z.; Xu, Y. Mechanism of immune evasion in breast cancer. Onco Targets Ther. 2017, 10, 1561–1573. [Google Scholar] [CrossRef] [Green Version]
- Nicolini, A.; Ferrari, P.; Rossi, G.; Carpi, A. Tumour growth and immune evasion as targets for a new strategy in advanced cancer. Endocr. Relat. Cancer 2018, 25, R577–R604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.; Jene, N.; Byrne, D.; Millar, E.K.; O’Toole, S.A.; McNeil, C.M.; Bates, G.J.; Harris, A.L.; Banham, A.H.; Sutherland, R.L.; et al. Recruitment of regulatory T cells is correlated with hypoxia-induced CXCR4 expression, and is associated with poor prognosis in basal-like breast cancers. Breast Cancer Res. 2011, 13, R47. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Shao, N.; Aierken, N.; Xie, C.; Ye, R.; Qian, X.; Hu, Z.; Zhang, J.; Lin, Y. Prognostic value of tumor-infiltrating Foxp3+ regulatory T cells in patients with breast cancer: A meta-analysis. J. Cancer 2017, 8, 4098–4105. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.L.; Li, L.; Guo, Y.W.; Yu, P.; Yin, X.J.; Wang, S.; Liu, C.P. CD8(+) cytotoxic and FoxP3(+) regulatory T lymphocytes serve as prognostic factors in breast cancer. Am. J. Transl. Res. 2019, 11, 5039–5053. [Google Scholar] [PubMed]
- Olkhanud, P.B.; Damdinsuren, B.; Bodogai, M.; Gress, R.E.; Sen, R.; Wejksza, K.; Malchinkhuu, E.; Wersto, R.P.; Biragyn, A. Tumor-evoked regulatory B cells promote breast cancer metastasis by converting resting CD4(+) T cells to T-regulatory cells. Cancer Res. 2011, 71, 3505–3515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohling, S.D.; Allison, K.H. Immunosuppressive regulatory T cells are associated with aggressive breast cancer phenotypes: A potential therapeutic target. Mod. Pathol. 2008, 21, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Ohara, M.; Yamaguchi, Y.; Matsuura, K.; Murakami, S.; Arihiro, K.; Okada, M. Possible involvement of regulatory T cells in tumor onset and progression in primary breast cancer. Cancer Immunol. Immunother. 2009, 58, 441–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munir, M.T.; Kay, M.K.; Kang, M.H.; Rahman, M.M.; Al-Harrasi, A.; Choudhury, M.; Moustaid-Moussa, N.; Hussain, F.; Rahman, S.M. Tumor-Associated Macrophages as Multifaceted Regulators of Breast Tumor Growth. Int. J. Mol. Sci. 2021, 22, 6526. [Google Scholar] [CrossRef] [PubMed]
- Cendrowicz, E.; Sas, Z.; Bremer, E.; Rygiel, T.P. The Role of Macrophages in Cancer Development and Therapy. Cancers 2021, 13, 1946. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cheng, S.; Zhang, M.; Zhen, L.; Pang, D.; Zhang, Q.; Li, Z. High-infiltration of tumor-associated macrophages predicts unfavorable clinical outcome for node-negative breast cancer. PLoS ONE 2013, 8, e76147. [Google Scholar] [CrossRef]
- Hollmen, M.; Roudnicky, F.; Karaman, S.; Detmar, M. Characterization of macrophage--cancer cell crosstalk in estrogen receptor positive and triple-negative breast cancer. Sci. Rep. 2015, 5, 9188. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Sznol, M.; Chen, L. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer. Clin. Cancer Res. 2013, 19, 1021–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, B.; Woodward, W.A.; Wang, X.; Reuben, J.M.; Ueno, N.T. Inflammatory breast cancer biology: The tumour microenvironment is key. Nat. Rev. Cancer 2018, 18, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Luo, Y. Targeting macrophages in cancer immunotherapy. Signal. Transduct. Target. Ther. 2021, 6, 127. [Google Scholar] [CrossRef]
- Burugu, S.; Asleh-Aburaya, K.; Nielsen, T.O. Immune infiltrates in the breast cancer microenvironment: Detection, characterization and clinical implication. Breast Cancer 2017, 24, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Pinton, L.; Solito, S.; Damuzzo, V.; Francescato, S.; Pozzuoli, A.; Berizzi, A.; Mocellin, S.; Rossi, C.R.; Bronte, V.; Mandruzzato, S. Activated T cells sustain myeloid-derived suppressor cell-mediated immune suppression. Oncotarget 2016, 7, 1168–1184. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Wang, Y.; Yan, F.; Zhang, P.; Li, H.; Zhao, H.; Yan, C.; Yan, F.; Ren, X. Noncanonical NF-kappaB activation mediates STAT3-stimulated IDO upregulation in myeloid-derived suppressor cells in breast cancer. J. Immunol. 2014, 193, 2574–2586. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Wilkes, D.W.; Samuel, N.; Blanco, M.A.; Nayak, A.; Alicea-Torres, K.; Gluck, C.; Sinha, S.; Gabrilovich, D.; Chakrabarti, R. DeltaNp63-driven recruitment of myeloid-derived suppressor cells promotes metastasis in triple-negative breast cancer. J. Clin. Investig. 2018, 128, 5095–5109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, K.; Sakurai, M.; Yamamoto, Y.; Suzuki, E.; Tsuda, M.; Kataoka, T.R.; Hirata, M.; Nishie, M.; Nojiri, T.; Kumazoe, M.; et al. Alteration of specific cytokine expression patterns in patients with breast cancer. Sci. Rep. 2019, 9, 2924. [Google Scholar] [CrossRef]
- Ohl, K.; Tenbrock, K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front. Immunol. 2018, 9, 2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaul, M.E.; Fridlender, Z.G. Neutrophils as active regulators of the immune system in the tumor microenvironment. J. Leukoc. Biol. 2017, 102, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraiva, D.P.; Correia, B.F.; Salvador, R.; de Sousa, N.; Jacinto, A.; Braga, S.; Cabral, M.G. Circulating Low Density Neutrophils of Breast Cancer Patients are Associated with their Worse Prognosis due to the Impairment of T cell Responses. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wu, L.; Saxena, S.; Goel, P.; Prajapati, D.R.; Wang, C.; Singh, R.K. Breast Cancer Cell-Neutrophil Interactions Enhance Neutrophil Survival and Pro-Tumorigenic Activities. Cancers 2020, 12, 2884. [Google Scholar] [CrossRef] [PubMed]
- Rotondo, R.; Bertolotto, M.; Barisione, G.; Astigiano, S.; Mandruzzato, S.; Ottonello, L.; Dallegri, F.; Bronte, V.; Ferrini, S.; Barbieri, O. Exocytosis of azurophil and arginase 1-containing granules by activated polymorphonuclear neutrophils is required to inhibit T lymphocyte proliferation. J. Leukoc. Biol. 2011, 89, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houghton, A.M.; Rzymkiewicz, D.M.; Ji, H.; Gregory, A.D.; Egea, E.E.; Metz, H.E.; Stolz, D.B.; Land, S.R.; Marconcini, L.A.; Kliment, C.R.; et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat. Med. 2010, 16, 219–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavel, C.; Polette, M.; Doco, M.; Binninger, I.; Birembaut, P. Immunolocalization of matrix metallo-proteinases and their tissue inhibitor in human mammary pathology. Bull. Cancer 1992, 79, 261–270. [Google Scholar]
- Hurt, B.; Schulick, R.; Edil, B.; El Kasmi, K.C.; Barnett, C., Jr. Cancer-promoting mechanisms of tumor-associated neutrophils. Am. J. Surg. 2017, 214, 938–944. [Google Scholar] [CrossRef]
- Spiegel, A.; Brooks, M.W.; Houshyar, S.; Reinhardt, F.; Ardolino, M.; Fessler, E.; Chen, M.B.; Krall, J.A.; DeCock, J.; Zervantonakis, I.K.; et al. Neutrophils Suppress Intraluminal NK Cell-Mediated Tumor Cell Clearance and Enhance Extravasation of Disseminated Carcinoma Cells. Cancer Discov. 2016, 6, 630–649. [Google Scholar] [CrossRef] [Green Version]
- Granot, Z.; Henke, E.; Comen, E.A.; King, T.A.; Norton, L.; Benezra, R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell 2011, 20, 300–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ethier, J.L.; Desautels, D.; Templeton, A.; Shah, P.S.; Amir, E. Prognostic role of neutrophil-to-lymphocyte ratio in breast cancer: A systematic review and meta-analysis. Breast Cancer Res. 2017, 19, 2. [Google Scholar] [CrossRef] [Green Version]
- Koh, C.H.; Bhoo-Pathy, N.; Ng, K.L.; Jabir, R.S.; Tan, G.H.; See, M.H.; Jamaris, S.; Taib, N.A. Utility of pre-treatment neutrophil-lymphocyte ratio and platelet-lymphocyte ratio as prognostic factors in breast cancer. Br. J. Cancer 2015, 113, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, R.P.; Sharma, S.; Watabe, K. The Confounders of Cancer Immunotherapy: Roles of Lifestyle, Metabolic Disorders and Sociological Factors. Cancers 2020, 12, 2983. [Google Scholar] [CrossRef]
- Nishijima, T.F.; Muss, H.B.; Shachar, S.S.; Moschos, S.J. Comparison of efficacy of immune checkpoint inhibitors (ICIs) between younger and older patients: A systematic review and meta-analysis. Cancer Treat. Rev. 2016, 45, 30–37. [Google Scholar] [CrossRef]
- Zhao, C.; Hu, W.; Xu, Y.; Wang, D.; Wang, Y.; Lv, W.; Xiong, M.; Yi, Y.; Wang, H.; Zhang, Q.; et al. Current Landscape: The Mechanism and Therapeutic Impact of Obesity for Breast Cancer. Front. Oncol. 2021, 11, 704893. [Google Scholar] [CrossRef]
- Dai, X.; Bu, X.; Gao, Y.; Guo, J.; Hu, J.; Jiang, C.; Zhang, Z.; Xu, K.; Duan, J.; He, S.; et al. Energy status dictates PD-L1 protein abundance and anti-tumor immunity to enable checkpoint blockade. Mol. Cell 2021, 81, 2317–2331. [Google Scholar] [CrossRef] [PubMed]
- Ferrere, G.; Tidjani Alou, M.; Liu, P.; Goubet, A.G.; Fidelle, M.; Kepp, O.; Durand, S.; Iebba, V.; Fluckiger, A.; Daillere, R.; et al. Ketogenic diet and ketone bodies enhance the anticancer effects of PD-1 blockade. JCI Insight 2021, 6, e145207. [Google Scholar] [CrossRef] [PubMed]
- Khodabakhshi, A.; Akbari, M.E.; Mirzaei, H.R.; Seyfried, T.N.; Kalamian, M.; Davoodi, S.H. Effects of Ketogenic metabolic therapy on patients with breast cancer: A randomized controlled clinical trial. Clin. Nutr. 2021, 40, 751–758. [Google Scholar] [CrossRef]
- Marhelava, K.; Pilch, Z.; Bajor, M.; Graczyk-Jarzynka, A.; Zagozdzon, R. Targeting Negative and Positive Immune Checkpoints with Monoclonal Antibodies in Therapy of Cancer. Cancers 2019, 11, 1756. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Zou, X.; Zheng, S.; Tang, H.; Zhang, L.; Liu, P.; Xie, X. Efficacy and predictive factors of immune checkpoint inhibitors in metastatic breast cancer: A systematic review and meta-analysis. Ther. Adv. Med. Oncol. 2020, 12, 1758835920940928. [Google Scholar] [CrossRef] [PubMed]
- Albanell, J.; Baselga, J. Trastuzumab, a humanized anti-HER2 monoclonal antibody, for the treatment of breast cancer. Drugs Today (Barc) 1999, 35, 931–946. [Google Scholar] [PubMed]
- Hudis, C.A. Trastuzumab-mechanism of action and use in clinical practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarantino, P.; Morganti, S.; Uliano, J.; Giugliano, F.; Crimini, E.; Curigliano, G. Margetuximab for the treatment of HER2-positive metastatic breast cancer. Expert Opin. Biol. Ther. 2021, 21, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.; Winer, E.P. Trastuzumab emtansine: A novel antibody-drug conjugate for HER2-positive breast cancer. Clin. Cancer Res. 2014, 20, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Skidmore, L.; Sakamuri, S.; Knudsen, N.A.; Hewet, A.G.; Milutinovic, S.; Barkho, W.; Biroc, S.L.; Kirtley, J.; Marsden, R.; Storey, K.; et al. ARX788, a Site-specific Anti-HER2 Antibody-Drug Conjugate, Demonstrates Potent and Selective Activity in HER2-low and T-DM1-resistant Breast and Gastric Cancers. Mol. Cancer Ther. 2020, 19, 1833–1843. [Google Scholar] [CrossRef] [PubMed]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, A Novel HER2-Targeting ADC with a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy with Differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef] [Green Version]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- Murthy, R.K.; Loi, S.; Okines, A.; Paplomata, E.; Hamilton, E.; Hurvitz, S.A.; Lin, N.U.; Borges, V.; Abramson, V.; Anders, C.; et al. Tucatinib, Trastuzumab, and Capecitabine for HER2-Positive Metastatic Breast Cancer. N. Engl. J. Med. 2020, 382, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Braiteh, F.S.; Cassier, P.; Delord, J.P.; Eder, J.P.; Fasso, M.; Xiao, Y.; Wang, Y.; Molinero, L.; Chen, D.S.; et al. Abstract 2859: Inhibition of PD-L1 by MPDL3280A leads to clinical activity in patients with metastatic triple-negative breast cancer (TNBC). In Proceedings of the American Association for Cancer Research, Philadelphia, PA, USA, 18–22 April 2015; p. 2859. [Google Scholar]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kummel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef]
- Yuan, Y.; Lee, J.S.; Yost, S.E.; Frankel, P.H.; Ruel, C.; Egelston, C.A.; Guo, W.; Gillece, J.D.; Folkerts, M.; Reining, L.; et al. A Phase II Clinical Trial of Pembrolizumab and Enobosarm in Patients with Androgen Receptor-Positive Metastatic Triple-Negative Breast Cancer. Oncologist 2021, 26, e99–e217. [Google Scholar] [CrossRef] [PubMed]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B.; et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Kalinsky, K.; Diamond, J.R.; Vahdat, L.T.; Tolaney, S.M.; Juric, D.; O’Shaughnessy, J.; Moroose, R.L.; Mayer, I.A.; Abramson, V.G.; Goldenberg, D.M.; et al. Sacituzumab govitecan in previously treated hormone receptor-positive/HER2-negative metastatic breast cancer: Final results from a phase I/II, single-arm, basket trial. Ann. Oncol. 2020, 31, 1709–1718. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Bardia, A.; Tolaney, S.M.; Arteaga, C.; Cortes, J.; Sohn, J.; Marme, F.; Hong, Q.; Delaney, R.J.; Hafeez, A.; et al. TROPiCS-02: A Phase III study investigating sacituzumab govitecan in the treatment of HR+/HER2- metastatic breast cancer. Future Oncol. 2020, 16, 705–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagayama, A.; Vidula, N.; Ellisen, L.; Bardia, A. Novel antibody-drug conjugates for triple negative breast cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920915980. [Google Scholar] [CrossRef]
- McGuinness, J.E.; Kalinsky, K. Antibody-drug conjugates in metastatic triple negative breast cancer: A spotlight on sacituzumab govitecan, ladiratuzumab vedotin, and trastuzumab deruxtecan. Expert Opin. Biol. Ther. 2021, 21, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Neidhardt-Berard, E.M.; Berard, F.; Banchereau, J.; Palucka, A.K. Dendritic cells loaded with killed breast cancer cells induce differentiation of tumor-specific cytotoxic T lymphocytes. Breast Cancer Res. 2004, 6, R322–R328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avigan, D.; Vasir, B.; Gong, J.; Borges, V.; Wu, Z.; Uhl, L.; Atkins, M.; Mier, J.; McDermott, D.; Smith, T.; et al. Fusion cell vaccination of patients with metastatic breast and renal cancer induces immunological and clinical responses. Clin. Cancer Res. 2004, 10, 4699–4708. [Google Scholar] [CrossRef] [Green Version]
- Baek, S.; Kim, C.S.; Kim, S.B.; Kim, Y.M.; Kwon, S.W.; Kim, Y.; Kim, H.; Lee, H. Combination therapy of renal cell carcinoma or breast cancer patients with dendritic cell vaccine and IL-2: Results from a phase I/II trial. J. Transl. Med. 2011, 9, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowenfeld, L.; Mick, R.; Datta, J.; Xu, S.; Fitzpatrick, E.; Fisher, C.S.; Fox, K.R.; DeMichele, A.; Zhang, P.J.; Weinstein, S.P.; et al. Dendritic Cell Vaccination Enhances Immune Responses and Induces Regression of HER2(pos) DCIS Independent of Route: Results of Randomized Selection Design Trial. Clin. Cancer Res. 2017, 23, 2961–2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, E.P.; Kaklamani, V.; Falkson, C.; Vidal, G.A.; Ward, P.J.; Patre, M.; Chui, S.Y.; Rotmensch, J.; Gupta, K.; Molinero, L.; et al. Impact of Anti-HER2 Treatments Combined With Atezolizumab on the Tumor Immune Microenvironment in Early or Metastatic Breast Cancer: Results From a Phase Ib Study. Clin. Breast Cancer 2021, 21, 539–551. [Google Scholar] [CrossRef]
- Li, Z.L.; Zhang, H.L.; Huang, Y.; Huang, J.H.; Sun, P.; Zhou, N.N.; Chen, Y.H.; Mai, J.; Wang, Y.; Yu, Y.; et al. Autophagy deficiency promotes triple-negative breast cancer resistance to T cell-mediated cytotoxicity by blocking tenascin-C degradation. Nat. Commun. 2020, 11, 3806. [Google Scholar] [CrossRef] [PubMed]
- Vito, A.; Salem, O.; El-Sayes, N.; MacFawn, I.P.; Portillo, A.L.; Milne, K.; Harrington, D.; Ashkar, A.A.; Wan, Y.; Workenhe, S.T.; et al. Immune checkpoint blockade in triple negative breast cancer influenced by B cells through myeloid-derived suppressor cells. Commun. Biol. 2021, 4, 859. [Google Scholar] [CrossRef]
- Ho, A.Y.; Barker, C.A.; Arnold, B.B.; Powell, S.N.; Hu, Z.I.; Gucalp, A.; Lebron-Zapata, L.; Wen, H.Y.; Kallman, C.; D’Agnolo, A.; et al. A phase 2 clinical trialassessing theefficacy and safety of pembrolizumab and radiotherapy in patients with metastatic triple-negative breast cancer. Cancer 2020, 126, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Barroso-Sousa, R.; Krop, I.E.; Trippa, L.; Tan-Wasielewski, Z.; Li, T.; Osmani, W.; Andrews, C.; Dillon, D.; Richardson, E.T., 3rd; Pastorello, R.G.; et al. A Phase II Study of Pembrolizumab in Combination With Palliative Radiotherapy for Hormone Receptor-positive Metastatic Breast Cancer. Clin. Breast Cancer 2020, 20, 238–245. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Winer, E.; Lipatov, O.; Im, S.-A.; Goncalves, A.; Cortes, J.; Lee, K.S.; Schmid, P.; Testa, L.; Witzel, I.; et al. Abstract PD5-03: Relationship between tumor-infiltrating lymphocytes (TILs) and outcomes in the KEYNOTE-119 study of pembrolizumab vs chemotherapy for previously treated metastatic triple-negative breast cancer (mTNBC). In Proceedings of the 2019 San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 10–14 December 2019; p. PD5-03. [Google Scholar]
- Schmid, P.; Salgado, R.; Park, Y.H.; Munoz-Couselo, E.; Kim, S.B.; Sohn, J.; Im, S.A.; Foukakis, T.; Kuemmel, S.; Dent, R.; et al. Pembrolizumab plus chemotherapy as neoadjuvant treatment of high-risk, early-stage triple-negative breast cancer: Results from the phase 1b open-label, multicohort KEYNOTE-173 study. Ann. Oncol. 2020, 31, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Huang, C.-S.; Egle, D.; Bermejo, B.; Zamagni, C.; Thill, M.; Anton, A.; Zambelli, S.; Russo, S.; Ciruelos, E.M.; et al. LBA13 Tumour infiltrating lymphocytes (TILs), PD-L1 expression and their dynamics in the NeoTRIPaPDL1 trial. Ann. Oncol. 2020, 31, S1145–S1146. [Google Scholar] [CrossRef]
- Loibl, S.; Untch, M.; Burchardi, N.; Huober, J.; Sinn, B.V.; Blohmer, J.U.; Grischke, E.M.; Furlanetto, J.; Tesch, H.; Hanusch, C.; et al. A randomised phase II study investigating durvalumab in addition to an anthracycline taxane-based neoadjuvant therapy in early triple-negative breast cancer: Clinical results and biomarker analysis of GeparNuevo study. Ann. Oncol. 2019, 30, 1279–1288. [Google Scholar] [CrossRef] [Green Version]
- Klopfenstein, Q.; Derangere, V.; Arnould, L.; Thibaudin, M.; Limagne, E.; Ghiringhelli, F.; Truntzer, C.; Ladoire, S. Evaluation of tumor immune contexture among intrinsic molecular subtypes helps to predict outcome in early breast cancer. J. Immunother. Cancer 2021, 9, e002036. [Google Scholar] [CrossRef]
- Xu, Q.; Chen, S.; Hu, Y.; Huang, W. Landscape of Immune Microenvironment Under Immune Cell Infiltration Pattern in Breast Cancer. Front. Immunol. 2021, 12, 711433. [Google Scholar] [CrossRef]
- Gruosso, T.; Gigoux, M.; Manem, V.S.K.; Bertos, N.; Zuo, D.; Perlitch, I.; Saleh, S.M.I.; Zhao, H.; Souleimanova, M.; Johnson, R.M.; et al. Spatially distinct tumor immune microenvironments stratify triple-negative breast cancers. J. Clin. Investig. 2019, 129, 1785–1800. [Google Scholar] [CrossRef] [Green Version]
- Ali, H.R.; Provenzano, E.; Dawson, S.J.; Blows, F.M.; Liu, B.; Shah, M.; Earl, H.M.; Poole, C.J.; Hiller, L.; Dunn, J.A.; et al. Association between CD8+ T-cell infiltration and breast cancer survival in 12,439 patients. Ann. Oncol. 2014, 25, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Muul, L.M.; Solomon, D.; Rosenberg, S.A. Expansion of human tumor infiltrating lymphocytes for use in immunotherapy trials. J. Immunol. Methods 1987, 102, 127–141. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevanovic, S.; Draper, L.M.; Langhan, M.M.; Campbell, T.E.; Kwong, M.L.; Wunderlich, J.R.; Dudley, M.E.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J. Clin. Oncol. 2015, 33, 1543–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Avi, R.; Farhi, R.; Ben-Nun, A.; Gorodner, M.; Greenberg, E.; Markel, G.; Schachter, J.; Itzhaki, O.; Besser, M.J. Establishment of adoptive cell therapy with tumor infiltrating lymphocytes for non-small cell lung cancer patients. Cancer Immunol. Immunother. 2018, 67, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kim, Y.A.; Sim, C.K.; Heo, S.H.; Song, I.H.; Park, H.S.; Park, S.Y.; Bang, W.S.; Park, I.A.; Lee, M.; et al. Expansion of tumor-infiltrating lymphocytes and their potential for application as adoptive cell transfer therapy in human breast cancer. Oncotarget 2017, 8, 113345–113359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazdanifar, M.; Barbarito, G.; Bertaina, A.; Airoldi, I. gammadelta T Cells: The Ideal Tool for Cancer Immunotherapy. Cells 2020, 9, 1305. [Google Scholar] [CrossRef]
- Capietto, A.H.; Martinet, L.; Fournie, J.J. Stimulated gammadelta T cells increase the in vivo efficacy of trastuzumab in HER-2+ breast cancer. J. Immunol. 2011, 187, 1031–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Kyle-Cezar, F.; Woolf, R.T.; Naceur-Lombardelli, C.; Owen, J.; Biswas, D.; Lorenc, A.; Vantourout, P.; Gazinska, P.; Grigoriadis, A.; et al. An innate-like Vdelta1(+) gammadelta T cell compartment in the human breast is associated with remission in triple-negative breast cancer. Sci. Transl. Med. 2019, 11, eaax9364. [Google Scholar] [CrossRef] [PubMed]
- Geller, M.A.; Cooley, S.; Judson, P.L.; Ghebre, R.; Carson, L.F.; Argenta, P.A.; Jonson, A.L.; Panoskaltsis-Mortari, A.; Curtsinger, J.; McKenna, D.; et al. A phase II study of allogeneic.c natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy 2011, 13, 98–107. [Google Scholar] [CrossRef] [Green Version]
- Chiba, T.; Ohtani, H.; Mizoi, T.; Naito, Y.; Sato, E.; Nagura, H.; Ohuchi, A.; Ohuchi, K.; Shiiba, K.; Kurokawa, Y.; et al. Intraepithelial CD8+ T-cell-count becomes a prognostic factor after a longer follow-up period in human colorectal carcinoma: Possible association with suppression of micrometastasis. Br. J. Cancer 2004, 91, 1711–1717. [Google Scholar] [CrossRef] [PubMed]
- Maiti, S.N.; Huls, H.; Singh, H.; Dawson, M.; Figliola, M.; Olivares, S.; Rao, P.; Zhao, Y.J.; Multani, A.; Yang, G.; et al. Sleeping beauty system to redirect T-cell specificity for human applications. J. Immunother. 2013, 36, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvak, Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Deniger, D.C.; Switzer, K.; Mi, T.; Maiti, S.; Hurton, L.; Singh, H.; Huls, H.; Olivares, S.; Lee, D.A.; Champlin, R.E.; et al. Bispecific T-cells expressing polyclonal repertoire of endogenous gammadelta T-cell receptors and introduced CD19-specific chimeric antigen receptor. Mol. Ther. 2013, 21, 638–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef]
- Zhu, H.; Blum, R.H.; Bjordahl, R.; Gaidarova, S.; Rogers, P.; Lee, T.T.; Abujarour, R.; Bonello, G.B.; Wu, J.; Tsai, P.F.; et al. Pluripotent stem cell-derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood 2020, 135, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.W.; Cho, J.Y. Recent Advances in Allogeneic CAR-T Cells. Biomolecules 2020, 10, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.; Li, X.; He, Y.; Zhu, W.; Gao, L.; Liu, Y.; Gao, L.; Wen, Q.; Zhong, J.F.; Zhang, C.; et al. Recent advances in CAR-T cell engineering. J. Hematol. Oncol. 2020, 13, 86. [Google Scholar] [CrossRef] [PubMed]
- Zhylko, A.; Winiarska, M.; Graczyk-Jarzynka, A. The Great War of Today: Modifications of CAR-T Cells to Effectively Combat Malignancies. Cancers 2020, 12, 2030. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riviere, I.; Sadelain, M. Chimeric Antigen Receptors: A Cell and Gene Therapy Perspective. Mol. Ther. 2017, 25, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Del Zotto, G.; Marcenaro, E.; Vacca, P.; Sivori, S.; Pende, D.; Della Chiesa, M.; Moretta, F.; Ingegnere, T.; Mingari, M.C.; Moretta, A.; et al. Markers and function of human NK cells in normal and pathological conditions. Cytom. B Clin. Cytom. 2017, 92, 100–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchou, J.; Zhao, Y.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.; Lacey, S.F.; et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol. Res. 2017, 5, 1152–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.C.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carioca, A.A.; Verde, S.M.; Luzia, L.A.; Rondo, P.H.; Latorre, M.R.; Ellery, T.H.; Damasceno, N.R. Association of oxidative stress biomarkers with adiposity and clinical staging in women with breast cancer. Eur. J. Clin. Nutr. 2015, 69, 1256–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firczuk, M.; Bajor, M.; Graczyk-Jarzynka, A.; Fidyt, K.; Goral, A.; Zagozdzon, R. Harnessing altered oxidative metabolism in cancer by augmented prooxidant therapy. Cancer Lett. 2020, 471, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bajor, M.; Graczyk-Jarzynka, A.; Marhelava, K.; Kurkowiak, M.; Rahman, A.; Aura, C.; Russell, N.; Zych, A.O.; Firczuk, M.; Winiarska, M.; et al. Triple Combination of Ascorbate, Menadione and the Inhibition of Peroxiredoxin-1 Produces Synergistic Cytotoxic Effects in Triple-Negative Breast Cancer Cells. Antioxidants 2020, 9, 320. [Google Scholar] [CrossRef] [PubMed]
- Bajor, M.; Zych, A.O.; Graczyk-Jarzynka, A.; Muchowicz, A.; Firczuk, M.; Trzeciak, L.; Gaj, P.; Domagala, A.; Siernicka, M.; Zagozdzon, A.; et al. Targeting peroxiredoxin 1 impairs growth of breast cancer cells and potently sensitises these cells to prooxidant agents. Br. J. Cancer 2018, 119, 873–884. [Google Scholar] [CrossRef] [Green Version]
- Catania, A.; Barrajon-Catalan, E.; Nicolosi, S.; Cicirata, F.; Micol, V. Immunoliposome encapsulation increases cytotoxic activity and selectivity of curcumin and resveratrol against HER2 overexpressing human breast cancer cells. Breast Cancer Res. Treat. 2013, 141, 55–65. [Google Scholar] [CrossRef]
- Zhang, H.G.; Kim, H.; Liu, C.; Yu, S.; Wang, J.; Grizzle, W.E.; Kimberly, R.P.; Barnes, S. Curcumin reverses breast tumor exosomes mediated immune suppression of NK cell tumor cytotoxicity. Biochim. Biophys. Acta 2007, 1773, 1116–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Tian, W.; Cai, X.; Wang, X.; Dang, W.; Tang, H.; Cao, H.; Wang, L.; Chen, T. Hydrazinocurcumin Encapsuled nanoparticles “re-educate” tumor-associated macrophages and exhibit anti-tumor effects on breast cancer following STAT3 suppression. PLoS ONE 2013, 8, e65896. [Google Scholar] [CrossRef]
- Lim, S.O.; Li, C.W.; Xia, W.; Cha, J.H.; Chan, L.C.; Wu, Y.; Chang, S.S.; Lin, W.C.; Hsu, J.M.; Hsu, Y.H.; et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazdi, M.H.; Mahdavi, M.; Faghfuri, E.; Faramarzi, M.A.; Sepehrizadeh, Z.; Hassan, Z.M.; Gholami, M.; Shahverdi, A.R. Th1 Immune Response Induction by Biogenic Selenium Nanoparticles in Mice with Breast Cancer: Preliminary Vaccine Model. Iran. J. Biotechnol. 2015, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Liu, T.; Li, J.; Mai, F.; Li, J.; Chen, Y.; Jing, Y.; Dong, X.; Lin, L.; He, J.; et al. Selenium nanoparticles as new strategy to potentiate gammadelta T cell anti-tumor cytotoxicity through upregulation of tubulin-alpha acetylation. Biomaterials 2019, 222, 119397. [Google Scholar] [CrossRef]
- Liu, X.; Feng, Z.; Wang, C.; Su, Q.; Song, H.; Zhang, C.; Huang, P.; Liang, X.J.; Dong, A.; Kong, D.; et al. Co-localized delivery of nanomedicine and nanovaccine augments the postoperative cancer immunotherapy by amplifying T-cell responses. Biomaterials 2020, 230, 119649. [Google Scholar] [CrossRef] [PubMed]
- Magri, A.; Germano, G.; Lorenzato, A.; Lamba, S.; Chila, R.; Montone, M.; Amodio, V.; Ceruti, T.; Sassi, F.; Arena, S.; et al. High-dose vitamin C enhances cancer immunotherapy. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Raninga, P.V.; Lee, A.C.; Sinha, D.; Shih, Y.Y.; Mittal, D.; Makhale, A.; Bain, A.L.; Nanayakarra, D.; Tonissen, K.F.; Kalimutho, M.; et al. Therapeutic cooperation between auranofin, a thioredoxin reductase inhibitor and anti-PD-L1 antibody for treatment of triple-negative breast cancer. Int. J. Cancer 2020, 146, 123–136. [Google Scholar] [CrossRef]
- Olesch, C.; Sirait-Fischer, E.; Berkefeld, M.; Fink, A.F.; Susen, R.M.; Ritter, B.; Michels, B.E.; Steinhilber, D.; Greten, F.R.; Savai, R.; et al. S1PR4 ablation reduces tumor growth and improves chemotherapy via CD8+ T cell expansion. J. Clin. Investig. 2020, 130, 5461–5476. [Google Scholar] [CrossRef]
- Guo, S.; Smeltz, R.B.; Nanajian, A.; Heller, R. IL-15/IL-15Ralpha Heterodimeric Complex as Cancer Immunotherapy in Murine Breast Cancer Models. Front. Immunol. 2020, 11, 614667. [Google Scholar] [CrossRef] [PubMed]
- Gurusamy, D.; Henning, A.N.; Yamamoto, T.N.; Yu, Z.; Zacharakis, N.; Krishna, S.; Kishton, R.J.; Vodnala, S.K.; Eidizadeh, A.; Jia, L.; et al. Multi-phenotype CRISPR-Cas9 Screen Identifies p38 Kinase as a Target for Adoptive Immunotherapies. Cancer Cell 2020, 37, 818–833.e9. [Google Scholar] [CrossRef] [PubMed]
- Zonneville, J.; Colligan, S.; Grant, S.; Miller, A.; Wallace, P.; Abrams, S.I.; Bakin, A.V. Blockade of p38 kinase impedes the mobilization of protumorigenic myeloid populations to impact breast cancer metastasis. Int. J. Cancer 2020, 147, 2279–2292. [Google Scholar] [CrossRef]
- Zhang, M.; Shi, Y.; Zhang, Y.; Wang, Y.; Alotaibi, F.; Qiu, L.; Wang, H.; Peng, S.; Liu, Y.; Li, Q.; et al. miRNA-5119 regulates immune checkpoints in dendritic cells to enhance breast cancer immunotherapy. Cancer Immunol. Immunother. 2020, 69, 951–967. [Google Scholar] [CrossRef]
- Zhou, W.; Yu, M.; Pan, H.; Qiu, W.; Wang, H.; Qian, M.; Che, N.; Zhang, K.; Mao, X.; Li, L.; et al. Microwave ablation induces Th1-type immune response with activation of ICOS pathway in early-stage breast cancer. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Bolis, M.; Paroni, G.; Fratelli, M.; Vallerga, A.; Guarrera, L.; Zanetti, A.; Kurosaki, M.; Garattini, S.K.; Gianni, M.; Lupi, M.; et al. All-Trans Retinoic Acid Stimulates Viral Mimicry, Interferon Responses and Antigen Presentation in Breast-Cancer Cells. Cancers 2020, 12, 1169. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Liu, Y.; Zhang, X.; Kong, D.; Kong, J.; Zhao, D.; Guo, Y.; Sun, L.; Chu, L.; Liu, S.; et al. The anti-B7-H4 checkpoint synergizes trastuzumab treatment to promote phagocytosis and eradicate breast cancer. Neoplasia 2020, 22, 539–553. [Google Scholar] [CrossRef]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front. Immunol. 2021, 12, 636568. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Udono, H.; Tanahashi, N.; Hamada, N.; Watanabe, K.; Adachi, K.; Yamano, T.; Yui, K.; Kobayashi, N.; Kasahara, M.; et al. Immunoproteasome assembly and antigen presentation in mice lacking both PA28alpha and PA28beta. EMBO J. 2001, 20, 5898–5907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Dai, X.; Gong, K.; Song, K.; Tai, F.; Shi, J. PA28alpha/beta Promote Breast Cancer Cell Invasion and Metastasis via Down-Regulation of CDK15. Front. Oncol. 2019, 9, 1283. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Wang, L.; Wan, C.; Sun, Y.; Van der Jeught, K.; Zhou, Z.; Dong, T.; So, K.M.; Yu, T.; Li, Y.; et al. MAL2 drives immune evasion in breast cancer by suppressing tumor antigen presentation. J. Clin. Investig. 2021, 131, e140837. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, G.; Mullan, K.A.; Duscharla, D.; Ayala, R.; Croft, N.P.; Faridi, P.; Purcell, A.W. IFNgamma Modulates the Immunopeptidome of Triple Negative Breast Cancer Cells by Enhancing and Diversifying Antigen Processing and Presentation. Front. Immunol. 2021, 12, 645770. [Google Scholar] [CrossRef]
- Li, Z.; Li, Y.; Wang, X.; Yang, Q. PPP2R2B downregulation is associated with immune evasion and predicts poor clinical outcomes in triple-negative breast cancer. Cancer Cell Int. 2021, 21, 13. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Wen, H.; Liang, L.; Dong, X.; Du, R.; Zhhou, W.; Zhang, X.; Zhang, C.; Xiang, R.; Li, N. IL20RA signaling enhances stemness and promotes the formation of an immunosuppressive microenvironment in breast cancer. Theranostics 2021, 11, 2564–2580. [Google Scholar] [CrossRef]
- Kheshtchin, N.; Arab, S.; Ajami, M.; Mirzaei, R.; Ashourpour, M.; Mousavi, N.; Khosravianfar, N.; Jadidi-Niaragh, F.; Namdar, A.; Noorbakhsh, F.; et al. Inhibition of HIF-1alpha enhances anti-tumor effects of dendritic cell-based vaccination in a mouse model of breast cancer. Cancer Immunol. Immunother. 2016, 65, 1159–1167. [Google Scholar] [CrossRef]
- Goyette, M.A.; Elkholi, I.E.; Apcher, C.; Kuasne, H.; Rothlin, C.V.; Muller, W.J.; Richard, D.E.; Park, M.; Gratton, J.P.; Cote, J.F. Targeting Axl favors an antitumorigenic microenvironment that enhances immunotherapy responses by decreasing Hif-1alpha levels. Proc. Natl. Acad. Sci. USA 2021, 118, e2023868118. [Google Scholar] [CrossRef] [PubMed]
- Varikuti, S.; Singh, B.; Volpedo, G.; Ahirwar, D.K.; Jha, B.K.; Saljoughian, N.; Viana, A.G.; Verma, C.; Hamza, O.; Halsey, G.; et al. Ibrutinib treatment inhibits breast cancer progression and metastasis by inducing conversion of myeloid-derived suppressor cells to dendritic cells. Br. J. Cancer 2020, 122, 1005–1013. [Google Scholar] [CrossRef]
- Gupta, N.; Gaikwad, S.; Kaushik, I.; Wright, S.E.; Markiewski, M.M.; Srivastava, S.K. Atovaquone Suppresses Triple-Negative Breast Tumor Growth by Reducing Immune-Suppressive Cells. Int. J. Mol. Sci. 2021, 22, 5150. [Google Scholar] [CrossRef]
- Cao, Y.; Feng, Y.H.; Gao, L.W.; Li, X.Y.; Jin, Q.X.; Wang, Y.Y.; Xu, Y.Y.; Jin, F.; Lu, S.L.; Wei, M.J. Artemisinin enhances the anti-tumor immune response in 4T1 breast cancer cells in vitro and in vivo. Int. Immunopharmacol. 2019, 70, 110–116. [Google Scholar] [CrossRef]
- Cresswell, G.M.; Wang, B.; Kischuk, E.M.; Broman, M.M.; Alfar, R.A.; Vickman, R.E.; Dimitrov, D.S.; Kularatne, S.A.; Sundaram, C.P.; Singhal, S.; et al. Folate Receptor Beta Designates Immunosuppressive Tumor-Associated Myeloid Cells That Can Be Reprogrammed with Folate-Targeted Drugs. Cancer Res. 2021, 81, 671–684. [Google Scholar] [CrossRef]
- Deng, Y.; Hu, J.C.; He, S.H.; Lou, B.; Ding, T.B.; Yang, J.T.; Mo, M.G.; Ye, D.Y.; Zhou, L.; Jiang, X.C.; et al. Sphingomyelin synthase 2 facilitates M2-like macrophage polarization and tumor progression in a mouse model of triple-negative breast cancer. Acta Pharmacol. Sin. 2021, 42, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Holen, I.; Coleman, R.E. Anti-tumour activity of bisphosphonates in preclinical models of breast cancer. Breast Cancer Res. 2010, 12, 214. [Google Scholar] [CrossRef] [Green Version]
- Bai, F.; Zhang, P.; Fu, Y.; Chen, H.; Zhang, M.; Huang, Q.; Li, D.; Li, B.; Wu, K. Targeting ANXA1 abrogates Treg-mediated immune suppression in triple-negative breast cancer. J. Immunother. Cancer 2020, 8, e000169. [Google Scholar] [CrossRef]
- Robbins, Y.; Greene, S.; Friedman, J.; Clavijo, P.E.; Van Waes, C.; Fabian, K.P.; Padget, M.R.; Abdul Sater, H.; Lee, J.H.; Soon-Shiong, P.; et al. Tumor control via targeting PD-L1 with chimeric antigen receptor modified NK cells. Elife 2020, 9, e54854. [Google Scholar] [CrossRef]
- Xie, Y.J.; Dougan, M.; Jailkhani, N.; Ingram, J.; Fang, T.; Kummer, L.; Momin, N.; Pishesha, N.; Rickelt, S.; Hynes, R.O.; et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc. Natl. Acad. Sci. USA 2019, 116, 7624–7631. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Garcia, A.; Lynn, R.C.; Poussin, M.; Eiva, M.A.; Shaw, L.C.; O’Connor, R.S.; Minutolo, N.G.; Casado-Medrano, V.; Lopez, G.; Matsuyama, T.; et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat. Commun. 2021, 12, 877. [Google Scholar] [CrossRef] [PubMed]
- Szoor, A.; Toth, G.; Zsebik, B.; Szabo, V.; Eshhar, Z.; Abken, H.; Vereb, G. Trastuzumab derived HER2-specific CARs for the treatment of trastuzumab-resistant breast cancer: CAR T cells penetrate and eradicate tumors that are not accessible to antibodies. Cancer Lett. 2020, 484, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bajgain, P.; Tawinwung, S.; D’Elia, L.; Sukumaran, S.; Watanabe, N.; Hoyos, V.; Lulla, P.; Brenner, M.K.; Leen, A.M.; Vera, J.F. CAR T cell therapy for breast cancer: Harnessing the tumor milieu to drive T cell activation. J. Immunother. Cancer 2018, 6, 34. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, Y.; Liu, W.; Li, X. Engineered IL-7 Receptor Enhances the Therapeutic Effect of AXL-CAR-T Cells on Triple-Negative Breast Cancer. BioMed Res. Int. 2020, 2020, 4795171. [Google Scholar] [CrossRef] [PubMed]
- Uhl, M.; Aulwurm, S.; Wischhusen, J.; Weiler, M.; Ma, J.Y.; Almirez, R.; Mangadu, R.; Liu, Y.W.; Platten, M.; Herrlinger, U.; et al. SD-208, a novel transforming growth factor beta receptor I kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo. Cancer Res. 2004, 64, 7954–7961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuber, T.; Monjezi, R.; Wallstabe, L.; Kuhnemundt, J.; Nietzer, S.L.; Dandekar, G.; Wockel, A.; Einsele, H.; Wischhusen, J.; Hudecek, M. Inhibition of TGF-beta-receptor signaling augments the antitumor function of ROR1-specific CAR T-cells against triple-negative breast cancer. J. Immunother. Cancer 2020, 8, e000676. [Google Scholar] [CrossRef]
- Du, L.; Nai, Y.; Shen, M.; Li, T.; Huang, J.; Han, X.; Wang, W.; Pang, D.; Jin, A. IL-21 Optimizes the CAR-T Cell Preparation Through Improving Lentivirus Mediated Transfection Efficiency of T Cells and Enhancing CAR-T Cell Cytotoxic Activities. Front. Mol. Biosci. 2021, 8, 675179. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xiao, F.; Zhang, A.; Zhang, D.; Nie, W.; Xu, T.; Han, B.; Seth, P.; Wang, H.; Yang, Y.; et al. Oncolytic adenovirus targeting TGF-beta enhances anti-tumor responses of mesothelin-targeted chimeric antigen receptor T cell therapy against breast cancer. Cell Immunol. 2020, 348, 104041. [Google Scholar] [CrossRef]
- Li, H.; Yuan, W.; Bin, S.; Wu, G.; Li, P.; Liu, M.; Yang, J.; Li, X.; Yang, K.; Gu, H. Overcome trastuzumab resistance of breast cancer using anti-HER2 chimeric antigen receptor. T cells and PD1 blockade. Am. J. Cancer Res. 2020, 10, 688–703. [Google Scholar] [PubMed]
- Lei, X.; Ou, Z.; Yang, Z.; Zhong, J.; Zhu, Y.; Tian, J.; Wu, J.; Deng, H.; Lin, X.; Peng, Y.; et al. A Pan-Histone Deacetylase Inhibitor Enhances the Antitumor Activity of B7-H3-Specific CAR T Cells in Solid Tumors. Clin. Cancer Res. 2021, 27, 3757–3771. [Google Scholar] [CrossRef]
- Sun, R.; Luo, H.; Su, J.; Di, S.; Zhou, M.; Shi, B.; Sun, Y.; Du, G.; Zhang, H.; Jiang, H.; et al. Olaparib Suppresses MDSC Recruitment via SDF1alpha/CXCR4 Axis to Improve the Anti-tumor Efficacy of CAR-T Cells on Breast Cancer in Mice. Mol. Ther. 2021, 29, 60–74. [Google Scholar] [CrossRef]
- Xu, N.; Palmer, D.C.; Robeson, A.C.; Shou, P.; Bommiasamy, H.; Laurie, S.J.; Willis, C.; Dotti, G.; Vincent, B.G.; Restifo, N.P.; et al. STING agonist promotes CAR T cell trafficking and persistence in breast cancer. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef]
- Xia, L.; Zheng, Z.; Liu, J.Y.; Chen, Y.J.; Ding, J.; Hu, G.S.; Hu, Y.H.; Liu, S.; Luo, W.X.; Xia, N.S.; et al. Targeting Triple-Negative Breast Cancer with Combination Therapy of EGFR CAR T Cells and CDK7 Inhibition. Cancer Immunol. Res. 2021, 9, 707–722. [Google Scholar] [CrossRef]
- Thakur, A.; Scholler, J.; Kubicka, E.; Bliemeister, E.T.; Schalk, D.L.; June, C.H.; Lum, L.G. Bispecific Antibody Armed Metabolically Enhanced Headless CAR T Cells. Front. Immunol. 2021, 12, 690437. [Google Scholar] [CrossRef]
- Seitz, C.M.; Schroeder, S.; Knopf, P.; Krahl, A.C.; Hau, J.; Schleicher, S.; Martella, M.; Quintanilla-Martinez, L.; Kneilling, M.; Pichler, B.; et al. GD2-targeted chimeric antigen receptor T cells prevent metastasis formation by elimination of breast cancer stem-like cells. Oncoimmunology 2020, 9, 1683345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.; Cao, X.; Cai, H.; Feng, P.; Chen, X.; Zhu, Y.; Yang, Y.; An, W.; Yang, Y.; Jie, J. The exosomes derived from CAR-T cell efficiently target mesothelin and reduce triple-negative breast cancer growth. Cell Immunol. 2021, 360, 104262. [Google Scholar] [CrossRef]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed Catalase Protects Chimeric Antigen Receptor-Redirected T Cells as well as Bystander Cells from Oxidative Stress-Induced Loss of Antitumor Activity. J. Immunol. 2016, 196, 759–766. [Google Scholar] [CrossRef] [Green Version]
- Klopotowska, M.; Bajor, M.; Graczyk-Jarzynka, A.; Kraft, G.A.; Pilch, Z.; Zhylko, A.; Firczuk, M.; Baranowska, I.; Lazniewski, M.; Plewczynski, D.; et al. PRDX-1 supports the survival and antitumor activity of primary and CAR-modified NK cells under oxidative stress. Cancer Immunol. Res. 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Chen, H.; Wei, F.; Yin, M.; Zhao, Q.; Liu, Z.; Yu, B.; Huang, Z. CD27 enhances the killing effect of CAR T cells targeting trophoblast cell surface antigen 2 in the treatment of solid tumors. Cancer Immunol. Immunother. 2021, 70, 2059–2071. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z. Tissue factor as a new target for CAR-NK cell immunotherapy of triple-negative breast cancer. Sci. Rep. 2020, 10, 2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portillo, A.L.; Hogg, R.; Poznanski, S.M.; Rojas, E.A.; Cashell, N.J.; Hammill, J.A.; Chew, M.V.; Shenouda, M.M.; Ritchie, T.M.; Cao, Q.T.; et al. Expanded human NK cells armed with CAR uncouple potent anti-tumor activity from off-tumor toxicity against solid tumors. iScience 2021, 24, 102619. [Google Scholar] [CrossRef]
- Eitler, J.; Wotschel, N.; Miller, N.; Boissel, L.; Klingemann, H.G.; Wels, W.; Tonn, T. Inability of granule polarization by NK cells defines tumor resistance and can be overcome by CAR or ADCC mediated targeting. J. Immunother. Cancer 2021, 9, e001334. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, Y.; Huang, K.H.; Fang, X.; Li, Y.; Wang, F.; An, L.; Chen, Q.; Zhang, Y.; Shi, A.; et al. Targeting epidermal growth factor-overexpressing triple-negative breast cancer by natural killer cells expressing a specific chimeric antigen receptor. Cell Prolif 2020, 53, e12858. [Google Scholar] [CrossRef] [PubMed]
- Fabian, K.P.; Padget, M.R.; Donahue, R.N.; Solocinski, K.; Robbins, Y.; Allen, C.T.; Lee, J.H.; Rabizadeh, S.; Soon-Shiong, P.; Schlom, J.; et al. PD-L1 targeting high-affinity NK (t-haNK) cells induce direct antitumor effects and target suppressive MDSC populations. J. Immunother. Cancer 2020, 8, e000450. [Google Scholar] [CrossRef]
- Schonfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bonig, H.; Kohl, U.; Kloess, S.; et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol. Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Rezvani, K.; Rouce, R.; Liu, E.; Shpall, E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol. Ther. 2017, 25, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Grosser, R.; Cherkassky, L.; Chintala, N.; Adusumilli, P.S. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 2019, 36, 471–482. [Google Scholar] [CrossRef]
- Yoon, D.H.; Osborn, M.J.; Tolar, J.; Kim, C.J. Incorporation of Immune Checkpoint Blockade into Chimeric Antigen Receptor T Cells (CAR-Ts): Combination or Built-In CAR-T. Int. J. Mol. Sci. 2018, 19, 340. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Han, X.; Bo, J.; Han, W. Target selection for CAR-T therapy. J. Hematol. Oncol. 2019, 12, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Stasi, A.; De Angelis, B.; Rooney, C.M.; Zhang, L.; Mahendravada, A.; Foster, A.E.; Heslop, H.E.; Brenner, M.K.; Dotti, G.; Savoldo, B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 2009, 113, 6392–6402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slaney, C.Y.; Kershaw, M.H.; Darcy, P.K. Trafficking of T cells into tumors. Cancer Res. 2014, 74, 7168–7174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Rui, W.; Zheng, H.; Huang, D.; Yu, F.; Zhang, Y.; Dong, J.; Zhao, X.; Lin, X. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur. J. Immunol. 2020, 50, 712–724. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Role in TME | Cell Type | Immunomodulatory Factors | Function |
|---|---|---|---|
| Anti-tumor | T cell | IL-1, IFN-γ | Tumor antigen recognition, killing tumor cells, promotion of inflammation in TME |
| B cell | Antibodies, IL-6, IL-21 | Production of antibodies, T cell activation | |
| NK cell | Granzyme, Perforin, IFN-γ, TNF-α | Activation of immune cells, MHC class I non-restricted recognition of tumor cells, killing tumor cells | |
| DC | IL-12, CXCL9, CXCL10 | Ag presentation to CD4+ and CD8+ T cells, T cell activation, induction of immunological response | |
| M1-like Mφ | IL-1β, IL-6, IL-12, CXCL9, CXCL10, IFN-γ, TNF-α | Tumor cell phagocytosis, promotion of immune response, facilitating cancer cell disruption | |
| N1-TAN | IL-1β, IL-6, IL-12, CXCL9, CXCL10, CXCL11,TNF-α, ROS | Activation of immune cells, killing tumor cells, promotion of inflammation in the tumor microenvironment | |
| Pro-tumor | T cell | IL-4, IL-6, IL-10, IL-13 | Inhibition of immune response, activation of immune checkpoints |
| Treg | IL-10, TGF-β | Inhibition of immune response, promotion of tumor vascularization, effector cell cytotoxicity impairment, disruption of metabolism, and modulation of antigen-presenting cells | |
| NK cell (CD56brightCD16+) | MMP9, VEGF, angiogenin | Increase tumor vascularization, proliferation of immunosuppressive cells, T cell exhaustion, reduction of T cell recruitment | |
| DC | CXCL8, TNF-α, VEGF, TGF-β | Inhibition of cytotoxic T cells, upregulation of regulatory T cells, increase tumor vascularization | |
| N2-TAN | CXCL8, IDO, Arg1, iNOS, MMP9, NE, VEGF | Inhibition of T cells and NK cells, ECM degradation, promotion of angiogenesis | |
| M2-like Mφ | CCL2, CXCL8, CXCL12, IL-10, TGF-β, Arg1, MMP2/9, VEGF, PGE2, ROS | Promotion of tumor vascularization, inhibition of cytotoxic T cells, promotion T cell differentiation into T reg, ECM degradation | |
| MDSC | IL-10, TGF-β, IDO, Arg1, MMP9, VEGF, ROS | Inactivation of T cells and NK cells, ECM degradation, promotion of angiogenesis, inhibition of T cell proliferation and induction of T cell apoptosis, attracting immunosuppressive cells |
| Treatment | Additional Treatment | A Phase of the Study | Clinical Trial ID | No. of Patients | Posted Results |
|---|---|---|---|---|---|
| Pertuzumab | Trastuzumab, paclitaxel | Phase II | NCT01276041 | 70 | CR = 15, PR = 27, SD = 17, PD = 1 |
| Trastuzumab Emtansine | - | Phase III | NCT01702571 | 2185 | median OS 95% CI 27.2 (25.5 to 28.7) |
| Trastuzumab emtansine | - | Phase III | NCT01419197 | 602 | 6-Month Survival = 90.9 (87.79 to 94.01) 1-Year Survival = 68.6 (59.91 to 77.28) median OS 95% CI NA (13.14 to NA) |
| Trastuzumab emtansine | - | Phase II | NCT00509769 | 112 | median PFS 95% CI 4.6 (3.9 to 8.6) |
| Gemcitabine Trastuzumab Pertuzumab | - | Phase I, II | NCT02139358 | 15 | median PFS 95% CI 6.4883 (2.7807 to 9.0372) |
| DS-8201a | - | Phase II | NCT03248492 | 253 | median DR 95% CI NA (8.3 to NA) |
| Trastuzumab | - | Phase I, II | NCT01325207 | 34 | CR = 0, PR = 6, SD = 18, PD = 10; median OS 95% CI 8.7 (5.6 to 17.3) |
| Technology | Additional Treatment | Subtype of BC | A Phase of the Study | Clinical Trial ID/Reference | No. of Patients | Posted Results |
|---|---|---|---|---|---|---|
| TIL therapy | ||||||
| TILs | IL-2 | BC | Phase I | NCT01462903 | 20 | - |
| CD3+ or CD8+ TILs | Aldesleukin Cyclophosphamide Fludarabine Pembrolizumab | Metastatic BC | Phase II | NCT01174121 [126] | 93 | - |
| TILs after stem cell transplantation | Aldesleukin Trastuzumab Paclitaxel Surgery | BC | Phase I | NCT00301730 | 1 | - |
| TILs (LN-145) | - | Metastatic TNBC | Phase II | NCT04111510 | 10 | - |
| Autologous Lymphoid Effector Cells Specific Against Tumor cells (ALECSAT) | Carboplatin Gemcitabine | TNBC | Phase Ib | NCT04609215 | 20 | - |
| TCR therapy | ||||||
| Neoepitopes | Nivolumab IL-2 | HER2+ | Phase I | NCT03970382 | 148 | - |
| Neoepitopes | Fludarabine Cyclophosphamide Aldesleukin | BC | Phase II | NCT04102436 [127,128,129] | 210 | - |
| Neoepitopes | Pembrolizumab Aldesleukin Fludarabine Cyclophosphamide | BC | Phase II | NCT03412877 | 10 | - |
| NY ESO-1 | Cyclophosphamide Fludarabine Aldesleukin | BC | Phase II | NCT01967823 [130] | 10 | CR = 1, PR = 5 |
| NY ESO-1 | Fludarabine Cyclophosphamide | BC | Phase I | NCT02457650 | 36 | - |
| NY ESO-1 | - | BC | Phase I | NCT03159585 | 6 | - |
| TAA-specific CTLs | - | HER2+ | Phase II | NCT03093350 | 10 | median PFS = 69.5 days (13 to 72), median OS = 116 days (37 to NA) |
| MAGE-A3 | Aldesleukin Fludarabine Cyclophosphamide | BC | Phase I, II | NCT02111850 | 21 | - |
| NK cell therapy | ||||||
| Activated NK cells | - | BC | Phase I, II | NCT03634501 | 200 | - |
| NK cells (DF1001) | Nivolumab or Nab paclitaxel | HER2+ | Phase I, II | NCT04143711 | 220 | - |
| iPSC-derived NK cells (FT500) | Nivolumab Pembrolizumab Atezolizumab Cyclophosphamide Fludarabine IL-2 | HER2+ | Phase I | NCT03841110 NCT04106167 | 37 76 | - |
| iPSC-derived NK cells (FT516) | Avelumab Cyclophosphamide Fludarabine IL-2 | TNBC | Phase I | NCT04551885 [131] | 12 | - |
| Target | CAR Technology | Additional Treatment | Subtype of BC | A Phase of the Study | Clinical Trial ID/References | No. of Patients |
|---|---|---|---|---|---|---|
| HER2, GD2, CD44v6 | multi CAR-T | - | HER2+ | Phase II | NCT04430595 | 100 |
| CD44v6 | single CAR-T | - | BC | Phase II | NCT04427449 | 100 |
| HER2 | HER2 (EQ) BBζ/CD19t + | - | HER2+ with brain metastases | Phase I | NCT03696030 | 39 |
| dual-switch CAR-T | - | HER2+ | Phase I | NCT04650451 | 220 | |
| single CAR-T | oncolytic adenovirus CAdVEC | HER2+ | Phase I | NCT03740256 | 45 | |
| single CAR-macrophages | - | HER2+ | Phase I clinical trial | NCT04660929 [139] | 18 | |
| HER2, PD-L1 | dual CAR-T | - | HER2+ with serosal cavity metastases | Early Phase I | NCT04684459 | 18 |
| MUC1 | huMNC2-CAR44 MUC1 | - | metastatic BC | Phase I | NCT04020575 | 69 |
| single CAR-T | - | TNBC | Phase II | NCT02587689 | 20 | |
| single CAR-pNK | - | TNBC | Phase II | NCT02839954 [140] | 10 | |
| TnMUC1 | single CAR-T | Cyclophosphamide, Fludarabine | TNBC | Phase I | NCT04025216 | 112 |
| Mesothelin | single CAR-T | Cyclophosphamide, AP1903 | HER2- | Phase I | NCT02792114 | 186 |
| single CAR-T | Cyclophosphamide or pembrolizumab | BC | Phase II | NCT02414269 | 113 | |
| EpCAM | single CAR-T | - | HER2+, TNBC | Phase I | NCT02915445 | 30 |
| c-Met | mRNA CAR-T | - | TNBC, metastatic BC | Phase I | NCT01837602 [141] | 6 |
| Nectin4/FAP | single CAR-T | - | advanced BC | Phase I | NCT03932565 [142,143] | 30 |
| CEA | single CAR-T | - | BC | Phase I | NCT02349724 | 75 |
| single CAR-T | - | BC | Phase II | NCT04348643 | 40 | |
| ROR1 | single CAR-T | - | TNBC | Phase I | NCT02706392 | 60 |
| NKG2DL | single CAR-Tγδ | - | TNBC | Phase I | NCT04107142 | 10 |
| CT303-406 | single CAR-T | Cyclophosphamide, Fludarabine | HER2+ | Phase I | NCT04511871 | 15 |
| PSMA | UniCAR02-T-pPSMA | Cyclophosphamide, Fludarabine | PSMA+ BC | Phase I | NCT04633148 | 35 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Retecki, K.; Seweryn, M.; Graczyk-Jarzynka, A.; Bajor, M. The Immune Landscape of Breast Cancer: Strategies for Overcoming Immunotherapy Resistance. Cancers 2021, 13, 6012. https://doi.org/10.3390/cancers13236012
Retecki K, Seweryn M, Graczyk-Jarzynka A, Bajor M. The Immune Landscape of Breast Cancer: Strategies for Overcoming Immunotherapy Resistance. Cancers. 2021; 13(23):6012. https://doi.org/10.3390/cancers13236012
Chicago/Turabian StyleRetecki, Kuba, Milena Seweryn, Agnieszka Graczyk-Jarzynka, and Malgorzata Bajor. 2021. "The Immune Landscape of Breast Cancer: Strategies for Overcoming Immunotherapy Resistance" Cancers 13, no. 23: 6012. https://doi.org/10.3390/cancers13236012
APA StyleRetecki, K., Seweryn, M., Graczyk-Jarzynka, A., & Bajor, M. (2021). The Immune Landscape of Breast Cancer: Strategies for Overcoming Immunotherapy Resistance. Cancers, 13(23), 6012. https://doi.org/10.3390/cancers13236012