
Pediatric versus Adult Medulloblastoma: Towards a Definition That Goes beyond Age

Abstract
:Simple Summary
Abstract
1. Background
2. Materials and Methods
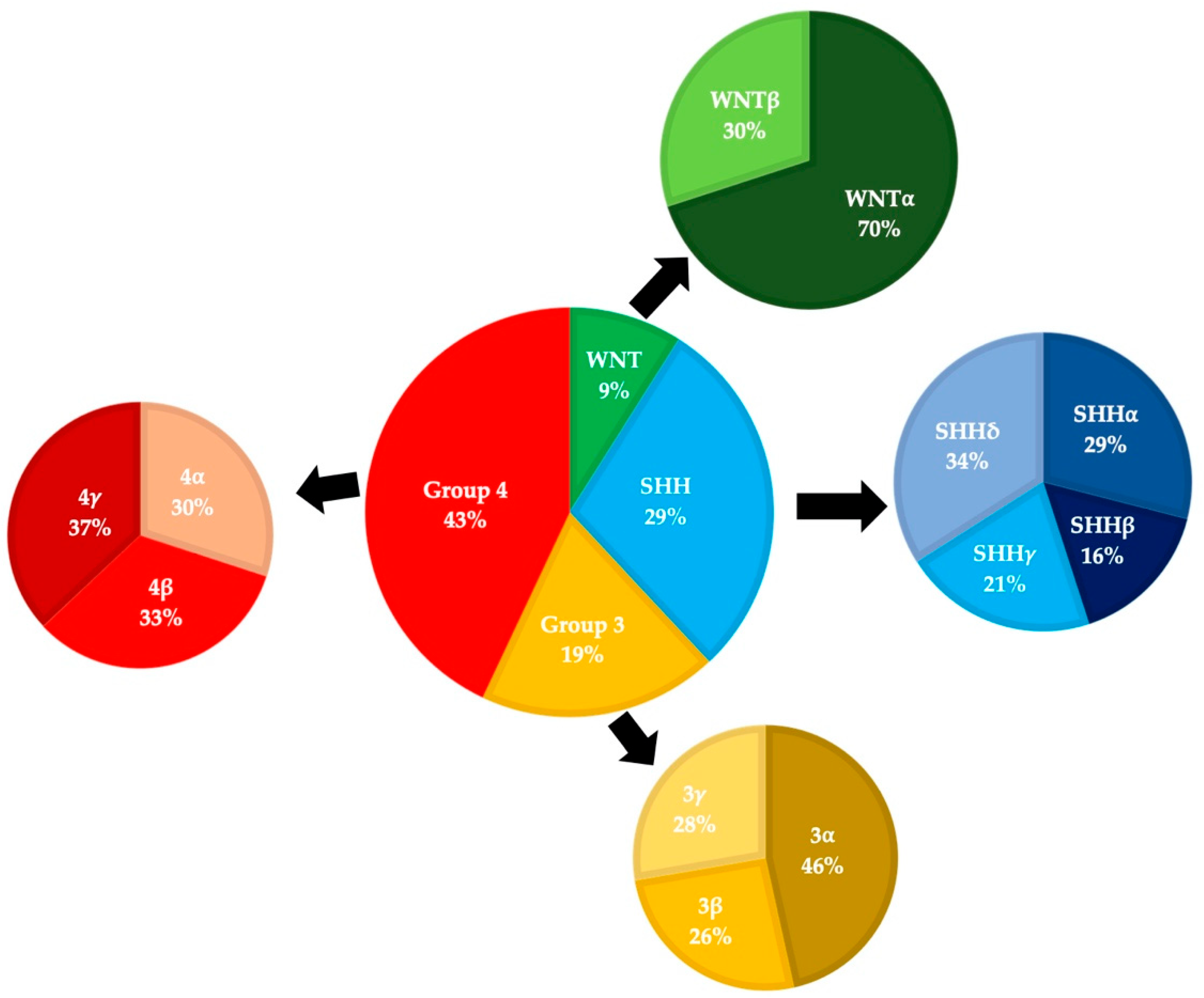
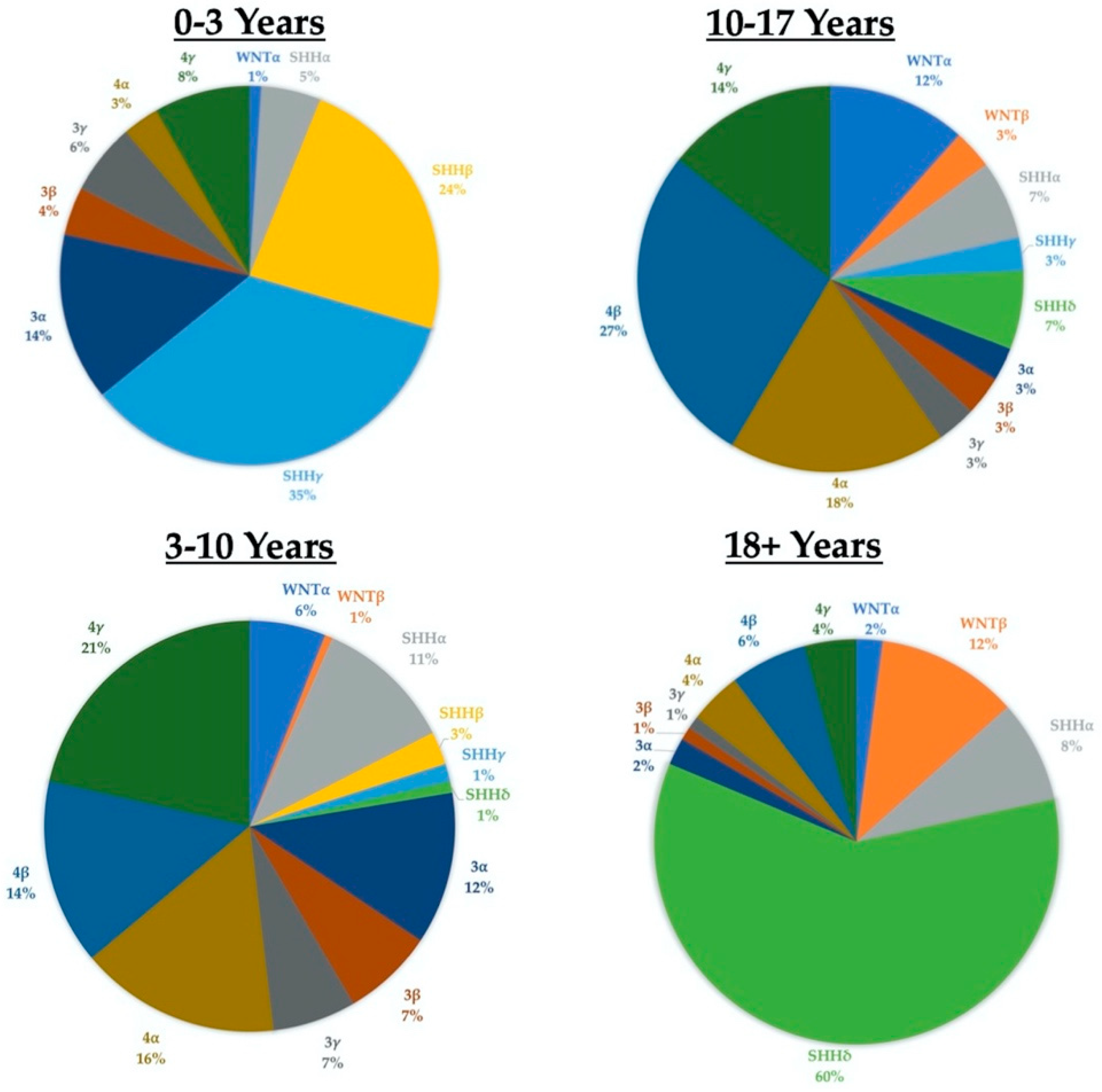
3. Epidemiology
4. Treatment and Outcomes
5. Clinical Trials
6. Models for Improvement
6.1. Patient Age and Childhood Leukemia
6.2. Pembrolizumab in MSI-Unstable/MMR-Deficient Tumors
6.3. Larotrectinib in TRK-Fusion Positive Tumors
7. Challenges
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bailey, P.; Cushing, H. Medulloblastoma Cerebelli: A common type of midcerebellar glioma of childhood. Arch. Neurol. Psychiatry 1925, 14, 192–224. [Google Scholar] [CrossRef]
- Rorke, L.B. The cerebellar medulloblastoma and its relationship to primitive neuroectodermal tumors. J. Neuropathol. Exp. Neurol. 1983, 42, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleihues, P.; Louis, D.N.; Scheithauer, B.W.; Rorke, L.B.; Reifenberger, G.; Burger, P.C.; Cavenee, W.K. The WHO classification of tumors of the nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Orr, B.A. Pathology, diagnostics, and classification of medulloblastoma. Brain Pathol. 2020, 30, 664–678. [Google Scholar] [CrossRef]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012, 123, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Robinson, G.W.; Gajjar, A. Genomics Paves the Way for Better Infant Medulloblastoma Therapy. J. Clin. Oncol. 2020, 38, 2010–2013. [Google Scholar] [CrossRef]
- Cavalli, F.M.G.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, A.S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754.e6. [Google Scholar] [CrossRef] [Green Version]
- Smoll, N.R.; Drummond, K.J. The incidence of medulloblastomas and primitive neurectodermal tumours in adults and children. J. Clin. Neurosci. 2012, 19, 1541–1544. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro-Oncology 2020, 22, iv1–iv96. [Google Scholar] [CrossRef]
- Botta, L.; Gatta, G.; Trama, A.; Bernasconi, A.; Sharon, E.; Capocaccia, R.; Mariotto, A.B.; the RARECAREnet Working Group. Incidence and survival of rare cancers in the US and Europe. Cancer Med. 2020, 9, 5632–5642. [Google Scholar] [CrossRef]
- Keinath, M.C.; Prior, D.E.; Prior, T.W. Spinal Muscular Atrophy: Mutations, Testing, and Clinical Relevance. Appl. Clin. Genet. 2021, 14, 11–25. [Google Scholar] [CrossRef]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Juraschka, K.; Taylor, M.D. Medulloblastoma in the age of molecular subgroups: A review. J. Neurosurg. Pediatr. 2019, 24, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Coltin, H.; Sundaresan, L.; Smith, K.S.; Skowron, P.; Massimi, L.; Eberhart, C.G.; Schreck, K.C.; Gupta, N.; Weiss, W.A.; Tirapelli, D.; et al. Subgroup and subtype-specific outcomes in adult medulloblastoma. Acta Neuropathol. 2021, 142, 859–871. [Google Scholar] [CrossRef]
- Remke, M.; Hielscher, T.; Northcott, P.A.; Witt, H.; Ryzhova, M.; Wittmann, A.; Benner, A.; von Deimling, A.; Scheurlen, W.; Perry, A.; et al. Adult medulloblastoma comprises three major molecular variants. J. Clin. Oncol. 2011, 29, 2717–2723. [Google Scholar] [CrossRef]
- Zhao, F.; Ohgaki, H.; Xu, L.; Giangaspero, F.; Li, C.; Li, P.; Yang, Z.; Wang, B.; Wang, X.; Wang, Z.; et al. Molecular subgroups of adult medulloblastoma: A long-term single-institution study. Neuro-Oncology 2016, 18, 982–990. [Google Scholar] [CrossRef]
- Franceschi, E.; Hofer, S.; Brandes, A.A.; Frappaz, D.; Kortmann, R.D.; Bromberg, J.; Dangouloff-Ros, V.; Boddaert, N.; Hattingen, E.; Wiestler, B.; et al. EANO-EURACAN clinical practice guideline for diagnosis, treatment, and follow-up of post-pubertal and adult patients with medulloblastoma. Lancet Oncol. 2019, 20, e715–e728. [Google Scholar] [CrossRef]
- Minh Thong, P.; Minh Duc, N. The Role of Apparent Diffusion Coefficient in the Differentiation between Cerebellar Medulloblastoma and Brainstem Glioma. Neurol. Int. 2020, 12, 34–40. [Google Scholar] [CrossRef]
- Chang, C.H.; Housepian, E.M.; Herbert, C., Jr. An operative staging system and a megavoltage radiotherapeutic technic for cerebellar medulloblastomas. Radiology 1969, 93, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Padovani, L.; Sunyach, M.P.; Perol, D.; Mercier, C.; Alapetite, C.; Haie-Meder, C.; Hoffstetter, S.; Muracciole, X.; Kerr, C.; Wagner, J.P.; et al. Common strategy for adult and pediatric medulloblastoma: A multicenter series of 253 adults. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Zeltzer, P.M.; Boyett, J.M.; Finlay, J.L.; Albright, A.L.; Rorke, L.B.; Milstein, J.M.; Allen, J.C.; Stevens, K.R.; Stanley, P.; Li, H.; et al. Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: Conclusions from the Children’s Cancer Group 921 randomized phase III study. J. Clin. Oncol. 1999, 17, 832–845. [Google Scholar] [CrossRef] [PubMed]
- Becker, R.L.; Becker, A.D.; Sobel, D.F. Adult medulloblastoma: Review of 13 cases with emphasis on MRI. Neuroradiology 1995, 37, 104–108. [Google Scholar] [CrossRef]
- Fruehwald-Pallamar, J.; Puchner, S.B.; Rossi, A.; Garre, M.L.; Cama, A.; Koelblinger, C.; Osborn, A.G.; Thurnher, M.M. Magnetic resonance imaging spectrum of medulloblastoma. Neuroradiology 2011, 53, 387–396. [Google Scholar] [CrossRef]
- Palmer, S.L.; Armstrong, C.; Onar-Thomas, A.; Wu, S.; Wallace, D.; Bonner, M.J.; Schreiber, J.; Swain, M.; Chapieski, L.; Mabbott, D.; et al. Processing speed, attention, and working memory after treatment for medulloblastoma: An international, prospective, and longitudinal study. J. Clin. Oncol. 2013, 31, 3494–3500. [Google Scholar] [CrossRef] [Green Version]
- Beier, D.; Proescholdt, M.; Reinert, C.; Pietsch, T.; Jones, D.T.W.; Pfister, S.M.; Hattingen, E.; Seidel, C.; Dirven, L.; Luerding, R.; et al. Multicenter pilot study of radiochemotherapy as first-line treatment for adults with medulloblastoma (NOA-07). Neuro-Oncology 2018, 20, 400–410. [Google Scholar] [CrossRef]
- Moxon-Emre, I.; Bouffet, E.; Taylor, M.D.; Laperriere, N.; Scantlebury, N.; Law, N.; Spiegler, B.J.; Malkin, D.; Janzen, L.; Mabbott, D. Impact of craniospinal dose, boost volume, and neurologic complications on intellectual outcome in patients with medulloblastoma. J. Clin. Oncol. 2014, 32, 1760–1768. [Google Scholar] [CrossRef] [Green Version]
- Packer, R.J.; Goldwein, J.; Nicholson, H.S.; Vezina, L.G.; Allen, J.C.; Ris, M.D.; Muraszko, K.; Rorke, L.B.; Wara, W.M.; Cohen, B.H.; et al. Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy: A Children’s Cancer Group Study. J. Clin. Oncol. 1999, 17, 2127–2136. [Google Scholar] [CrossRef]
- Tian, S.; Sudmeier, L.J.; Zhang, C.; Madden, N.A.; Buchwald, Z.S.; Shu, H.G.; Curran, W.J.; Eaton, B.R.; Esiashvili, N. Reduced-volume tumor-bed boost is not associated with inferior local control and survival outcomes in high-risk medulloblastoma. Pediatr. Blood Cancer 2020, 67, e28027. [Google Scholar] [CrossRef] [Green Version]
- Packer, R.J.; Sutton, L.N.; Elterman, R.; Lange, B.; Goldwein, J.; Nicholson, H.S.; Mulne, L.; Boyett, J.; D’Angio, G.; Wechsler-Jentzsch, K.; et al. Outcome for children with medulloblastoma treated with radiation and cisplatin, CCNU, and vincristine chemotherapy. J. Neurosurg. 1994, 81, 690–698. [Google Scholar] [CrossRef]
- Taylor, R.E.; Bailey, C.C.; Robinson, K.; Weston, C.L.; Ellison, D.; Ironside, J.; Lucraft, H.; Gilbertson, R.; Tait, D.M.; Walker, D.A.; et al. Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: The International Society of Paediatric Oncology/United Kingdom Children’s Cancer Study Group PNET-3 Study. J. Clin. Oncol. 2003, 21, 1581–1591. [Google Scholar] [CrossRef]
- Brandes, A.A.; Franceschi, E.; Tosoni, A.; Blatt, V.; Ermani, M. Long-term results of a prospective study on the treatment of medulloblastoma in adults. Cancer 2007, 110, 2035–2041. [Google Scholar] [CrossRef]
- Thompson, E.M.; Hielscher, T.; Bouffet, E.; Remke, M.; Luu, B.; Gururangan, S.; McLendon, R.E.; Bigner, D.D.; Lipp, E.S.; Perreault, S.; et al. Prognostic value of medulloblastoma extent of resection after accounting for molecular subgroup: A retrospective integrated clinical and molecular analysis. Lancet Oncol. 2016, 17, 484–495. [Google Scholar] [CrossRef] [Green Version]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.; Schlanstein, M.; Northcott, P.A.; Cho, Y.J.; Koster, J.; Schouten-van Meeteren, A.; van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [Green Version]
- Goschzik, T.; Schwalbe, E.C.; Hicks, D.; Smith, A.; Zur Muehlen, A.; Figarella-Branger, D.; Doz, F.; Rutkowski, S.; Lannering, B.; Pietsch, T.; et al. Prognostic effect of whole chromosomal aberration signatures in standard-risk, non-WNT/non-SHH medulloblastoma: A retrospective, molecular analysis of the HIT-SIOP PNET 4 trial. Lancet Oncol. 2018, 19, 1602–1616. [Google Scholar] [CrossRef] [Green Version]
- Kool, M.; Korshunov, A.; Pfister, S.M. Update on molecular and genetic alterations in adult medulloblastoma. Mag. Eur. Med. Oncol. 2012, 5, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Schwalbe, E.C.; Lindsey, J.C.; Nakjang, S.; Crosier, S.; Smith, A.J.; Hicks, D.; Rafiee, G.; Hill, R.M.; Iliasova, A.; Stone, T.; et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: A cohort study. Lancet Oncol. 2017, 18, 958–971. [Google Scholar] [CrossRef] [Green Version]
- Zhukova, N.; Ramaswamy, V.; Remke, M.; Pfaff, E.; Shih, D.J.; Martin, D.C.; Castelo-Branco, P.; Baskin, B.; Ray, P.N.; Bouffet, E.; et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J. Clin. Oncol. 2013, 31, 2927–2935. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Grobner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Jenkin, R.D. Medulloblastoma in childhood: Radiation therapy. Can. Med. Assoc. J. 1969, 100, 51–53. [Google Scholar]
- Lassman, L.P.; Pearce, G.W.; Gang, J. Sensitivity of Intracranial Gliomas to Vincristine Sulphate. Lancet 1965, 1, 296–298. [Google Scholar] [CrossRef]
- Lampkin, B.C.; Mauer, A.M.; McBride, B.H. Response of medulloblastoma to vincristine sulfate: A case report. Pediatrics 1967, 39, 761–763. [Google Scholar] [CrossRef]
- Broder, L.E.; Rall, D.P. Chemotherapy of brain tumors. Prog. Exp. Tumor Res. 1972, 17, 373–399. [Google Scholar] [CrossRef]
- Fewer, D.; Wilson, C.B.; Boldrey, E.B.; Enot, J.K. Phase II study of 1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea (CCNU; NSC-79037) in the treatment of brain tumors. Cancer Chemother. Rep. 1972, 56, 421–427. [Google Scholar]
- Wilson, C.B.; Boldrey, E.B.; Enot, K.J. 1,3-bis (2-chloroethyl)-1-nitrosourea (NSC-409962) in the treatment of brain tumors. Cancer Chemother. Rep. 1970, 54, 273–281. [Google Scholar]
- Evans, A.E.; Jenkin, R.D.; Sposto, R.; Ortega, J.A.; Wilson, C.B.; Wara, W.; Ertel, I.J.; Kramer, S.; Chang, C.H.; Leikin, S.L.; et al. The treatment of medulloblastoma. Results of a prospective randomized trial of radiation therapy with and without CCNU, vincristine, and prednisone. J. Neurosurg. 1990, 72, 572–582. [Google Scholar] [CrossRef]
- Tait, D.M.; Thornton-Jones, H.; Bloom, H.J.; Lemerle, J.; Morris-Jones, P. Adjuvant chemotherapy for medulloblastoma: The first multi-centre control trial of the International Society of Paediatric Oncology (SIOP I). Eur. J. Cancer 1990, 26, 464–469. [Google Scholar] [CrossRef]
- Packer, R.J.; Sutton, L.N.; Goldwein, J.W.; Perilongo, G.; Bunin, G.; Ryan, J.; Cohen, B.H.; D’Angio, G.; Kramer, E.D.; Zimmerman, R.A.; et al. Improved survival with the use of adjuvant chemotherapy in the treatment of medulloblastoma. J. Neurosurg. 1991, 74, 433–440. [Google Scholar] [CrossRef]
- Bailey, C.C.; Gnekow, A.; Wellek, S.; Jones, M.; Round, C.; Brown, J.; Phillips, A.; Neidhardt, M.K. Prospective randomised trial of chemotherapy given before radiotherapy in childhood medulloblastoma. International Society of Paediatric Oncology (SIOP) and the (German) Society of Paediatric Oncology (GPO): SIOP II. Med. Pediatr. Oncol. 1995, 25, 166–178. [Google Scholar] [CrossRef]
- Hoff, K.V.; Hinkes, B.; Gerber, N.U.; Deinlein, F.; Mittler, U.; Urban, C.; Benesch, M.; Warmuth-Metz, M.; Soerensen, N.; Zwiener, I.; et al. Long-term outcome and clinical prognostic factors in children with medulloblastoma treated in the prospective randomised multicentre trial HIT’91. Eur. J. Cancer 2009, 45, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Kortmann, R.D.; Kuhl, J.; Timmermann, B.; Mittler, U.; Urban, C.; Budach, V.; Richter, E.; Willich, N.; Flentje, M.; Berthold, F.; et al. Postoperative neoadjuvant chemotherapy before radiotherapy as compared to immediate radiotherapy followed by maintenance chemotherapy in the treatment of medulloblastoma in childhood: Results of the German prospective randomized trial HIT’91. Int. J. Radiat. Oncol. Biol. Phys. 2000, 46, 269–279. [Google Scholar] [CrossRef]
- Lannering, B.; Rutkowski, S.; Doz, F.; Pizer, B.; Gustafsson, G.; Navajas, A.; Massimino, M.; Reddingius, R.; Benesch, M.; Carrie, C.; et al. Hyperfractionated versus conventional radiotherapy followed by chemotherapy in standard-risk medulloblastoma: Results from the randomized multicenter HIT-SIOP PNET 4 trial. J. Clin. Oncol. 2012, 30, 3187–3193. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, S.; Bode, U.; Deinlein, F.; Ottensmeier, H.; Warmuth-Metz, M.; Soerensen, N.; Graf, N.; Emser, A.; Pietsch, T.; Wolff, J.E.; et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N. Engl. J. Med. 2005, 352, 978–986. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Ouyang, T.; Kang, H.; Long, W.; Thomas, B.; Zhu, S. Adult medulloblastoma: Clinical characters, prognostic factors, outcomes and patterns of relapse. J. Neurooncol. 2015, 124, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Brandes, A.A.; Ermani, M.; Amista, P.; Basso, U.; Vastola, F.; Gardiman, M.; Iuzzolino, P.; Turazzi, S.; Rotilio, A.; Volpin, L.; et al. The treatment of adults with medulloblastoma: A prospective study. Int. J. Radiat. Oncol. Biol. Phys. 2003, 57, 755–761. [Google Scholar] [CrossRef]
- Brandes, A.A.; Franceschi, E.; Tosoni, A.; Frezza, G.; Agati, R.; Maestri, A.; Ghimenton, C.; Mazzocchi, V.; Scopece, L.; Ermani, M. Efficacy of tailored treatment for high- and low-risk medulloblastoma in adults: A large prospective phase II trial. J. Clin. Oncol. 2010, 28, 2003. [Google Scholar] [CrossRef]
- Friedrich, C.; von Bueren, A.O.; von Hoff, K.; Kwiecien, R.; Pietsch, T.; Warmuth-Metz, M.; Hau, P.; Deinlein, F.; Kuehl, J.; Kortmann, R.D.; et al. Treatment of adult nonmetastatic medulloblastoma patients according to the paediatric HIT 2000 protocol: A prospective observational multicentre study. Eur. J. Cancer. 2013, 49, 893–903. [Google Scholar] [CrossRef]
- Kocakaya, S.; Beier, C.P.; Beier, D. Chemotherapy increases long-term survival in patients with adult medulloblastoma—A literature-based meta-analysis. Neuro-Oncology 2016, 18, 408–416. [Google Scholar] [CrossRef]
- Kann, B.H.; Lester-Coll, N.H.; Park, H.S.; Yeboa, D.N.; Kelly, J.R.; Baehring, J.M.; Becker, K.P.; Yu, J.B.; Bindra, R.S.; Roberts, K.B. Adjuvant chemotherapy and overall survival in adult medulloblastoma. Neuro-Oncology 2017, 19, 259–269. [Google Scholar] [CrossRef] [Green Version]
- Kieran, M.W.; Chisholm, J.; Casanova, M.; Brandes, A.A.; Aerts, I.; Bouffet, E.; Bailey, S.; Leary, S.; MacDonald, T.J.; Mechinaud, F.; et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro-Oncology 2017, 19, 1542–1552. [Google Scholar] [CrossRef]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef]
- Murthy, V.H.; Krumholz, H.M.; Gross, C.P. Participation in cancer clinical trials: Race-, sex-, and age-based disparities. JAMA 2004, 291, 2720–2726. [Google Scholar] [CrossRef]
- Unger, J.M.; Cook, E.; Tai, E.; Bleyer, A. The Role of Clinical Trial Participation in Cancer Research: Barriers, Evidence, and Strategies. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, 185–198. [Google Scholar] [CrossRef]
- Lee, E.Q.; Weller, M.; Sul, J.; Bagley, S.J.; Sahebjam, S.; van den Bent, M.; Ahluwalia, M.; Campian, J.L.; Galanis, E.; Gilbert, M.R.; et al. Optimizing eligibility criteria and clinical trial conduct to enhance clinical trial participation for primary brain tumor patients. Neuro-Oncology 2020, 22, 601–612. [Google Scholar] [CrossRef]
- Viale, P.H. The American Cancer Society’s Facts & Figures: 2020 Edition. J. Adv. Pract. Oncol. 2020, 11, 135–136. [Google Scholar] [CrossRef]
- Pui, C.H.; Yang, J.J.; Hunger, S.P.; Pieters, R.; Schrappe, M.; Biondi, A.; Vora, A.; Baruchel, A.; Silverman, L.B.; Schmiegelow, K.; et al. Childhood Acute Lymphoblastic Leukemia: Progress Through Collaboration. J. Clin. Oncol. 2015, 33, 2938–2948. [Google Scholar] [CrossRef]
- de Bont, J.M.; Holt, B.; Dekker, A.W.; van der Does-van den Berg, A.; Sonneveld, P.; Pieters, R. Significant difference in outcome for adolescents with acute lymphoblastic leukemia treated on pediatric vs adult protocols in the Netherlands. Leukemia 2004, 18, 2032–2035. [Google Scholar] [CrossRef]
- Stock, W.; Luger, S.M.; Advani, A.S.; Yin, J.; Harvey, R.C.; Mullighan, C.G.; Willman, C.L.; Fulton, N.; Laumann, K.M.; Malnassy, G.; et al. A pediatric regimen for older adolescents and young adults with acute lymphoblastic leukemia: Results of CALGB 10403. Blood 2019, 133, 1548–1559. [Google Scholar] [CrossRef] [Green Version]
- Hause, R.J.; Pritchard, C.C.; Shendure, J.; Salipante, S.J. Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 2016, 22, 1342–1350. [Google Scholar] [CrossRef]
- Andre, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site—When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef]
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 2014, 5, 4846. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Trial # | Year Opened | Phase | Patient # (Planned) | Ages (Years) | Eligibility | Treatment |
|---|---|---|---|---|---|---|
| NCT04315064 | 2020 | 1 | 5 | 1–80 | Recurrent/Progressive MB | Panobinostat (intrathecal) |
| NCT04315064 | 2019 | 1/2 | 60 | 3 and up | Recurrent SHH MB | CX-4945 |
| NCT01878617 | 2013 | 2 | 625 | 3–39 | Any MB age 3–22, SHH MB 22–39 | Chemo, RT, Vismodegib |
| NCT01857453 | 2013 | 2 | 97 | 18–70 | Standard risk adult MB | Chemo + reduced dose RT |
| NCT02962167 | 2017 | 1 | 46 | 12–39 | Recurrent MB or ATRT | Modified Measles virus (MV-NIS) |
| NCT04661384 | 2021 | 1 | 30 | 18+ | Recurrent Leptomeningeal MB, GB, or Ependymoma | IL-13Ralpha-2 CAR-T Cells |
| NCT03893487 | 2019 | 1 | 30 | 3–39 | DIPG, Recurrent MB of HGG | Fimepinostat |
| NCT03434262 | 2018 | 1 | 108 | 1–39 | Recurrent MB + other CNS Tumors | Ribociclib + gemcitabine, trametinib, or sonidegib |
| NCT03173950 | 2017 | 2 | 180 | 18+ | Recurrent MB + others | Nivolumab |
| NCT03734913 | 2019 | 1 | 65 | 18–75 | Advanced MB + others | ZSP1602 |
| NCT04541082 | 2020 | 1 | 102 | 18+ | Recurrent MB + others | ONC206 |
| NCT01505569 | 2011 | 2 | 20 | Up to 70 | Recurrent MB + others | Chemo + Autologous HSCT |
| NCT02905110 | 2016 | 1 | 10 | 1–80 | Recurrent MB + other PF Tumors | Etoposide and Methotrexate (intrathecal) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wooley, J.R.; Penas-Prado, M. Pediatric versus Adult Medulloblastoma: Towards a Definition That Goes beyond Age. Cancers 2021, 13, 6313. https://doi.org/10.3390/cancers13246313
Wooley JR, Penas-Prado M. Pediatric versus Adult Medulloblastoma: Towards a Definition That Goes beyond Age. Cancers. 2021; 13(24):6313. https://doi.org/10.3390/cancers13246313
Chicago/Turabian StyleWooley, Joseph R., and Marta Penas-Prado. 2021. "Pediatric versus Adult Medulloblastoma: Towards a Definition That Goes beyond Age" Cancers 13, no. 24: 6313. https://doi.org/10.3390/cancers13246313
APA StyleWooley, J. R., & Penas-Prado, M. (2021). Pediatric versus Adult Medulloblastoma: Towards a Definition That Goes beyond Age. Cancers, 13(24), 6313. https://doi.org/10.3390/cancers13246313