
Metabolic Rewiring Is Essential for AML Cell Survival to Overcome Autophagy Inhibition by Loss of ATG3

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. CRISPR/Cas9 Proliferation Screen
2.3. Flow Cytometry Analyses
2.4. Immunofluorescence Staining
2.5. Assessment of Mitochondrial Activity
2.6. NMR-Based Metabolomics and Spectrophotometry
2.7. Statistical Analysis
3. Results
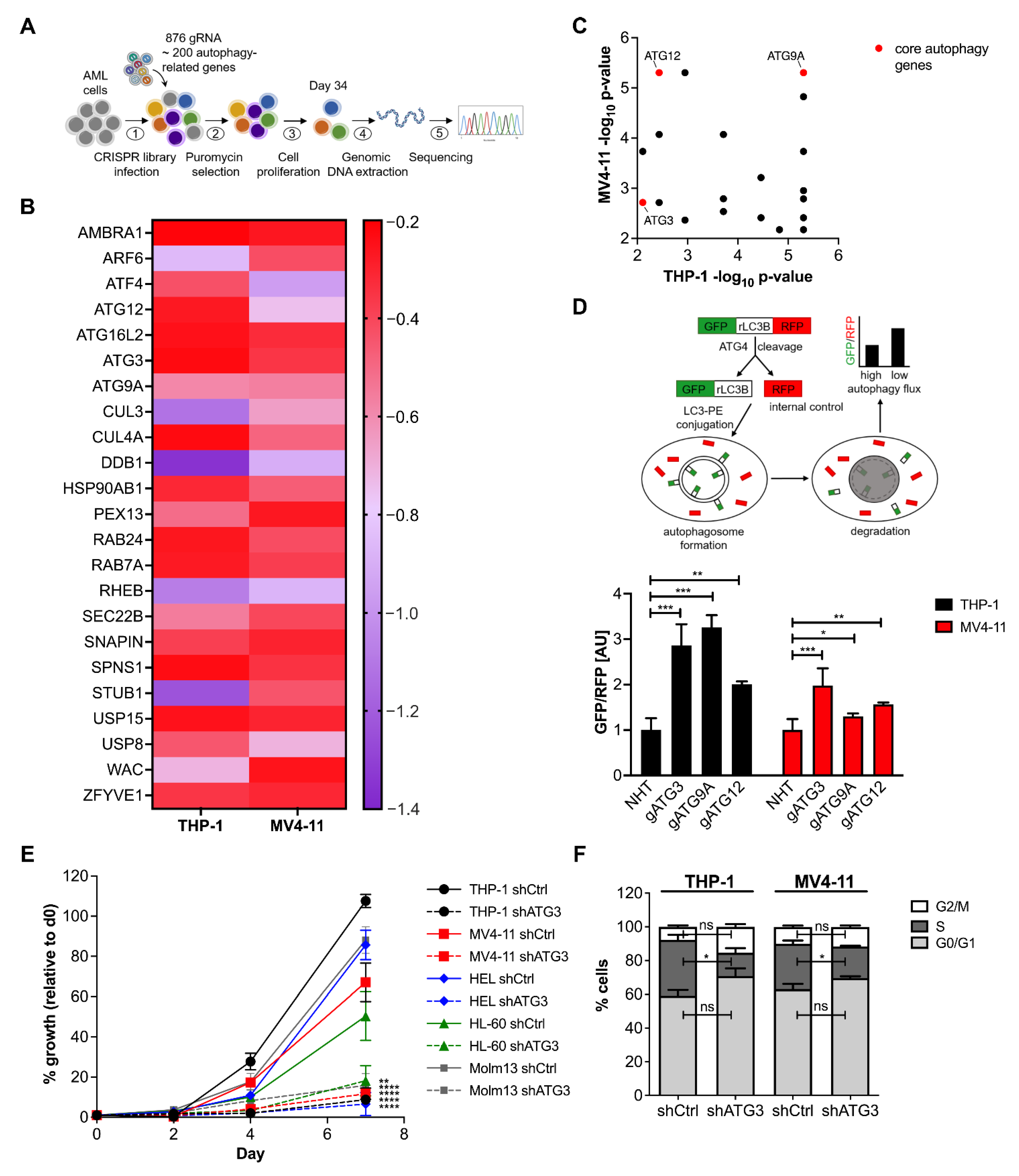
3.1. Identification of Critical Autophagy Genes for AML Survival by CRISPR/Cas9 Knockout Screen
3.2. Loss of ATG3 Effectively Inhibits Autophagy Flux Accompanied by Increased Cell Death While Retaining Cell Survival
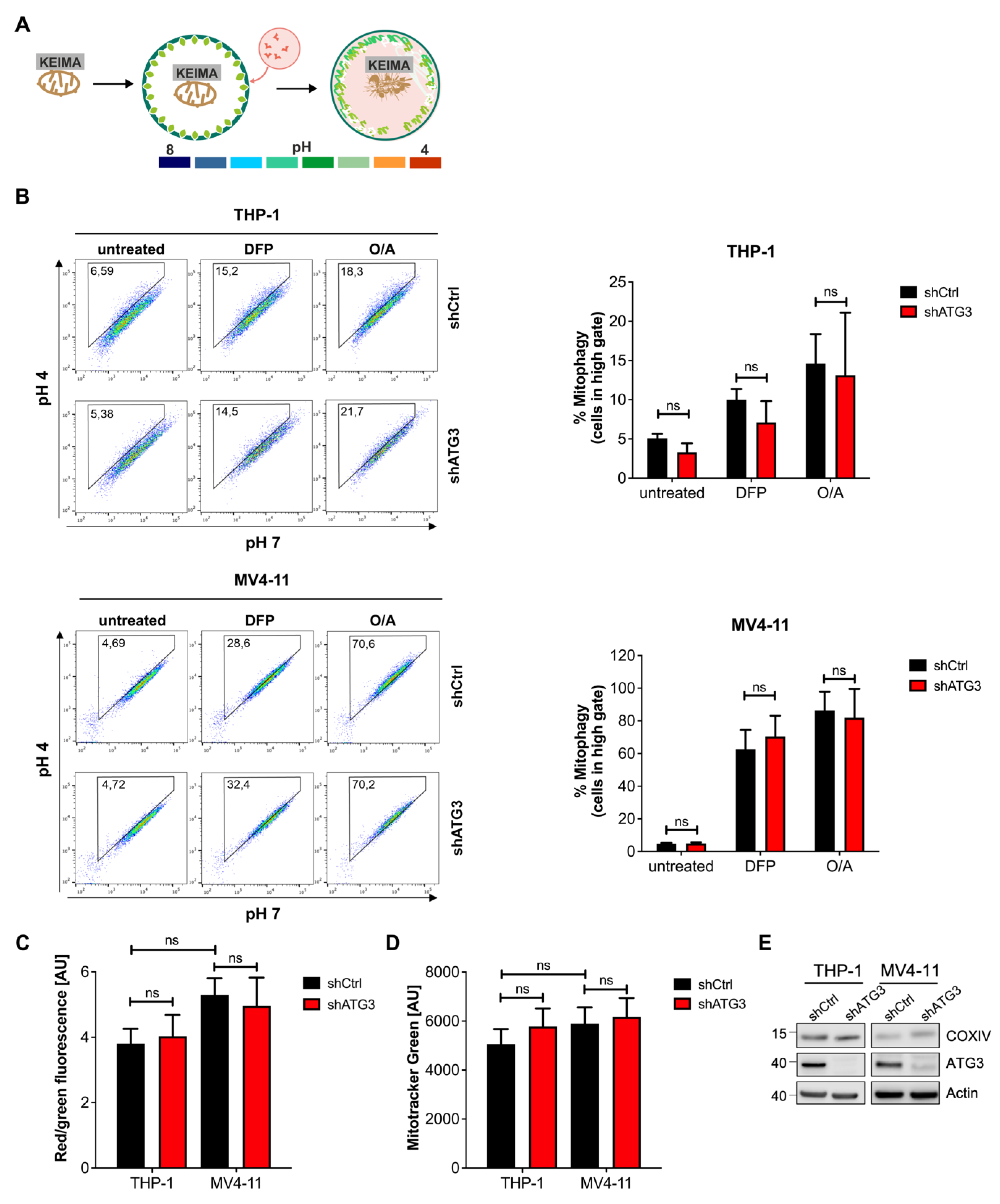
3.3. Mitochondrial Homeostasis Maintained by Mitophagy Is Not Disrupted by Inhibition of Autophagy upon Loss of ATG3
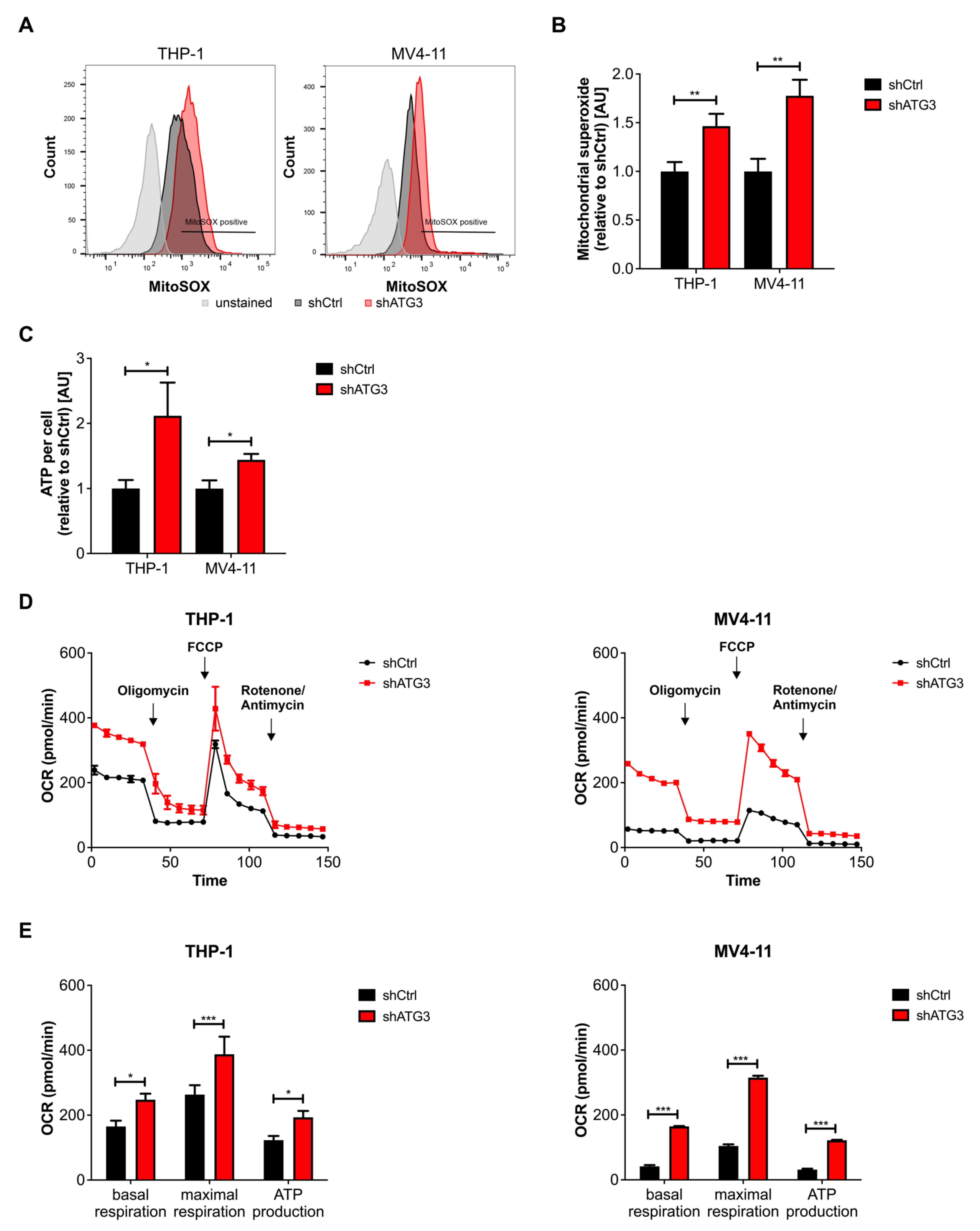
3.4. Loss of ATG3 Increases Mitochondrial Activity
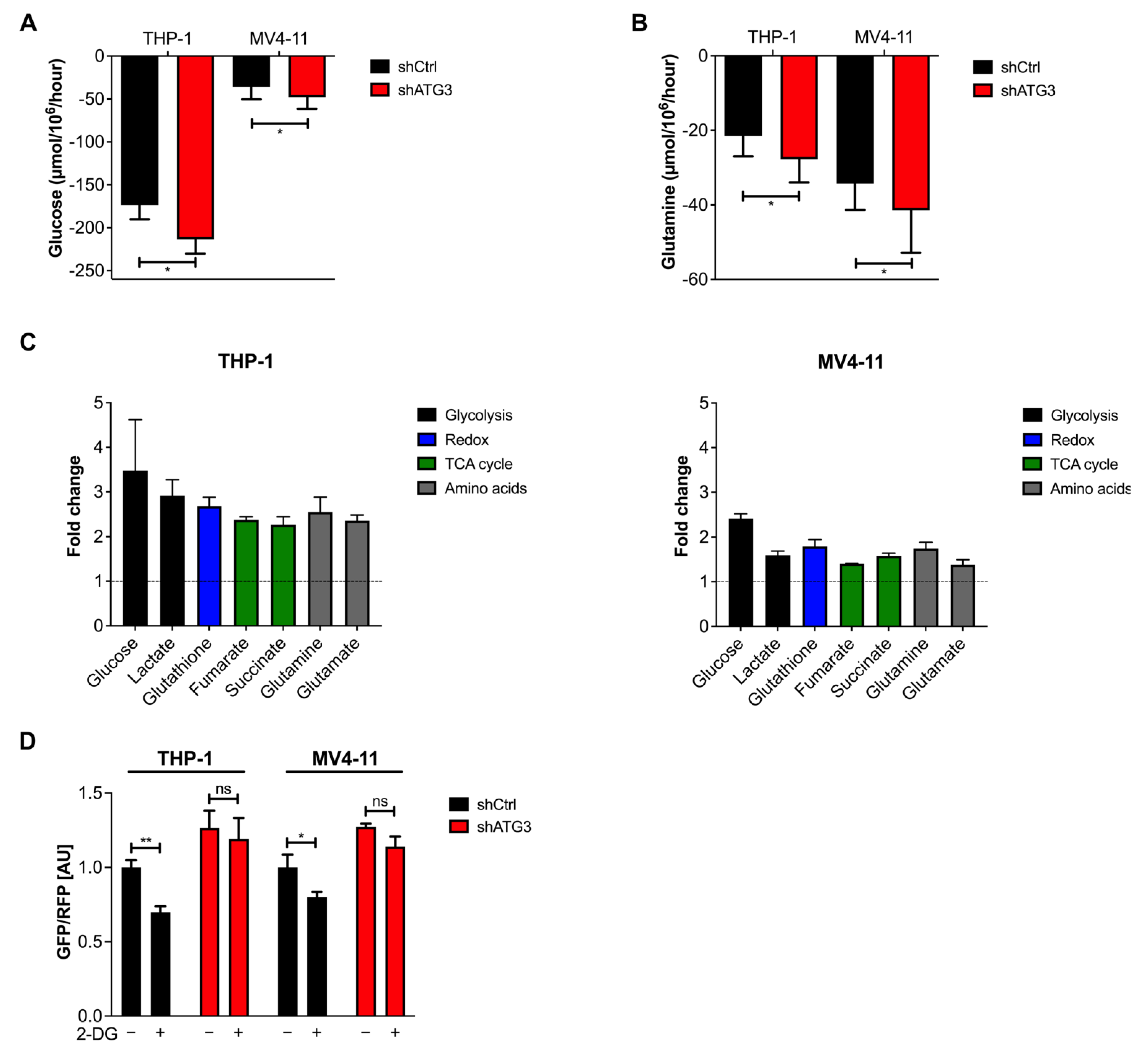
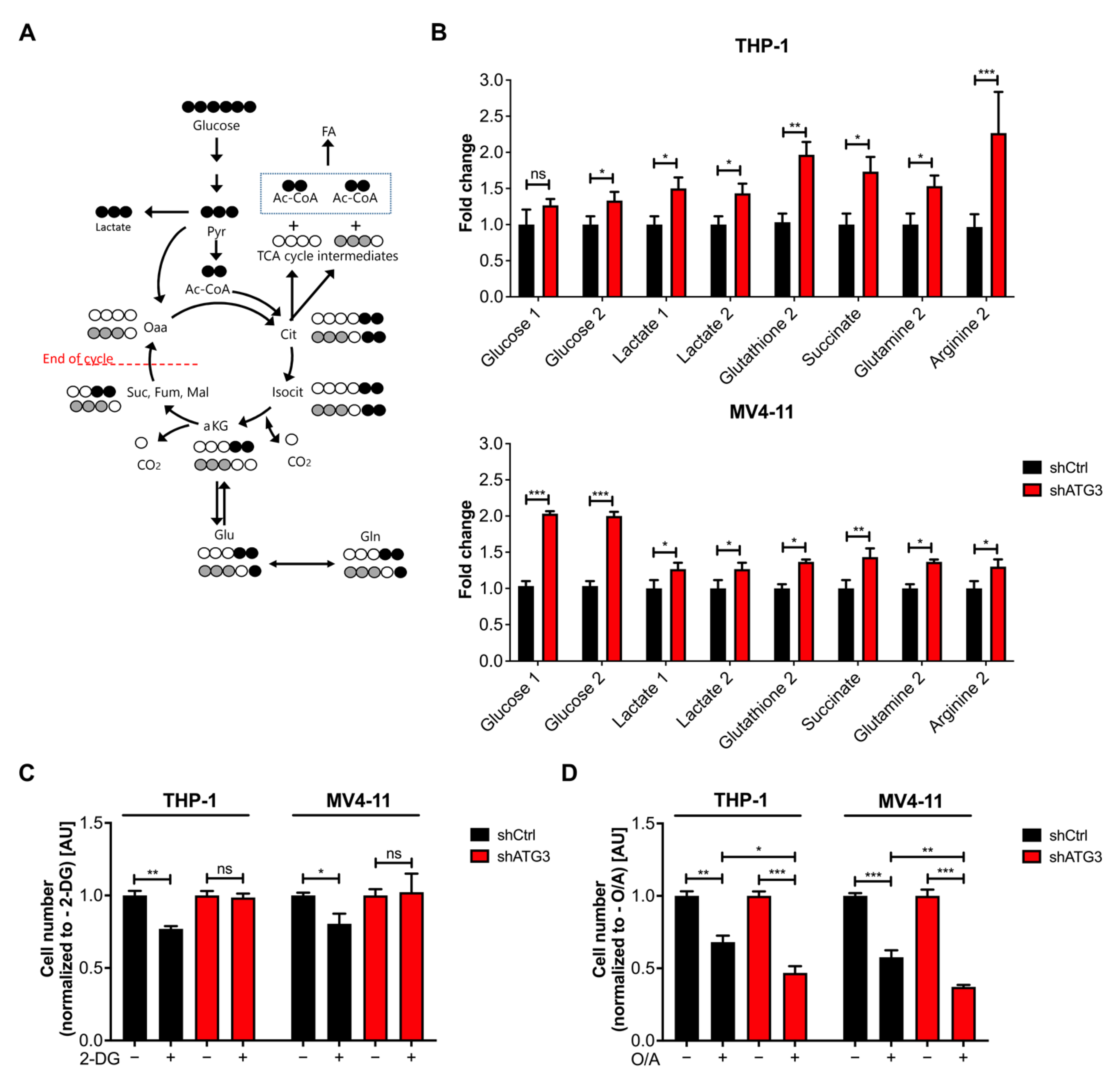
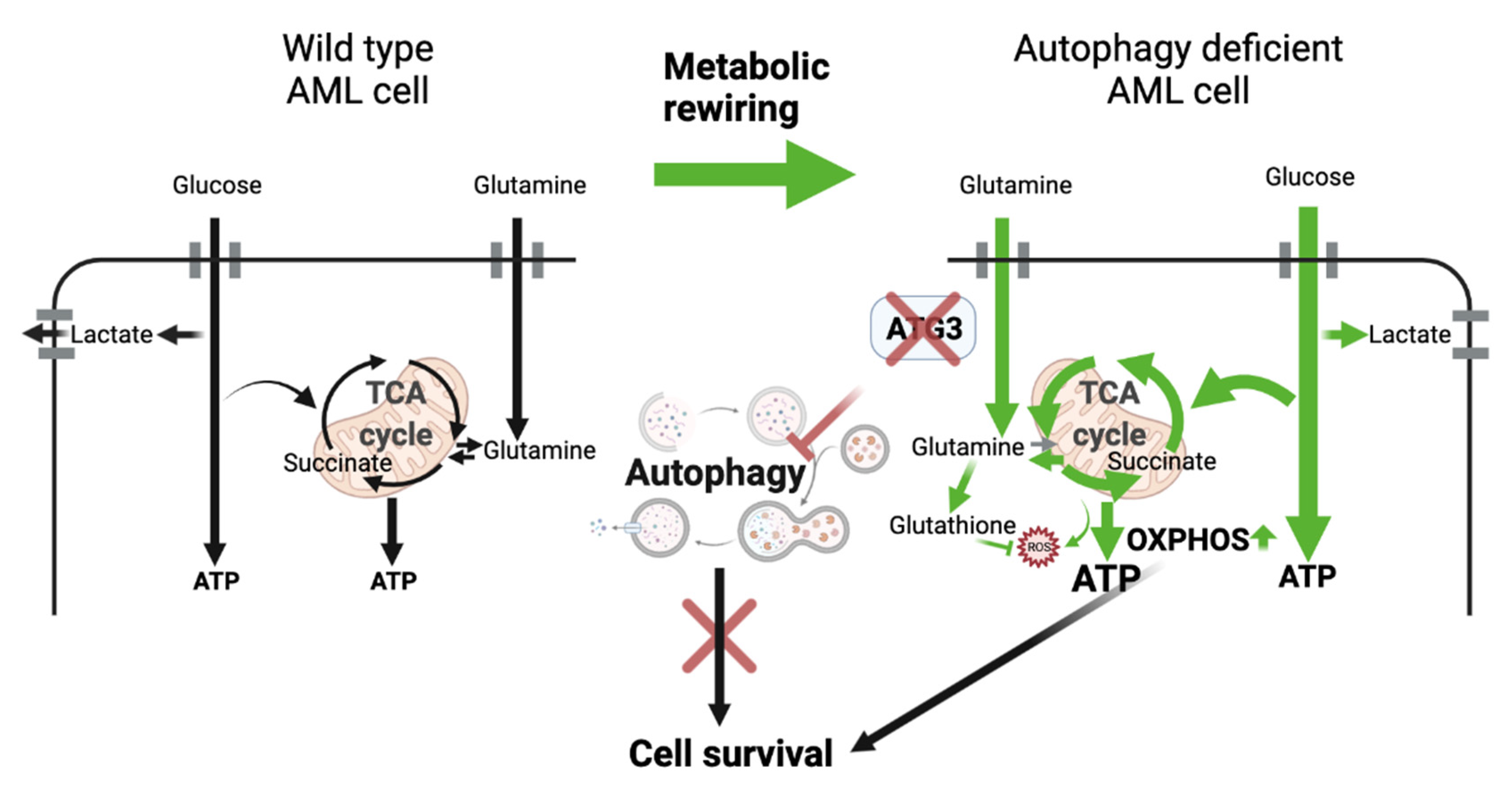
3.5. Loss of ATG3 Rewires Central Carbon Metabolism
3.6. The Glycolytic Pathway Is Highly Active in AML Cells upon Loss of ATG3
3.7. Survival of ATG3 Deficient AML Cells Relies on OXPHOS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Folkerts, H.; Hilgendorf, S.; Wierenga, A.T.J.; Jaques, J.; Mulder, A.B.; Coffer, P.J.; Schuringa, J.J.; Vellenga, E. Inhibition of autophagy as a treatment strategy for p53 wild-type acute myeloid leukemia. Cell Death Dis. 2017, 8, e2927. [Google Scholar] [CrossRef] [Green Version]
- Dykstra, K.M.; Fay, H.R.S.; Massey, A.C.; Yang, N.; Johnson, M.; Portwood, S.; Guzman, M.L.; Wang, E.S. Inhibiting autophagy targets human leukemic stem cells and hypoxic AML blasts by disrupting mitochondrial homeostasis. Blood Adv. 2021, 5, 2087–2100. [Google Scholar] [CrossRef]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Auberger, P.; Puissant, A. Autophagy, a key mechanism of oncogenesis and resistance in leukemia. Blood 2017, 129, 547–552. [Google Scholar] [CrossRef]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.W.; Lee, S.H. The roles of autophagy in cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.D.; Shaid, S.; Vakhrusheva, O.; Koschade, S.E.; Klann, K.; Thölken, M.; Baker, F.; Zhang, J.; Oellerich, T.; Sürün, D.; et al. Loss of the selective autophagy receptor p62 impairs murine myeloid leukemia progression and mitophagy. Blood 2019, 133, 168–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, S.; Minhajuddin, M.; Adane, B.; Khan, N.; Stevens, B.M.; Mack, S.C.; Lai, S.; Rich, J.N.; Inguva, A.; Shannon, K.M.; et al. AMPK/FIS1-Mediated Mitophagy Is Required for Self-Renewal of Human AML Stem Cells. Cell Stem Cell 2018, 23, 86–100. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, Y.; Yin, J.; Wang, C.; Yang, M.; Gu, J.; He, M.; Xu, H.; Fu, W.; Zhang, W.; et al. A mitophagy inhibitor targeting p62 attenuates the leukemia-initiation potential of acute myeloid leukemia cells. Cancer Lett. 2021, 510, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Perez-Hernandez, M.; Arias, A.; Martinez-Garcia, D.; Perez-Tomas, R.; Quesada, R.; Soto-Cerrato, V. Targeting Autophagy for Cancer Treatment and Tumor Chemosensitization. Cancers 2019, 11, 1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumitomo, Y.; Koya, J.; Nakazaki, K.; Kataoka, K.; Tsuruta-Kishino, T.; Morita, K.; Sato, T.; Kurokawa, M. Cytoprotective autophagy maintains leukemia-initiating cells in murine myeloid leukemia. Blood 2016, 128, 1614–1624. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Chen, L.; Atkinson, J.M.; Claxton, D.F.; Wang, H.G. Atg5-dependent autophagy contributes to the development of acute myeloid leukemia in an MLL-AF9-driven mouse model. Cell Death Dis. 2016, 7, e2361. [Google Scholar] [CrossRef]
- Martens, S.; Fracchiolla, D. Activation and targeting of ATG8 protein lipidation. Cell Discov. 2020, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Y.; Ma, C. Atg3 promotes Atg8 lipidation via altering lipid diffusion and rearrangement. Protein Sci. 2020, 29, 1511–1523. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Qiu, Y.; Grace, C.R.R.; Liu, X.; Klionsky, D.J.; Schulman, B.A. A switch element in the autophagy E2 Atg3 mediates allosteric regulation across the lipidation cascade. Nat. Commun. 2019, 10, 3600. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Jacobs, S.R.; Mason, E.F.; Michalek, R.D.; MacIntyre, A.N.; Coloff, J.L.; Ilkayeva, O.; Jia, W.; He, Y.W.; Rathmell, J.C. Autophagy is essential to suppress cell stress and to allow BCR-Abl-mediated leukemogenesis. Oncogene 2011, 30, 1855–1867. [Google Scholar] [CrossRef] [Green Version]
- Wegner, M.; Diehl, V.; Bittl, V.; De Bruyn, R.; Wiechmann, S.; Matthess, Y.; Hebel, M.; Hayes, M.G.B.; Schaubeck, S.; Benner, C.; et al. Circular synthesized CRISPR/Cas gRNAs for functional interrogations in the coding and noncoding genome. eLife 2019, 8, e42549. [Google Scholar] [CrossRef]
- Carrigan, J.B.; Reed, M.A.C.; Ludwig, C.; Khanim, F.L.; Bunce, C.M.; Günther, U.L. Tracer-Based Metabolic NMR-Based Flux Analysis in a Leukaemia Cell Line. Chempluschem 2016, 81, 453–459. [Google Scholar] [CrossRef]
- Ludwig, C.; Günther, U.L. MetaboLab—Advanced NMR data processing and analysis for metabolomics. BMC Bioinform. 2011, 12, 366. [Google Scholar] [CrossRef] [Green Version]
- Alshamleh, I.; Krause, N.; Richter, C.; Kurrle, N.; Serve, H.; Günther, U.L.; Schwalbe, H. Real-Time NMR Spectroscopy for Studying Metabolism. Angew. Chem. Int. Ed. 2020, 59, 2304–2308. [Google Scholar] [CrossRef]
- Diehl, V.; Wegner, M.; Grumati, P.; Husnjak, K.; Schaubeck, S.; Gubas, A.; Shah, V.J.; Polat, I.H.; Langschied, F.; Prieto-Garcia, C.; et al. Minimized combinatorial CRISPR screens identify genetic interactions in autophagy. Nucleic Acids Res. 2021, 49, 5684–5704. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.; Jang, I.; Han, H.; Kim, M.S.; Choi, J.; Lee, J.; Cho, S.Y.; Jun, Y.; Lee, C.; Kim, J.; et al. iCSDB: An integrated database of CRISPR screens. Nucleic Acids Res. 2021, 49, D956–D961. [Google Scholar] [CrossRef]
- Lebovitz, C.B.; Robertson, A.G.; Goya, R.; Jones, S.J.; Morin, R.D.; Marra, M.A.; Gorski, S.M. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy 2015, 11, 1668–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.G.; Zhang, H. Core autophagy genes and human diseases. Curr. Opin. Cell Biol. 2019, 61, 117–125. [Google Scholar] [CrossRef]
- Ishibashi, K.; Fujita, N.; Kanno, E.; Omori, H.; Yoshimori, T.; Itoh, T.; Fukuda, M. Atg16L2, a novel isoform of mammalian Atg16L that is not essential for canonical autophagy despite forming an Atg12-5-16L2 complex. Autophagy 2011, 7, 1500–1513. [Google Scholar] [CrossRef] [PubMed]
- Khor, B.; Conway, K.L.; Omar, A.S.; Biton, M.; Haber, A.L.; Rogel, N.; Baxt, L.A.; Begun, J.; Kuballa, P.; Gagnon, J.D.; et al. Distinct Tissue-Specific Roles for the Disease-Associated Autophagy Genes ATG16L2 and ATG16L1. J. Immunol. 2019, 203, 1820–1829. [Google Scholar] [CrossRef]
- Kaizuka, T.; Morishita, H.; Hama, Y.; Tsukamoto, S.; Matsui, T.; Toyota, Y.; Kodama, A.; Ishihara, T.; Mizushima, T.; Mizushima, N. An Autophagic Flux Probe that Releases an Internal Control. Mol. Cell 2016, 64, 835–849. [Google Scholar] [CrossRef] [Green Version]
- Tsuboyama, K.; Koyama-Honda, I.; Sakamaki, Y.; Koike, M.; Morishita, H.; Mizushima, N. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 2016, 354, 1036–1041. [Google Scholar] [CrossRef]
- Sou, Y.; Satoshi, W.; Jun-ichi, I.; Takashi, U.; Tsutomu, F.; Taichi, H.; Sawada, N.; Yamada, A.; Mizushima, N.; Uchiyama, Y.; et al. The Atg8 Conjugation System Is Indispensable for Proper Development of Autophagic Isolation Membranes in Mice. Mol. Biol. Cell 2008, 19, 4762–4775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, I.; Martín, G.; Bolufer, P.; Barragán, E.; Rueda, E.; Román, J.; Fernández, P.; León, P.; Mena, A.; Cervera, J.; et al. Incidence and prognostic value of FLT3 internal tandem duplication and D835 mutations in acute myeloid leukemia. Haematologica 2003, 88, 19–24. [Google Scholar]
- Dutta, S.; Pregartner, G.; Rücker, F.G.; Heitzer, E.; Zebisch, A.; Bullinger, L.; Berghold, A.; Döhner, K.; Sill, H. Functional classification of tp53 mutations in acute myeloid leukemia. Cancers 2020, 12, 637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, T.; Jiang, N.; Liao, H.; Shuai, X.; Su, J.; Zheng, Q. The FLT3-ITD mutation and the expression of its downstream signaling intermediates STAT5 and Pim-1 are positively correlated with CXCR4 expression in patients with acute myeloid leukemia. Sci. Rep. 2019, 9, 12209. [Google Scholar] [CrossRef] [PubMed]
- Reiter, K.; Polzer, H.; Krupka, C.; Maiser, A.; Vick, B.; Rothenberg-Thurley, M.; Metzeler, K.H.; Dörfel, D.; Salih, H.R.; Jung, G.; et al. Tyrosine kinase inhibition increases the cell surface localization of FLT3-ITD and enhances FLT3-directed immunotherapy of acute myeloid leukemia. Leukemia 2018, 32, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Elmeliegy, M.; Den Haese, J.; Talati, C.; Wetzler, M.; Jusko, W.J. Towards better combination regimens of cytarabine and FLT3 inhibitors in acute myeloid leukemia. Cancer Chemother. Phramacol. 2020, 86, 325–337. [Google Scholar] [CrossRef]
- Yan, B.; Chen, Q.; Xu, J.; Li, W.; Xu, B.; Qiu, Y. Low-frequency TP53 hotspot mutation contributes to chemoresistance through clonal expansion in acute myeloid leukemia. Leukemia 2020, 34, 1816–1827. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Halilovic, E.; Cooke, V.G.; Nonami, A.; Ren, T.; Sanda, T.; Simkin, I.; Yuan, J.; Antonakos, B.; Barys, L.; et al. Inhibition of wild-type p53-expressing AML by the novel small molecule HDM2 inhibitor CGM097. Mol. Cancer Ther. 2015, 14, 2249–2259. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, K.; Toyoshima, H.; Sakai, R.; Miyagawa, K.; Hagiwara, K.; Ishikawa, F.; Takaku, F.; Yazaki, Y.; Hirai, H. Frequent mutations in the p53 gene in human myeloid leukemia cell lines. Blood 1992, 79, 2378–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, H.; Kogure, T.; Mizushima, N.; Yoshimori, T.; Miyawaki, A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem. Biol. 2011, 18, 1042–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Chu, Y.; Chang, Y.; Lu, W.; Sheng, X.; Wang, S.; Xu, H.; Ma, J. Regulation of autophagy by glycolysis in cancer. Cancer Manag. Res. 2020, 12, 13259–13271. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, F.; Fan, N.; Zhou, C.; Li, D.; Macvicar, T.; Dong, Q.; Bruns, C.J.; Zhao, Y. Targeting Glutaminolysis: New Perspectives to Understand Cancer Development and Novel Strategies for Potential Target Therapies. Front. Oncol. 2020, 10, 589508. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.; Riffelmacher, T.; Stranks, A.; Williams, O.; De Boer, J.; Cain, K.; MacFarlane, M.; McGouran, J.; Kessler, B.; Khandwala, S.; et al. Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discov. 2015, 1, 15008. [Google Scholar] [CrossRef]
- Evangelisti, C.; Evangelisti, C.; Chiarini, F.; Lonetti, A.; Buontempo, F.; Neri, L.M.; McCubrey, J.A.; Martelli, A.M. Autophagy in acute leukemias: A double-edged sword with important therapeutic implications. Biochim. Biophys. Acta—Mol. Cell Res. 2015, 1853, 14–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, K.; Hama, Y.; Izume, T.; Tamura, N.; Ueno, T.; Yamashita, Y.; Sakamaki, Y.; Mimura, K.; Morishita, H.; Shihoya, W.; et al. Genome-wide CRISPR screen identifies TMEM41B as a gene required for autophagosome formation. J. Cell Biol. 2018, 217, 3817–3828. [Google Scholar] [CrossRef] [Green Version]
- Radoshevich, L.; Murrow, L.; Chen, N.; Fernandez, E.; Roy, S.; Fung, C.; Debnath, J. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell 2010, 142, 590–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Zhao, Q.; Liu, P.; Cao, J.; Gong, J.; Wang, C.; Wang, W.; Li, X.; Sun, H.; Zhang, C.; et al. ATG3-dependent autophagy mediates mitochondrial homeostasis in pluripotency acquirement and maintenance. Autophagy 2016, 12, 2000–2008. [Google Scholar] [CrossRef] [Green Version]
- Besteiro, S.; Brooks, C.F.; Striepen, B.; Dubremetz, J.F. Autophagy protein Atg3 is essential for maintaining mitochondrial integrity and for normal intracellular development of toxoplasma gondii tachyzoites. PLoS Pathog. 2011, 7, e1002416. [Google Scholar] [CrossRef] [Green Version]
- Strohecker, A.M.; Guo, J.Y.; Karsli-Uzunbas, G.; Price, S.M.; Chen, G.; Mathew, R.; McMahon, M.; White, E. Autophagy Sustains Mitochondrial Glutamine Metabolism and Growth of BRAFV600E–Driven Lung Tumors. Cancer Discov. 2013, 3, 1272–1285. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016, 30, 1704–1717. [Google Scholar] [CrossRef] [Green Version]
- Hirota, Y.; Yamashita, S.-i.; Kurihara, Y.; Jin, X.; Aihara, M.; Saigusa, T.; Kang, D.; Kanki, T. Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy 2015, 11, 332–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Heredia, J.M.; Carnero, A. Role of Mitochondria in Cancer Stem Cell Resistance. Cells 2020, 9, 1693. [Google Scholar] [CrossRef]
- Karvela, M.; Baquero, P.; Kuntz, E.M.; Mukhopadhyay, A.; Mitchell, R.; Allan, E.K.; Chan, E.; Kranc, K.R.; Calabretta, B.; Salomoni, P.; et al. ATG7 regulates energy metabolism, differentiation and survival of Philadelphia-chromosome-positive cells. Autophagy 2016, 12, 936–948. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, D.; Hirata, N.; Miyashita, R.; Kawaguchi, R.; Yamakage, M. Substrate-dependent modulation of oxidative phosphorylation in isolated mitochondria following in vitro hypoxia and reoxygenation injury. Exp. Clin. Cardiol. 2013, 18, 158–160. [Google Scholar]
- Leverve, X.M.; Fontaine, E. Role of substrates in the regulation of mitochondrial function in situ. IUBMB Life 2001, 52, 221–229. [Google Scholar] [CrossRef]
- Sundqvist, M.; Christenson, K.; Björnsdottir, H.; Osla, V.; Karlsson, A.; Dahlgren, C.; Speert, D.P.; Fasth, A.; Brown, K.L.; Bylund, J. Elevated mitochondrial reactive oxygen species and cellular redox imbalance in human NADPH-oxidase-deficient phagocytes. Front. Immunol. 2017, 8, 1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanzo, R.; Bartkova, J.; Merchut-Maya, J.M.; Hall, A.; Bouchal, J.; Dyrskjøt, L.; Frankel, L.B.; Gorgoulis, V.; Maya-Mendoza, A.; Jäättelä, M.; et al. Autophagy role(s) in response to oncogenes and DNA replication stress. Cell Death Differ. 2019, 27, 1134–1153. [Google Scholar] [CrossRef]
- Hill, B.G.; Benavides, G.A.; Lancaster, J.J.R.; Ballinger, S.; Dell’Italia, L.; Zhang, J.; Darley-Usmar, V.M. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol. Chem. 2012, 393, 1485–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divakaruni, A.S.; Brand, M.D. The regulation and physiology of mitochondrial proton leak. Physiology 2011, 26, 192–205. [Google Scholar] [CrossRef]
- Demarquoy, J.; Le Borgne, F. Crosstalk between mitochondria and peroxisomes. World J. Biol. Chem. 2015, 6, 301–309. [Google Scholar] [CrossRef]
- Edlund, J.; Fasching, A.; Liss, P.; Hansell, P.; Palm, F. The roles of NADPH-oxidase and nNOS for the increased oxidative stress and the oxygen consumption in the diabetic kidney. Diabetes Metab. Res. Rev. 2010, 26, 349–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polat, I.H.; Tarrado-castellarnau, M.; Benito, A.; Hernandez-carro, C. Glutamine Modulates Expression and Function of Glucose 6-Phosphate Dehydrogenase via NRF2 in Colon Cancer Cells. Antioxidants 2021, 10, 1349. [Google Scholar] [CrossRef]
- Pei, S.; Minhajuddin, M.; Callahan, K.P.; Balys, M.; Ashton, J.M.; Neering, S.J.; Lagadinou, E.D.; Corbett, C.; Ye, H.; Liesveld, J.L.; et al. Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J. Biol. Chem. 2013, 288, 33542–33558. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, L.; Sandhu, J.K.; Harper, M.E.; Cuperlovic-culf, M. Role of glutathione in cancer: From mechanisms to therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Zhang, Z.; Hoshino, A.; Zheng, H.D.; Morley, M.; Arany, Z.; Rabinowitz, J.D. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat. Metab. 2019, 1, 404–415. [Google Scholar] [CrossRef]
- Jia, D.; Lu, M.; Jung, K.H.; Park, J.H.; Yu, L.; Onuchic, J.N.; Kaipparettu, B.A.; Levine, H. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 3909–3918. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Wu, H.; Dai, C.; Pan, Q.; Ding, Z.; Hu, D.; Ji, B.; Luo, Y.; Hu, X. Beyond Warburg effect—Dual metabolic nature of cancer cells. Sci. Rep. 2014, 4, 4927. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.J.; Mahieu, N.G.; Huang, X.; Singh, M.; Crawford, P.A.; Johnson, S.L.; Gross, R.W.; Schaefer, J.; Patti, G.J. Lactate metabolism is associated with mammalian mitochondria. Nat. Chem. Biol. 2016, 12, 937–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic control of Autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiratori, R.; Furuichi, K.; Yamaguchi, M.; Miyazaki, N.; Aoki, H.; Chibana, H.; Ito, K.; Aoki, S. Glycolytic suppression dramatically changes the intracellular metabolic profile of multiple cancer cell lines in a mitochondrial metabolism-dependent manner. Sci. Rep. 2019, 9, 18699. [Google Scholar] [CrossRef] [Green Version]
- Škrtić, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriskanthadevan, S.; Jeyaraju, D.V.; Chung, T.E.; Prabha, S.; Xu, W.; Skrtic, M.; Jhas, B.; Hurren, R.; Gronda, M.; Wang, X.; et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood 2015, 125, 2120–2130. [Google Scholar] [CrossRef] [Green Version]
- Panina, S.B.; Baran, N.; Brasil da Costa, F.H.; Konopleva, M.; Kirienko, N.V. A mechanism for increased sensitivity of acute myeloid leukemia to mitotoxic drugs. Cell Death Dis. 2019, 10, 617. [Google Scholar] [CrossRef] [Green Version]
- Panina, S.B.; Pei, J.; Kirienko, N.V. Mitochondrial metabolism as a target for acute myeloid leukemia treatment. Cancer Metab. 2021, 9, 17. [Google Scholar] [CrossRef]
- Suganuma, K.; Miwa, H.; Imai, N.; Shikami, M.; Gotou, M.; Goto, M.; Mizuno, S.; Takahashi, M.; Yamamoto, H.; Hiramatsu, A.; et al. Energy metabolism of leukemia cells: Glycolysis versus oxidative phosphorylation. Leuk. Lymphoma 2010, 51, 2112–2119. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Neuzil, J. Targeting mitochondria as an anticancer strategy. Cancer Commun. 2019, 39, 63. [Google Scholar] [CrossRef] [Green Version]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.N.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, S.; Piya, S.; McQueen, T.; Konopleva, M.; Andreeff, M.; Borthakur, G. Inhibition of Unc-51 like Autophagy Activating Kinase 1 (ULK1) Is Highly Synergistic with Chemotherapy and Bcl2 Inhibition in Acute Myeloid Leukemia (AML). Blood 2017, 130, 1248. [Google Scholar] [CrossRef]
- Mirabelli, P.; Coppola, L.; Salvatore, M. Cancer cell lines are useful model systems for medical research. Cancers 2019, 11, 1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baker, F.; Polat, I.H.; Abou-El-Ardat, K.; Alshamleh, I.; Thoelken, M.; Hymon, D.; Gubas, A.; Koschade, S.E.; Vischedyk, J.B.; Kaulich, M.; et al. Metabolic Rewiring Is Essential for AML Cell Survival to Overcome Autophagy Inhibition by Loss of ATG3. Cancers 2021, 13, 6142. https://doi.org/10.3390/cancers13236142
Baker F, Polat IH, Abou-El-Ardat K, Alshamleh I, Thoelken M, Hymon D, Gubas A, Koschade SE, Vischedyk JB, Kaulich M, et al. Metabolic Rewiring Is Essential for AML Cell Survival to Overcome Autophagy Inhibition by Loss of ATG3. Cancers. 2021; 13(23):6142. https://doi.org/10.3390/cancers13236142
Chicago/Turabian StyleBaker, Fatima, Ibrahim H. Polat, Khalil Abou-El-Ardat, Islam Alshamleh, Marlyn Thoelken, Daniel Hymon, Andrea Gubas, Sebastian E. Koschade, Jonas B. Vischedyk, Manuel Kaulich, and et al. 2021. "Metabolic Rewiring Is Essential for AML Cell Survival to Overcome Autophagy Inhibition by Loss of ATG3" Cancers 13, no. 23: 6142. https://doi.org/10.3390/cancers13236142
APA StyleBaker, F., Polat, I. H., Abou-El-Ardat, K., Alshamleh, I., Thoelken, M., Hymon, D., Gubas, A., Koschade, S. E., Vischedyk, J. B., Kaulich, M., Schwalbe, H., Shaid, S., & Brandts, C. H. (2021). Metabolic Rewiring Is Essential for AML Cell Survival to Overcome Autophagy Inhibition by Loss of ATG3. Cancers, 13(23), 6142. https://doi.org/10.3390/cancers13236142