
Current Knowledge about Mechanisms of Drug Resistance against ALK Inhibitors in Non-Small Cell Lung Cancer


 ,
, 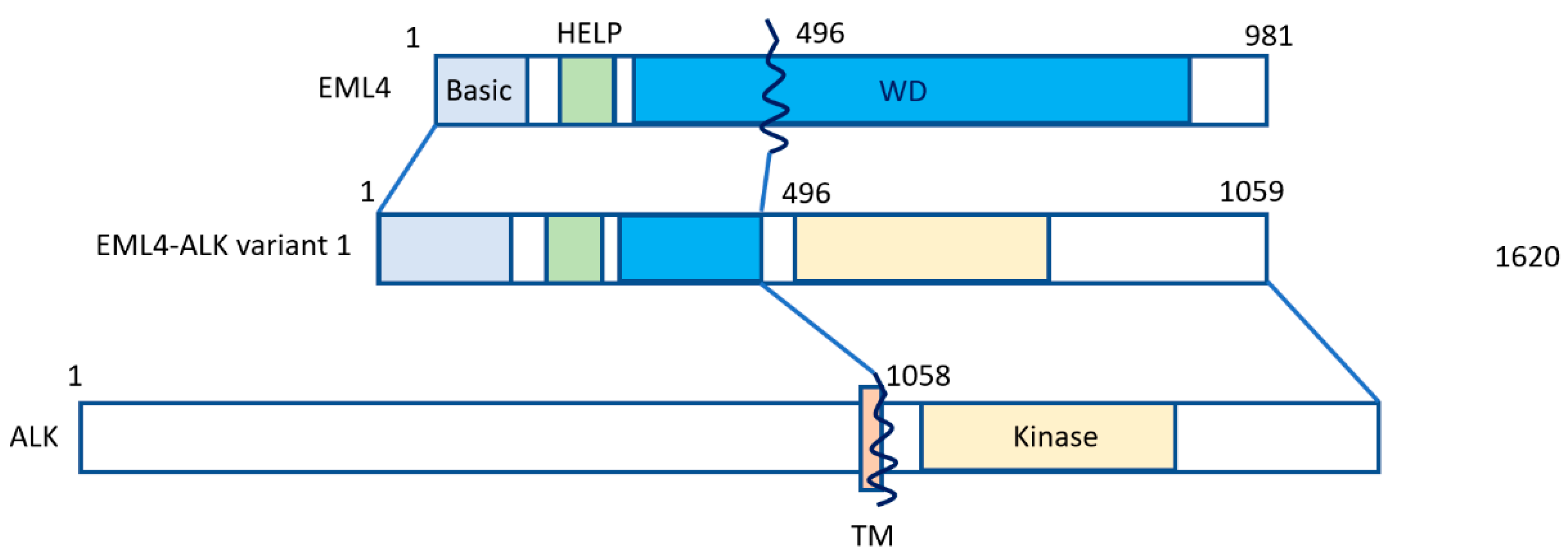{kind=link}
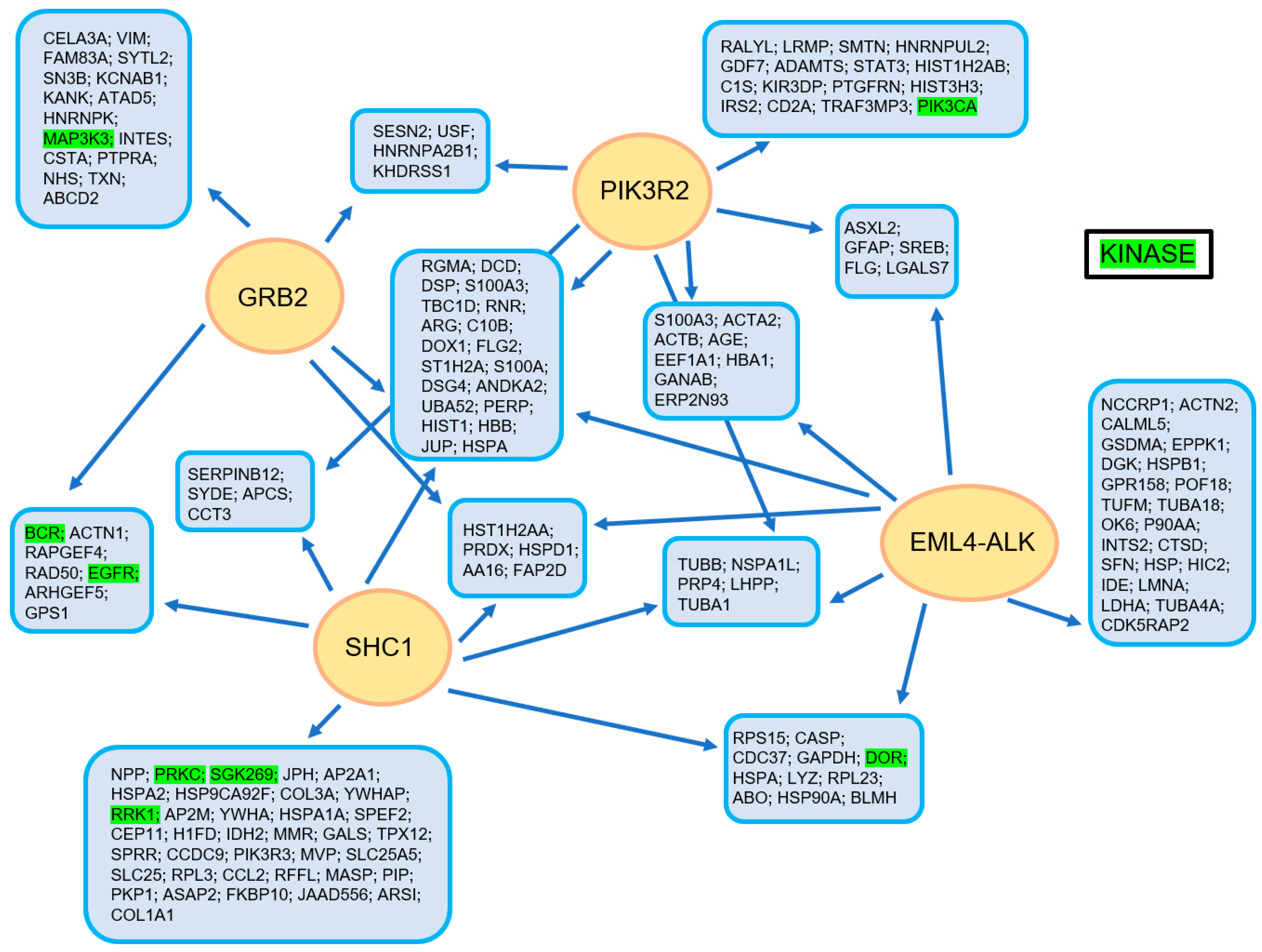{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Acquired ALK Resistance Mutations
3. Epithelial-to-Mesenchymal Transition
4. Genetic Assessment of ALK-Resistant Cancers
5. Alectinib and Brigatinib
6. Effectiveness of Lorlatinib
7. Conclusions
Funding
Conflicts of Interest
References
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global Survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.-I.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nat. Cell Biol. 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Marsilje, T.H.; Pei, W.; Chen, B.; Lu, W.; Uno, T.; Jin, Y.; Jiang, T.; Kim, S.; Li, N.; Warmuth, M.; et al. Synthesis, Structure–Activity Relationships, and in Vivo Efficacy of the Novel Potent and Selective Anaplastic Lymphoma Kinase (ALK) Inhibitor 5-Chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) Currently in Phase 1 and Phase 2 Clinical Trials. J. Med. Chem. 2013, 56, 5675–5690. [Google Scholar]
- Li, G.; Dai, W.-R.; Shao, F.-C. Effect of ALK-inhibitors in the treatment of non-small cell lung cancer: A systematic review and meta-analysis. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3496–3503. [Google Scholar]
- Wong, D.; Yip, S.; Sorensen, P.H. Methods for Identifying Patients with Tropomyosin Receptor Kinase (TRK) Fusion Cancer. Pathol. Oncol. Res. 2020, 26, 1385–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixido, C.; Giménez-Capitán, A.; Molina-Vila, M.A.; Peg, V.; Karachaliou, N.; Rodríguez-Capote, A.; Castellvi, J.; Rosell, R. RNA Analysis as a Tool to Determine Clinically Relevant Gene Fusions and Splice Variants. Arch. Pathol. Lab. Med. 2018, 142, 474–479. [Google Scholar] [CrossRef] [Green Version]
- Vendrell, J.A.; Taviaux, S.; Béganton, B.; Godreuil, S.; Audran, P.; Grand, D.; Clermont, E.; Serre, I.; Szablewski, V.; Coopman, P.; et al. Detection of known and novel ALK fusion transcripts in lung cancer patients using next-generation sequencing approaches. Sci. Rep. 2017, 7, 12510. [Google Scholar] [CrossRef] [Green Version]
- Duruisseaux, M.; Besse, B.; Cadranel, J.; Pérol, M.; Mennecier, B.; Bigay-Game, L.; Descourt, R.; Dansin, E.; Audigier-Valette, C.; Moreau, L.; et al. Overall survival with crizotinib and next-generation ALK inhibitors in ALK-positive non-small-cell lung cancer (IFCT-1302 CLINALK): A French nationwide cohort retrospective study. Oncotarget 2017, 8, 21903–21917. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, J.M.; Gao, D.; Smith, D.; Purcell, T.; Hancock, M.; Bunn, P.; Robin, T.; Liu, A.; Karam, S.; Gaspar, L.; et al. Natural History and Factors Associated with Overall Survival in Stage IV ALK-Rearranged Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2019, 14, 691–700. [Google Scholar] [CrossRef]
- Golding, B.; Luu, A.; Jones, R.; Viloria-Petit, A.M. The function and therapeutic targeting of anaplastic lymphoma kinase (ALK) in non-small cell lung cancer (NSCLC). Mol. Cancer 2018, 17, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Umapathy, G.; Mendoza-Garcia, P.; Hallberg, B.; Palmer, R.H. Targeting anaplastic lymphoma kinase in neuroblastoma. APMIS 2019, 127, 288–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Li, J.; Wu, P.; Zhou, L.; Lu, B.; Ying, K.; Chen, E.; Lu, Y.; Liu, P. Smoker and non-smoker lung adenocarcinoma is characterized by distinct tumor immune microenvironments. OncoImmunology 2018, 7, e1494677. [Google Scholar] [CrossRef]
- Lin, J.J.; Zhu, V.W.; Yoda, S.; Yeap, B.Y.; Schrock, A.B.; Dagogo-Jack, I.; Jessop, N.A.; Jiang, G.Y.; Le, L.P.; Gowen, K.; et al. Impact of EML4-ALK Variant on Resistance Mechanisms and Clinical Outcomes in ALK-Positive Lung Cancer. J. Clin. Oncol. 2018, 36, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef]
- Carles, F.; Bourg, S.; Meyer, C.; Bonnet, P. PKIDB: A Curated, Annotated and Updated Database of Protein Kinase Inhibitors in Clinical Trials. Molecules 2018, 23, 908. [Google Scholar] [CrossRef] [Green Version]
- Fischer, P.M. Approved and Experimental Small-Molecule Oncology Kinase Inhibitor Drugs: A Mid-2016 Overview. Med. Res. Rev. 2017, 37, 314–367. [Google Scholar] [CrossRef] [Green Version]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 2008, 8, 11–23. [Google Scholar] [CrossRef]
- Zhang, G.; Scarborough, H.; Kim, J.; Rozhok, A.I.; Chen, Y.A.; Zhang, X.; Song, L.; Bai, Y.; Fang, B.; Liu, R.Z.; et al. Coupling an EML4-ALK-centric interactome with RNA interference identifies sensitizers to ALK inhibitors. Sci. Signal. 2016, 9, rs12. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.J.; Riely, G.J.; Shaw, A.T. Targeting ALK: Precision Medicine Takes on Drug Resistance. Cancer Discov. 2017, 7, 137–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, E.L.; Bang, Y.-J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.-H.I.; Dezube, B.J.; Jänne, P.A.; Costa, D.B.; et al. Anaplastic Lymphoma Kinase Inhibition in Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Kim, D.-W.; Nakagawa, K.; Seto, T.; Crinó, L.; Ahn, M.-J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus Chemotherapy in AdvancedALK-Positive Lung Cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [Green Version]
- Solomon, B.J.; Mok, T.; Kim, D.-W.; Wu, Y.-L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-Line Crizotinib versus Chemotherapy in ALK-Positive Lung Cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [Green Version]
- Lucena-Araujo, A.R.; Moran, J.P.; VanderLaan, P.A.; Dias-Santagata, R.; Folch, E.; Majid, A.; Kent, M.S.; Gangadharan, S.; Rangachari, D.; Huberman, M.S.; et al. De novo ALK kinase domain mutations are uncommon in kinase inhibitor-naïve ALK rearranged lung cancers. Lung Cancer 2016, 99, 17–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, K.; Takeuchi, S.; Arai, S.; Katayama, R.; Nanjo, S.; Tanimoto, A.; Nishiyama, A.; Nakagawa, T.; Taniguchi, H.; Suzuki, T.; et al. Epithelial-to-Mesenchymal Transition Is a Mechanism of ALK Inhibitor Resistance in Lung Cancer Independent of ALK Mutation Status. Cancer Res. 2019, 79, 1658–1670. [Google Scholar] [CrossRef] [PubMed]
- Gadgeel, S.M.; Gandhi, L.; Riely, G.J.; Chiappori, A.A.; West, H.L.; Azada, M.C.; Morcos, P.N.; Lee, R.; Garcia, L.; Yu, L.; et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): Results from the dose-finding portion of a phase 1/2 study. Lancet Oncol. 2014, 15, 1119–1128. [Google Scholar] [CrossRef]
- Seto, T.; Kiura, K.; Nishio, M.; Nakagawa, K.; Maemondo, M.; Inoue, A.; Hida, T.; Yamamoto, N.; Yoshioka, H.; Harada, M.; et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): A single-arm, open-label, phase 1–2 study. Lancet Oncol. 2013, 14, 590–598. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kim, T.M.; Crinò, L.; Gridelli, C.; Kiura, K.; Liu, G.; Novello, S.; Bearz, A.; Gautschi, O.; Mok, T.; et al. Ceritinib versus chemotherapy in patients with ALK-rearranged non-small-cell lung cancer previously given chemotherapy and crizotinib (ASCEND-5): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 874–886. [Google Scholar] [CrossRef]
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci. Transl. Med. 2012, 4, 120ra17. [Google Scholar] [CrossRef] [Green Version]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular mechanisms of resistance to first- and second-generation alk inhibitors in alk-rearranged lung cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovly, C.M.; McDonald, N.T.; Chen, H.; Ortiz-Cuaran, S.; Heukamp, L.C.; Yan, Y.; Florin, A.; Ozretić, L.; Lim, D.; Wang, L.; et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion–positive lung cancer. Nat. Med. 2014, 20, 1027–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, T.; Takeuchi, S.; Nakade, J.; Kita, K.; Nakagawa, T.; Nanjo, S.; Nakamura, T.; Matsumoto, K.; Soda, M.; Mano, H.; et al. Paracrine receptor activation by microenvironment triggers bypass survival signals and alk inhibitor resistance in eml4-alk lung cancer cells. Clin. Cancer Res. 2012, 18, 3592–3602. [Google Scholar] [CrossRef] [Green Version]
- Doebele, R.C.; Pilling, A.B.; Aisner, D.L.; Kutateladze, T.G.; Le, A.T.; Weickhardt, A.J.; Kondo, K.L.; Linderman, D.J.; Heasley, L.E.; Franklin, W.A.; et al. Mechanisms of resistance to crizotinib in patients with alk gene rearranged non–small cell lung cancer. Clin. Cancer Res. 2012, 18, 1472–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, R.; Sakashita, T.; Yanagitani, N.; Ninomiya, H.; Horiike, A.; Friboulet, L.; Gainor, J.F.; Motoi, N.; Dobashi, A.; Sakata, S.; et al. P-glycoprotein Mediates Ceritinib Resistance in Anaplastic Lymphoma Kinase-rearranged Non-small Cell Lung Cancer. EBioMedicine 2016, 3, 54–66. [Google Scholar] [CrossRef] [Green Version]
- Costa, D.B.; Shaw, A.T.; Ou, S.-H.I.; Solomon, B.J.; Riely, G.J.; Ahn, M.-J.; Zhou, C.; Shreeve, S.M.; Selaru, P.; Polli, A.; et al. Clinical Experience With Crizotinib in Patients With Advanced ALK-Rearranged Non–Small-Cell Lung Cancer and Brain Metastases. J. Clin. Oncol. 2015, 33, 1881–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friboulet, L.; Li, N.; Katayama, R.; Lee, C.C.; Gainor, J.F.; Crystal, A.S.; Michellys, P.-Y.; Awad, M.M.; Yanagitani, N.; Kim, S.; et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non–small cell lung cancer. Cancer Discov. 2014, 4, 662–673. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.Y.; Friboulet, L.; Kodack, D.P.; Engstrom, L.D.; Li, Q.; West, M.; Tang, R.W.; Wang, H.; Tsaparikos, K.; Wang, J.; et al. PF-06463922, an ALK/ROS1 Inhibitor, Overcomes Resistance to First and Second Generation ALK Inhibitors in Preclinical Models. Cancer Cell 2015, 28, 70–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Kent, J.W.; Olivier, M.; Ali, O.; Cerjak, D.; Broeckel, U.; Abdou, R.M.; Dyer, T.D.; Comuzzie, A.G.; Curran, J.E.; et al. A comprehensive analysis of adiponectin QTLs using SNP association, SNP cis-effects on peripheral blood gene expression and gene expression correlation identified novel metabolic syndrome (MetS) genes with potential role in carcinogenesis and systemic inflammation. BMC Med. Genom. 2013, 6, 14. [Google Scholar]
- Ignatius Ou, S.H.; Azada, M.; Hsiang, D.J.; Herman, J.M.; Kain, T.S.; Siwak-Tapp, C.; Casey, C.; He, J.; Ali, S.M.; Klempner, S.J.; et al. Next-generation sequencing reveals a Novel NSCLC ALK F1174V mutation and confirms ALK G1202R mutation confers high-level resistance to alectinib (CH5424802/RO5424802) in ALK-rearranged NSCLC patients who progressed on crizotinib. J. Thorac. Oncol. 2014, 9, 549–553. [Google Scholar]
- Sakamoto, H.; Tsukaguchi, T.; Hiroshima, S.; Kodama, T.; Kobayashi, T.; Fukami, T.A.; Oikawa, N.; Tsukuda, T.; Ishii, N.; Aoki, Y. CH5424802, a selective alk inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 2011, 19, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Lan, S.; Li, H.; Liu, Y.; Xu, J.; Huang, Z.; Yan, S.; Zhang, Q.; Cheng, Y. A Novel ROS1-FBXL17 Fusion Co-Existing with CD74-ROS1 Fusion May Improve Sensitivity to Crizotinib and Prolong Progression-Free Survival of Patients with Lung Adenocarcinoma. OncoTargets Ther. 2020, 13, 11499–11504. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Kiesslich, T.; Pichler, M.; Neureiter, D. Epigenetic control of epithelial-mesenchymal-transition in human cancer. Mol. Clin. Oncol. 2013, 1, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Uramoto, H.; Yamada, T.; Yano, S.; Kondo, N.; Hasegawa, S.; Tanaka, F. Prognostic value of acquired resistance-related molecules in Japanese patients with NSCLC treated with an EGFR-TKI. Anticancer Res. 2012, 32, 2785–3790. [Google Scholar]
- Guarino, M.; Rubino, B.; Ballabio, G. The role of epithelial-mesenchymal transition in cancer pathology. Pathology 2007, 39, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Nanjo, S.; Nakagawa, T.; Takeuchi, S.; Kita, K.; Fukuda, K.; Nakada, M.; Uehara, H.; Nishihara, H.; Hara, E.; Uramoto, H.; et al. In vivo imaging models of bone and brain metastases and pleural carcinomatosis with a novel human EML4-ALK lung cancer cell line. Cancer Sci. 2015, 106, 244–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recondo, G.; Mezquita, L.; Facchinetti, F.; Planchard, D.; Gazzah, A.; Bigot, L.; Rizvi, A.Z.; Frias, R.L.; Thiery, J.P.; Scoazec, J.-Y.; et al. Diverse Resistance Mechanisms to the Third-Generation ALK Inhibitor Lorlatinib in ALK-Rearranged Lung Cancer. Clin. Cancer Res. 2020, 26, 242–255. [Google Scholar] [CrossRef] [Green Version]
- Tyner, J.W.; Fletcher, L.B.; Wang, E.Q.; Yang, W.F.; Rutenberg-Schoenberg, M.L.; Beadling, C.; Mori, M.; Heinrich, M.C.; Deininger, M.W.; Druker, B.J.; et al. MET receptor sequence variants R970C and T992I lack transforming capacity. Cancer Res. 2010, 70, 6233–6237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to egfr inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crystal, A.S.; Shaw, A.T.; Sequist, L.V.; Friboulet, L.; Niederst, M.J.; Lockerman, E.L.; Frias, R.L.; Gainor, J.F.; Amzallag, A.; Greninger, P.; et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014, 346, 1480–1486. [Google Scholar] [CrossRef] [Green Version]
- Dias-Santagata, D.; Akhavanfard, S.; David, S.S.; Vernovsky, K.; Kuhlmann, G.; Boisvert, S.L.; Stubbs, H.; McDermott, U.; Settleman, J.; Kwak, E.L.; et al. Rapid targeted mutational analysis of human tumours: A clinical platform to guide personalized cancer medicine. EMBO Mol. Med. 2010, 2, 146–158. [Google Scholar] [CrossRef]
- Rudd, M.L.; Price, J.C.; Fogoros, S.; Godwin, A.K.; Sgroi, D.C.; Merino, M.J.; Bell, D.W. A unique spectrum of somatic PIK3CA (p110α) mutations within primary endometrial carcinomas. Clin. Cancer Res. 2011, 17, 1331–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, R.; Sasaki, T.; Minami, Y.; Hibino, Y.; Okumura, S.; Sado, M.; Miyokawa, N.; Hayashi, S.; Kitada, M.; Ohsaki, Y. Activation of Src signaling mediates acquired resistance to ALK inhibition in lung cancer. Int. J. Oncol. 2017, 51, 1533–1540. [Google Scholar] [CrossRef] [Green Version]
- Hrustanovic, G.; Olivas, V.; Pazarentzos, E.; Tulpule, A.; Asthana, S.; Blakely, C.M.; Okimoto, R.A.; Lin, L.; Neel, D.S.; Sabnis, A.J.; et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK–positive lung cancer. Nat. Med. 2015, 21, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Madsen, A.T.; Winther-Larsen, A.; McCulloch, T.; Meldgaard, P.; Sorensen, B.S. Genomic Profiling of Circulating Tumor DNA Predicts Outcome and Demonstrates Tumor Evolution in ALK-Positive Non-Small Cell Lung Cancer Patients. Cancers 2020, 12, 947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodama, T.; Tsukaguchi, T.; Satoh, Y.; Yoshida, M.; Watanabe, Y.; Kondoh, O.; Sakamoto, H. Alectinib shows potent antitumor activity against ret-rearranged non–small cell lung cancer. Mol. Cancer Ther. 2014, 13, 2910–2918. [Google Scholar] [CrossRef] [Green Version]
- Kodama, T.; Tsukaguchi, T.; Yoshida, M.; Kondoh, O.; Sakamoto, H. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett. 2014, 351, 215–221. [Google Scholar] [CrossRef]
- Kodama, T.; Hasegawa, M.; Takanashi, K.; Sakurai, Y.; Kondoh, O.; Sakamoto, H. Antitumor activity of the selective ALK inhibitor alectinib in models of intracranial metastases. Cancer Chemother. Pharmacol. 2014, 74, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Gandhi, L.; Gadgeel, S.; Riely, G.J.; Cetnar, J.; West, H.; Camidge, D.R.; Socinski, M.A.; Chiappori, A.; Mekhail, T.; et al. Alectinib in ALK-positive, crizotinib-resistant, non-small-cell lung cancer: A single-group, multicentre, phase 2 trial. Lancet Oncol. 2016, 17, 234–242. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.-H.I.; Ahn, J.S.; De Petris, L.; Govindan, R.; Yang, J.C.-H.; Hughes, B.; Lena, H.; Moro-Sibilot, D.; Bearz, A.; Ramirez, S.V.; et al. Alectinib in Crizotinib-Refractory ALK-Rearranged Non–Small-Cell Lung Cancer: A Phase II Global Study. J. Clin. Oncol. 2016, 34, 661–668. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.C.-H.; Ou, S.-H.I.; De Petris, L.; Gadgeel, S.; Gandhi, L.; Kim, D.-W.; Barlesi, F.; Govindan, R.; Dingemans, A.-M.C.; Crino, L.; et al. Pooled Systemic Efficacy and Safety Data from the Pivotal Phase II Studies (NP28673 and NP28761) of Alectinib in ALK-positive Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1552–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novello, S.; Mazières, J.; Oh, I.-J.; De Castro, J.; Migliorino, M.; Helland, Å.; Dziadziuszko, R.; Griesinger, F.; Kotb, A.; Zeaiter, A.; et al. Alectinib versus chemotherapy in crizotinib-pretreated anaplastic lymphoma kinase (ALK)-positive non-small-cell lung cancer: Results from the phase III ALUR study. Ann. Oncol. 2018, 29, 1409–1416. [Google Scholar] [CrossRef]
- Hida, T.; Nokihara, H.; Kondo, M.; Kim, Y.H.; Azuma, K.; Seto, T.; Takiguchi, Y.; Nishio, M.; Yoshioka, H.; Imamura, F.; et al. Alectinib versus crizotinib in patients with ALK -positive non-small-cell lung cancer (J-ALEX): An open-label, randomised phase 3 trial. Lancet 2017, 390, 29–39. [Google Scholar] [CrossRef]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.-W.; Ou, S.-H.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus Crizotinib in Untreated ALK-Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef]
- Zhang, S.; Anjum, R.; Squillace, R.; Nadworny, S.; Zhou, T.; Keats, J.; Ning, Y.; Wardwell, S.D.; Miller, D.; Song, Y.; et al. The Potent ALK Inhibitor Brigatinib (AP26113) Overcomes Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in Preclinical Models. Clin. Cancer Res. 2016, 22, 5527–5538. [Google Scholar] [CrossRef] [Green Version]
- Gettinger, S.N.; Bazhenova, L.A.; Langer, C.J.; Salgia, R.; Gold, K.A.; Rosell, R.; Shaw, A.T.; Weiss, G.J.; Tugnait, M.; Narasimhan, N.I.; et al. Activity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: A single-arm, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 1683–1696. [Google Scholar] [CrossRef]
- Kim, D.-W.; Tiseo, M.; Ahn, M.-J.; Reckamp, K.L.; Hansen, K.H.; Kim, S.-W.; Huber, R.M.; West, H.L.; Groen, H.J.; Hochmair, M.J.; et al. Brigatinib in Patients With Crizotinib-Refractory Anaplastic Lymphoma Kinase–Positive Non–Small-Cell Lung Cancer: A Randomized, Multicenter Phase II Trial. J. Clin. Oncol. 2017, 35, 2490–2498. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Yoshida, T.; Kumagai, T.; Hida, T.; Toyozawa, R.; Shimokawaji, T.; Goto, K.; Nakagawa, K.; Ohe, Y.; Seto, T.; et al. Brigatinib in Japanese Patients With ALK-Positive NSCLC Previously Treated With Alectinib and Other Tyrosine Kinase Inhibitors: Outcomes of the Phase 2 J-ALTA Trial. J. Thorac. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Kim, H.R.; Ahn, M.-J.; Yang, J.C.-H.; Han, J.-Y.; Lee, J.-S.; Hochmair, M.J.; Li, J.Y.-C.; Chang, G.-C.; Lee, K.H.; et al. Brigatinib versus Crizotinib in ALK-Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2027–2039. [Google Scholar] [CrossRef]
- Johnson, T.W.; Richardson, P.F.; Bailey, S.; Brooun, A.; Burke, B.J.; Collins, M.R.; Cui, J.J.; Deal, J.G.; Deng, Y.-L.; Dinh, D.; et al. Discovery of (10r)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2h-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (pf-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (alk) and c-ros oncogene 1 (ros1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J. Med. Chem. 2013, 57, 4720–4744. [Google Scholar]
- Shaw, A.T.; Solomon, B.J.; Besse, B.; Bauer, T.M.; Lin, C.-C.; Soo, R.A.; Riely, G.J.; Ou, S.-H.I.; Clancy, J.S.; Li, S.; et al. ALK Resistance Mutations and Efficacy of Lorlatinib in Advanced Anaplastic Lymphoma Kinase-Positive Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2019, 37, 1370–1379. [Google Scholar] [CrossRef]
- Shaw, A.T.; Felip, E.; Bauer, T.M.; Besse, B.; Navarro, A.; Postel-Vinay, S.; Gainor, J.F.; Johnson, M.; Dietrich, J.; James, L.P.; et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: An international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 2017, 18, 1590–1599. [Google Scholar] [CrossRef]
- Solomon, B.J.; Besse, B.; Bauer, T.M.; Felip, E.; Soo, R.A.; Camidge, D.R.; Chiari, R.; Bearz, A.; Lin, C.-C.; Gadgeel, S.M.; et al. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: Results from a global phase 2 study. Lancet Oncol. 2018, 19, 1654–1667. [Google Scholar] [CrossRef]
- Yoda, S.; Lin, J.J.; Lawrence, M.S.; Burke, B.J.; Friboulet, L.; Langenbucher, A.; Dardaei, L.; Prutisto-Chang, K.; Dagogo-Jack, I.; Timofeevski, S.; et al. Sequential ALK Inhibitors Can Select for Lorlatinib-Resistant Compound ALK Mutations in ALK-Positive Lung Cancer. Cancer Discov. 2018, 8, 714–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Seto, Y.; Okada, K.; Uematsu, S.; Uchibori, K.; Tsukahara, M.; Oh-Hara, T.; Fujita, N.; Yanagitani, N.; Nishio, M.; et al. Overcoming resistance by ALK compound mutation (I1171S + G1269A) after sequential treatment of multiple ALK inhibitors in non-small cell lung cancer. Thorac. Cancer 2020, 11, 581–587. [Google Scholar] [CrossRef] [Green Version]
- Dagogo-Jack, I.; Rooney, M.; Lin, J.J.; Nagy, R.J.; Yeap, B.Y.; Hubbeling, H.; Chin, E.; Ackil, J.; Farago, A.F.; Hata, A.N.; et al. Treatment with Next-Generation ALK Inhibitors Fuels Plasma ALK Mutation Diversity. Clin. Cancer Res. 2019, 25, 6662–6670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, J.K.; Keam, B.; Koh, J.; Cho, H.J.; Jeon, Y.K.; Kim, T.M.; Lee, S.H.; Lee, D.S.; Kim, D.W.; Chung, D.H. Concomitant ALK translocation and EGFR mutation in lung cancer: A comparison of direct sequencing and sensitive assays and the impact on responsiveness to tyrosine kinase inhibitor. Ann. Oncol. 2014, 26, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Nolan, P.M.; Peters, J.; Strivens, M.; Rogers, D.; Hagan, J.; Spurr, N.; Gray, I.C.; Vizor, L.; Brooker, D.; Whitehill, E.; et al. A systematic, genome-wide, phenotype-driven mutagenesis programme for gene function studies in the mouse. Nat. Genet. 2000, 25, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Kim, D.-W.; Kotsakis, A.; Deng, S.; Lira, P.; Ho, S.N.; Lee, N.V.; Vizcarra, P.; Cao, J.Q.; Christensen, J.G.; et al. Multiplexed deep sequencing analysis of ALK kinase domain identifies resistance mutations in relapsed patients following crizotinib treatment. Genomics 2013, 102, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Friboulet, L.; Leshchiner, I.; Gainor, J.F.; Bergqvist, S.; Brooun, A.; Burke, B.J.; Deng, Y.; Liu, W.; Dardaei, L.; et al. Resensitization to Crizotinib by the Lorlatinib ALK Resistance Mutation L1198F. N. Engl. J. Med. 2016, 374, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Bauer, T.M.; De Marinis, F.; Felip, E.; Goto, Y.; Liu, G.; Mazieres, J.; Kim, D.-W.; Mok, T.; Polli, A.; et al. First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer. N. Engl. J. Med. 2020, 383, 2018–2029. [Google Scholar] [CrossRef] [PubMed]
- Horn, L.; Infante, J.R.; Reckamp, K.L.; Blumenschein, G.R.; Leal, T.A.; Waqar, S.N.; Gitlitz, B.J.; Sanborn, R.E.; Whisenant, J.G.; Du, L.; et al. Ensartinib (X-396) in ALK-Positive Non-Small Cell Lung Cancer: Results from a First-in-Human Phase I/II, Multicenter Study. Clin. Cancer Res. 2018, 24, 2771–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, K.; Akimoto, K.; Sato, H.; Manabe, R.; Kishino, Y.; Homma, T.; Kusumoto, S.; Yamaoka, T.; Tanaka, A.; Ohmori, T.; et al. Brigatinib and Alectinib for ALK Rearrangement-Positive Advanced Non-Small Cell Lung Cancer with or without Central Nervous System Metastasis: A Systematic Review and Network Meta-Analysis. Cancers 2020, 12, 942. [Google Scholar] [CrossRef]
- Redaelli, S.; Ceccon, M.; Zappa, M.; Sharma, G.; Mastini, C.; Mauri, M.; Nigoghossian, M.; Massimino, L.; Cordani, N.; Farina, F.; et al. Lorlatinib Treatment Elicits Multiple On- and Off-Target Mechanisms of Resistance in ALK-Driven Cancer. Cancer Res. 2018, 78, 6866–6880. [Google Scholar] [CrossRef] [Green Version]
- Pailler, E.; Faugeroux, V.; Oulhen, M.; Mezquita, L.; Laporte, M.; Honoré, A.; Lecluse, Y.; Queffelec, P.; Ngo-Camus, M.; Nicotra, C.; et al. Acquired Resistance Mutations to ALK Inhibitors Identified by Single Circulating Tumor Cell Sequencing in ALK-Rearranged Non–Small-Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 6671–6682. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smolle, E.; Taucher, V.; Lindenmann, J.; Jost, P.J.; Pichler, M. Current Knowledge about Mechanisms of Drug Resistance against ALK Inhibitors in Non-Small Cell Lung Cancer. Cancers 2021, 13, 699. https://doi.org/10.3390/cancers13040699
Smolle E, Taucher V, Lindenmann J, Jost PJ, Pichler M. Current Knowledge about Mechanisms of Drug Resistance against ALK Inhibitors in Non-Small Cell Lung Cancer. Cancers. 2021; 13(4):699. https://doi.org/10.3390/cancers13040699
Chicago/Turabian StyleSmolle, Elisabeth, Valentin Taucher, Joerg Lindenmann, Philipp J. Jost, and Martin Pichler. 2021. "Current Knowledge about Mechanisms of Drug Resistance against ALK Inhibitors in Non-Small Cell Lung Cancer" Cancers 13, no. 4: 699. https://doi.org/10.3390/cancers13040699
APA StyleSmolle, E., Taucher, V., Lindenmann, J., Jost, P. J., & Pichler, M. (2021). Current Knowledge about Mechanisms of Drug Resistance against ALK Inhibitors in Non-Small Cell Lung Cancer. Cancers, 13(4), 699. https://doi.org/10.3390/cancers13040699