
Descriptive and Functional Genomics in Acute Myeloid Leukemia (AML): Paving the Road for a Cure

Abstract
:Simple Summary
Abstract
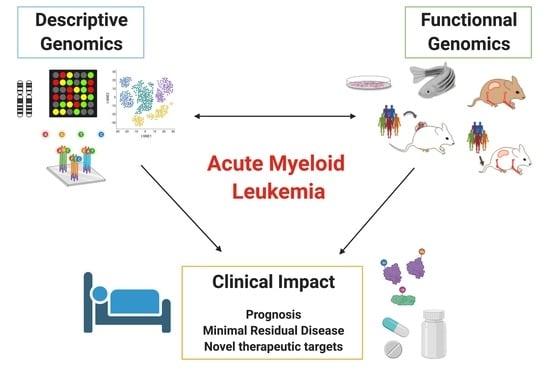
1. Introduction
2. The AML Genome: Descriptive Genomics
2.1. Genomic Technological Advances and Their Contribution to AML Molecular Characterization
2.2. The Main AML Recurrently Mutated Genes: Description and Function
2.3. Leukemogenesis and AML Clonal Evolution: From Preleukemic State to Relapse
3. Prognostic and Therapeutic Contributions of AML Genome Characterization
3.1. Molecular Subgroups of AML with Normal Karyotype
3.2. Minimal Residual Disease: An Independent Prognosis Factor
3.3. New Molecular Anomalies as Novel Therapeutic Targets
4. Functional Genomics: A Better Understanding of the Functionality and Interactions between Different Oncogenes in Aml, towards New Therapeutic Avenues
4.1. Therapeutic Limitations of Descriptive Genomics in AML
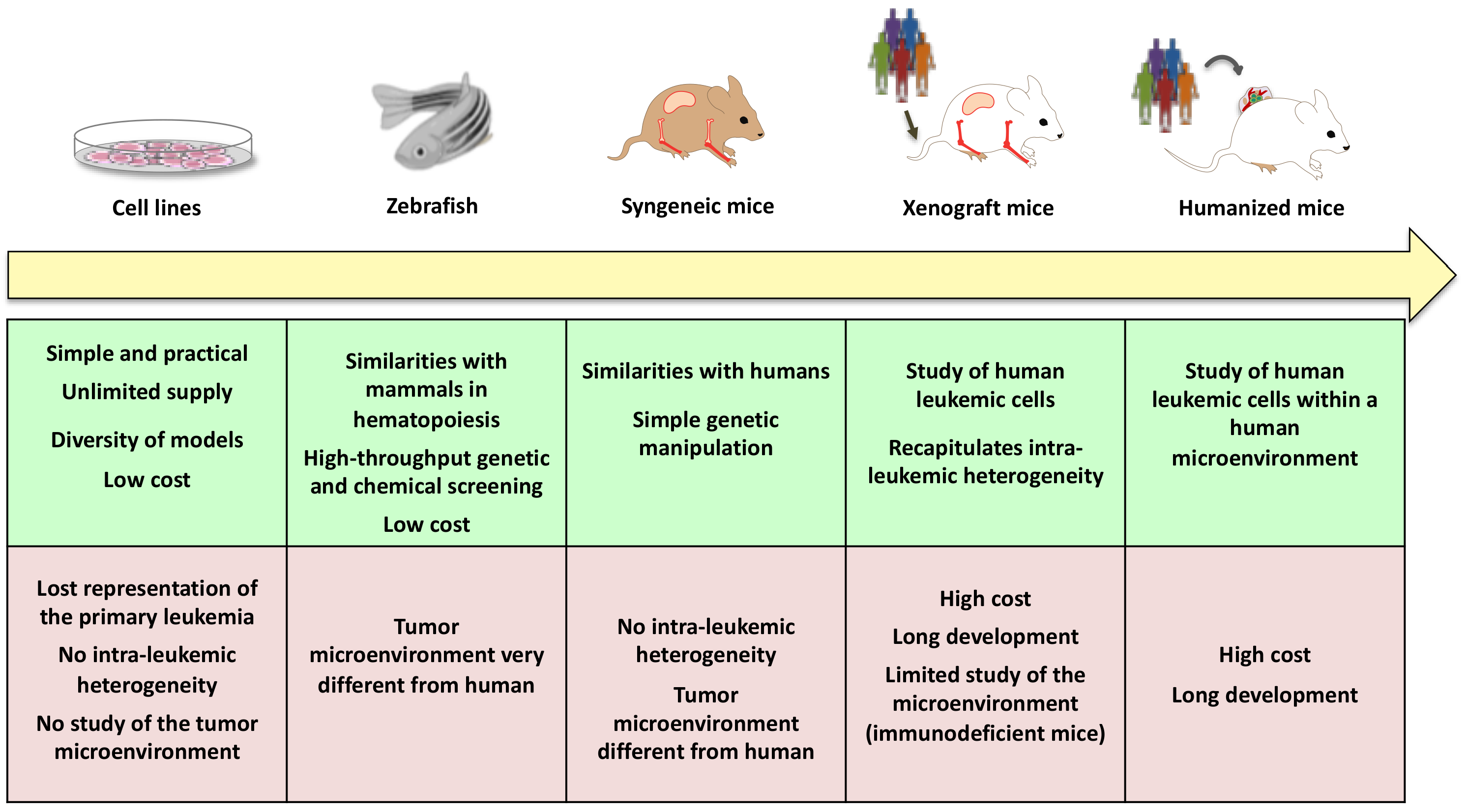
4.2. New Models and New Technologies to Better Understand AML Leukemogenesis and Ultimately Improve Treatment
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ostgård, L.S.G.; Kjeldsen, E.; Holm, M.S.; Brown, P.D.N.; Pedersen, B.B.; Bendix, K.; Johansen, P.; Kristensen, J.S.; Nørgaard, J.M. Reasons for Treating Secondary AML as de Novo AML. Eur. J. Haematol. 2010, 85, 217–226. [Google Scholar] [CrossRef]
- Visser, O.; Trama, A.; Maynadié, M.; Stiller, C.; Marcos-Gragera, R.; De Angelis, R.; Mallone, S.; Tereanu, C.; Allemani, C.; Ricardi, U.; et al. Incidence, Survival and Prevalence of Myeloid Malignancies in Europe. Eur. J. Cancer 2012, 48, 3257–3266. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.; Gralnick, H.R.; Sultan, C. Proposals for the Classification of the Acute Leukaemias French-American-British (FAB) Co-Operative Group. Br. J. Haematol. 1976, 33, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Vardiman, J.W.; Harris, N.L.; Brunning, R.D. The World Health Organization (WHO) Classification of the Myeloid Neoplasms. Blood 2002, 100, 2292–2302. [Google Scholar] [CrossRef] [PubMed]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 Revision of the World Health Organization (WHO) Classification of Myeloid Neoplasms and Acute Leukemia: Rationale and Important Changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and Management of Acute Myeloid Leukemia in Adults: Recommendations from an International Expert Panel, on Behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and Management of AML in Adults: 2017 ELN Recommendations from an International Expert Panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [Green Version]
- Oshimura, M.; Hayata, I.; Kakati, S.; Sandberg, A.A. Chromosomes and Causation of Human Cancer and Leukemia. XVII. Banding Studies in Acute Myeloblastic Leukemia (AML). Cancer 1976, 38, 748–761. [Google Scholar] [CrossRef]
- Larson, R.A.; Le Beau, M.M.; Vardiman, J.W.; Testa, J.R.; Golomb, H.M.; Rowley, J.D. The Predictive Value of Initial Cytogenetic Studies in 148 Adults with Acute Nonlymphocytic Leukemia: A 12-Year Study (1970–1982). Cancer Genet. Cytogenet. 1983, 10, 219–236. [Google Scholar] [CrossRef]
- Nakao, M.; Yokota, S.; Iwai, T.; Kaneko, H.; Horiike, S.; Kashima, K.; Sonoda, Y.; Fujimoto, T.; Misawa, S. Internal Tandem Duplication of the Flt3 Gene Found in Acute Myeloid Leukemia. Leukemia 1996, 10, 1911–1918. [Google Scholar]
- Nakagawa, T.; Saitoh, S.; Imoto, S.; Itoh, M.; Tsutsumi, M.; Hikiji, K.; Nakamura, H.; Matozaki, S.; Ogawa, R.; Nakao, Y.; et al. Multiple Point Mutation of N-ras and K-ras Oncogenes in Myelodysplastic Syndrome and Acute Myelogenous Leukemia. Oncology 1992, 49, 114–122. [Google Scholar] [CrossRef]
- Sanford, J.P.; Sait, S.N.; Pan, L.; Nowak, N.J.; Gill, H.J.; Le Beau, M.M.; Diaz, M.O.; Zabel, B.; Shows, T.B. Characterization of Two 11q23.3–11q24 Deletions and Mapping of Associated Anonymous DNA Markers. Genes Chromosomes Cancer 1993, 7, 67–73. [Google Scholar] [CrossRef] [PubMed]
- King-Underwood, L.; Pritchard-Jones, K. Wilms’ Tumor (WT1) Gene Mutations Occur Mainly in Acute Myeloid Leukemia and May Confer Drug Resistance. Blood 1998, 91, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
- Arland, M.; Fiedler, W.; Samalecos, A.; Hossfeld, D.K. Absence of Point Mutations in a Functionally Important Part of the Extracellular Domain of the C-Kit Proto-Oncogene in a Series of Patients with Acute Myeloid Leukemia (AML). Leukemia 1994, 8, 498–501. [Google Scholar]
- Pabst, T.; Eyholzer, M.; Haefliger, S.; Schardt, J.; Mueller, B.U. Somatic CEBPA Mutations Are a Frequent Second Event in Families with Germline CEBPA Mutations and Familial Acute Myeloid Leukemia. J. Clin. Oncol. 2008, 26, 5088–5093. [Google Scholar] [CrossRef]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic Nucleophosmin in Acute Myelogenous Leukemia with a Normal Karyotype. N. Engl. J. Med. 2005, 352, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in Myeloid Cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating Mutations of the Histone Methyltransferase Gene EZH2 in Myeloid Disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef]
- Ley, T.J.; Mardis, E.R.; Ding, L.; Fulton, B.; McLellan, M.D.; Chen, K.; Dooling, D.; Dunford-Shore, B.H.; McGrath, S.; Hickenbotham, M.; et al. DNA Sequencing of a Cytogenetically Normal Acute Myeloid Leukaemia Genome. Nature 2008, 456, 66–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring Mutations Found by Sequencing an Acute Myeloid Leukemia Genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, C.N.; Chong, C.-E.; Carmichael, C.L.; Wilkins, E.J.; Brautigan, P.J.; Li, X.-C.; Babic, M.; Lin, M.; Carmagnac, A.; Lee, Y.K.; et al. Heritable GATA2 Mutations Associated with Familial Myelodysplastic Syndrome and Acute Myeloid Leukemia. Nat. Genet. 2011, 43, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Van Vlierberghe, P.; Patel, J.; Abdel-Wahab, O.; Lobry, C.; Hedvat, C.V.; Balbin, M.; Nicolas, C.; Payer, A.R.; Fernandez, H.F.; Tallman, M.S.; et al. PHF6 Mutations in Adult Acute Myeloid Leukemia. Leukemia 2011, 25, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, V.; Tiacci, E.; Holmes, A.B.; Kohlmann, A.; Martelli, M.P.; Kern, W.; Spanhol-Rosseto, A.; Klein, H.-U.; Dugas, M.; Schindela, S.; et al. Whole-Exome Sequencing Identifies Somatic Mutations of BCOR in Acute Myeloid Leukemia with Normal Karyotype. Blood 2011, 118, 6153–6163. [Google Scholar] [CrossRef] [Green Version]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour Evolution Inferred by Single-Cell Sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [Green Version]
- van Galen, P.; Hovestadt, V.; Wadsworth, M.H., II; Hughes, T.K.; Griffin, G.K.; Battaglia, S.; Verga, J.A.; Stephansky, J.; Pastika, T.J.; Story, J.L.; et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 2019, 176, 1265–1281.e24. [Google Scholar] [CrossRef] [Green Version]
- Potter, N.; Miraki-Moud, F.; Ermini, L.; Titley, I.; Vijayaraghavan, G.; Papaemmanuil, E.; Campbell, P.; Gribben, J.; Taussig, D.; Greaves, M. Single Cell Analysis of Clonal Architecture in Acute Myeloid Leukaemia. Leukemia 2019, 33, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Kelly, L.M.; Kutok, J.L.; Williams, I.R.; Boulton, C.L.; Amaral, S.M.; Curley, D.P.; Ley, T.J.; Gilliland, D.G. PML/RARalpha and FLT3-ITD Induce an APL-like Disease in a Mouse Model. Proc. Natl. Acad. Sci. USA 2002, 99, 8283–8288. [Google Scholar] [CrossRef] [Green Version]
- Boissel, N.; Leroy, H.; Brethon, B.; Philippe, N.; de Botton, S.; Auvrignon, A.; Raffoux, E.; Leblanc, T.; Thomas, X.; Hermine, O.; et al. Incidence and Prognostic Impact of c-Kit, FLT3, and Ras Gene Mutations in Core Binding Factor Acute Myeloid Leukemia (CBF-AML). Leukemia 2006, 20, 965–970. [Google Scholar] [CrossRef] [Green Version]
- Grove, C.S.; Vassiliou, G.S. Acute Myeloid Leukaemia: A Paradigm for the Clonal Evolution of Cancer? Dis. Model. Mech. 2014, 7, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Boddu, P.; Kantarjian, H.; Garcia-Manero, G.; Allison, J.; Sharma, P.; Daver, N. The Emerging Role of Immune Checkpoint Based Approaches in AML and MDS. Leuk. Lymphoma 2018, 59, 790–802. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Busque, L.; Mio, R.; Mattioli, J.; Brais, E.; Blais, N.; Lalonde, Y.; Maragh, M.; Gilliland, D.G. Nonrandom X-Inactivation Patterns in Normal Females: Lyonization Ratios Vary with Age. Blood 1996, 88, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Champion, K.M.; Gilbert, J.G.; Asimakopoulos, F.A.; Hinshelwood, S.; Green, A.R. Clonal Haemopoiesis in Normal Elderly Women: Implications for the Myeloproliferative Disorders and Myelodysplastic Syndromes. Br. J. Haematol. 1997, 97, 920–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busque, L.; Gilliland, D.G. X-Inactivation Analysis in the 1990s: Promise and Potential Problems. Leukemia 1998, 12, 128–135. [Google Scholar] [CrossRef] [Green Version]
- Buscarlet, M.; Tessier, A.; Provost, S.; Mollica, L.; Busque, L. Human Blood Cell Levels of 5-Hydroxymethylcytosine (5hmC) Decline with Age, Partly Related to Acquired Mutations in TET2. Exp. Hematol. 2016, 44, 1072–1084. [Google Scholar] [CrossRef]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-Related Mutations Associated with Clonal Hematopoietic Expansion and Malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [Green Version]
- Vosberg, S.; Greif, P.A. Clonal Evolution of Acute Myeloid Leukemia from Diagnosis to Relapse. Genes Chromosomes Cancer 2019, 58, 839–849. [Google Scholar] [CrossRef] [Green Version]
- Miles, L.A.; Bowman, R.L.; Merlinsky, T.R.; Csete, I.S.; Ooi, A.T.; Durruthy-Durruthy, R.; Bowman, M.; Famulare, C.; Patel, M.A.; Mendez, P.; et al. Single-Cell Mutation Analysis of Clonal Evolution in Myeloid Malignancies. Nature 2020, 587, 477–482. [Google Scholar] [CrossRef]
- Klco, J.M.; Spencer, D.H.; Miller, C.A.; Griffith, M.; Lamprecht, T.L.; O’Laughlin, M.; Fronick, C.; Magrini, V.; Demeter, R.T.; Fulton, R.S.; et al. Functional Heterogeneity of Genetically Defined Subclones in Acute Myeloid Leukemia. Cancer Cell 2014, 25, 379–392. [Google Scholar] [CrossRef] [Green Version]
- Krönke, J.; Bullinger, L.; Teleanu, V.; Tschürtz, F.; Gaidzik, V.I.; Kühn, M.W.M.; Rücker, F.G.; Holzmann, K.; Paschka, P.; Kapp-Schwörer, S.; et al. Clonal Evolution in Relapsed NPM1-Mutated Acute Myeloid Leukemia. Blood 2013, 122, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal Evolution in Relapsed Acute Myeloid Leukaemia Revealed by Whole-Genome Sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Mrózek, K.; Dodge, R.K.; Carroll, A.J.; Edwards, C.G.; Arthur, D.C.; Pettenati, M.J.; Patil, S.R.; Rao, K.W.; Watson, M.S.; et al. Pretreatment Cytogenetic Abnormalities Are Predictive of Induction Success, Cumulative Incidence of Relapse, and Overall Survival in Adult Patients with de Novo Acute Myeloid Leukemia: Results from Cancer and Leukemia Group B (CALGB 8461). Blood 2002, 100, 4325–4336. [Google Scholar] [CrossRef]
- Grimwade, D.; Walker, H.; Oliver, F.; Wheatley, K.; Harrison, C.; Harrison, G.; Rees, J.; Hann, I.; Stevens, R.; Burnett, A.; et al. The Importance of Diagnostic Cytogenetics on Outcome in AML: Analysis of 1612 Patients Entered into the MRC AML 10 Trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood 1998, 92, 2322–2333. [Google Scholar] [CrossRef] [Green Version]
- Mrózek, K.; Heerema, N.A.; Bloomfield, C.D. Cytogenetics in Acute Leukemia. Blood Rev. 2004, 18, 115–136. [Google Scholar] [CrossRef]
- Slovak, M.L.; Kopecky, K.J.; Cassileth, P.A.; Harrington, D.H.; Theil, K.S.; Mohamed, A.; Paietta, E.; Willman, C.L.; Head, D.R.; Rowe, J.M.; et al. Karyotypic Analysis Predicts Outcome of Preremission and Postremission Therapy in Adult Acute Myeloid Leukemia: A Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood 2000, 96, 4075–4083. [Google Scholar] [CrossRef] [PubMed]
- Schlenk, R.F.; Döhner, K.; Krauter, J.; Fröhling, S.; Corbacioglu, A.; Bullinger, L.; Habdank, M.; Späth, D.; Morgan, M.; Benner, A.; et al. Mutations and Treatment Outcome in Cytogenetically Normal Acute Myeloid Leukemia. N. Engl. J. Med. 2008, 358, 1909–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, J.P.; Gönen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic Relevance of Integrated Genetic Profiling in Acute Myeloid Leukemia. N. Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gale, R.E.; Green, C.; Allen, C.; Mead, A.J.; Burnett, A.K.; Hills, R.K.; Linch, D.C.; On behalf of the Medical Research Council Adult Leukaemia Working Party. The Impact of FLT3 Internal Tandem Duplication Mutant Level, Number, Size, and Interaction with NPM1 Mutations in a Large Cohort of Young Adult Patients with Acute Myeloid Leukemia. Blood 2008, 111, 2776–2784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratcorona, M.; Brunet, S.; Nomdedéu, J.; Ribera, J.M.; Tormo, M.; Duarte, R.; Escoda, L.; Guàrdia, R.; de Llano, M.P.Q.; Salamero, O.; et al. Favorable Outcome of Patients with Acute Myeloid Leukemia Harboring a Low-Allelic Burden FLT3-ITD Mutation and Concomitant NPM1 Mutation: Relevance to Post-Remission Therapy. Blood 2013, 121, 2734–2738. [Google Scholar] [CrossRef] [Green Version]
- Cavé, H.; van der Werff ten Bosch, J.; Suciu, S.; Guidal, C.; Waterkeyn, C.; Otten, J.; Bakkus, M.; Thielemans, K.; Grandchamp, B.; Vilmer, E.; et al. Clinical Significance of Minimal Residual Disease in Childhood Acute Lymphoblastic Leukemia. N. Engl. J. Med. 1998, 339, 591–598. [Google Scholar] [CrossRef]
- van Dongen, J.J.; Seriu, T.; Panzer-Grümayer, E.R.; Biondi, A.; Pongers-Willemse, M.J.; Corral, L.; Stolz, F.; Schrappe, M.; Masera, G.; Kamps, W.A.; et al. Prognostic Value of Minimal Residual Disease in Acute Lymphoblastic Leukaemia in Childhood. Lancet 1998, 352, 1731–1738. [Google Scholar] [CrossRef]
- Grimwade, D.; Freeman, S.D. Defining Minimal Residual Disease in Acute Myeloid Leukemia: Which Platforms Are Ready for “Prime Time”? Blood 2014, 124, 3345–3355. [Google Scholar] [CrossRef] [Green Version]
- Inaba, H.; Coustan-Smith, E.; Cao, X.; Pounds, S.B.; Shurtleff, S.A.; Wang, K.Y.; Raimondi, S.C.; Onciu, M.; Jacobsen, J.; Ribeiro, R.C.; et al. Comparative Analysis of Different Approaches to Measure Treatment Response in Acute Myeloid Leukemia. J. Clin. Oncol. 2012, 30, 3625–3632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivey, A.; Hills, R.K.; Simpson, M.A.; Jovanovic, J.V.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of Minimal Residual Disease in Standard-Risk AML. N. Engl. J. Med. 2016, 374, 422–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnittger, S.; Kern, W.; Tschulik, C.; Weiss, T.; Dicker, F.; Falini, B.; Haferlach, C.; Haferlach, T. Minimal Residual Disease Levels Assessed by NPM1 Mutation-Specific RQ-PCR Provide Important Prognostic Information in AML. Blood 2009, 114, 2220–2231. [Google Scholar] [CrossRef] [Green Version]
- Krönke, J.; Schlenk, R.F.; Jensen, K.-O.; Tschürtz, F.; Corbacioglu, A.; Gaidzik, V.I.; Paschka, P.; Onken, S.; Eiwen, K.; Habdank, M.; et al. Monitoring of Minimal Residual Disease in NPM1-Mutated Acute Myeloid Leukemia: A Study from the German-Austrian Acute Myeloid Leukemia Study Group. J. Clin. Oncol. 2011, 29, 2709–2716. [Google Scholar] [CrossRef]
- Shayegi, N.; Kramer, M.; Bornhäuser, M.; Schaich, M.; Schetelig, J.; Platzbecker, U.; Röllig, C.; Heiderich, C.; Landt, O.; Ehninger, G.; et al. The Level of Residual Disease Based on Mutant NPM1 Is an Independent Prognostic Factor for Relapse and Survival in AML. Blood 2013, 122, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Hubmann, M.; Köhnke, T.; Hoster, E.; Schneider, S.; Dufour, A.; Zellmeier, E.; Fiegl, M.; Braess, J.; Bohlander, S.K.; Subklewe, M.; et al. Molecular Response Assessment by Quantitative Real-Time Polymerase Chain Reaction after Induction Therapy in NPM1-Mutated Patients Identifies Those at High Risk of Relapse. Haematologica 2014, 99, 1317–1325. [Google Scholar] [CrossRef]
- Afrin, S.; Zhang, C.R.; Meyer, C.; Stinson, C.L.; Pham, T.; Bruxner, T.J.C.; Venn, N.C.; Trahair, T.N.; Sutton, R.; Marschalek, R.; et al. Targeted Next-Generation Sequencing for Detecting MLL Gene Fusions in Leukemia. Mol. Cancer Res. 2018, 16, 279–285. [Google Scholar] [CrossRef] [Green Version]
- Kohlmann, A.; Nadarajah, N.; Alpermann, T.; Grossmann, V.; Schindela, S.; Dicker, F.; Roller, A.; Kern, W.; Haferlach, C.; Schnittger, S.; et al. Monitoring of Residual Disease by Next-Generation Deep-Sequencing of RUNX1 Mutations Can Identify Acute Myeloid Leukemia Patients with Resistant Disease. Leukemia 2014, 28, 129–137. [Google Scholar] [CrossRef]
- Pløen, G.G.; Nederby, L.; Guldberg, P.; Hansen, M.; Ebbesen, L.H.; Jensen, U.B.; Hokland, P.; Aggerholm, A. Persistence of DNMT3A Mutations at Long-Term Remission in Adult Patients with AML. Br. J. Haematol. 2014, 167, 478–486. [Google Scholar] [CrossRef]
- Cilloni, D.; Saglio, G. WT1 as a Universal Marker for Minimal Residual Disease Detection and Quantification in Myeloid Leukemias and in Myelodysplastic Syndrome. Acta Haematol. 2004, 112, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.; Rahul, E.; Gupta, A.K.; Meena, J.P.; Chopra, A.; Ranjan, A.; Hussain, S.; Rath, G.K.; Tanwar, P. Molecular Update on Biology of Wilms Tumor 1 Gene and Its Applications in Acute Myeloid Leukemia. Am. J. Blood Res. 2020, 10, 151–160. [Google Scholar] [PubMed]
- Ogawa, H.; Tamaki, H.; Ikegame, K.; Soma, T.; Kawakami, M.; Tsuboi, A.; Kim, E.H.; Hosen, N.; Murakami, M.; Fujioka, T.; et al. The Usefulness of Monitoring WT1 Gene Transcripts for the Prediction and Management of Relapse Following Allogeneic Stem Cell Transplantation in Acute Type Leukemia. Blood 2003, 101, 1698–1704. [Google Scholar] [CrossRef]
- Rautenberg, C.; Pechtel, S.; Hildebrandt, B.; Betz, B.; Dienst, A.; Nachtkamp, K.; Kondakci, M.; Geyh, S.; Wieczorek, D.; Haas, R.; et al. Wilms’ Tumor 1 Gene Expression Using a Standardized European LeukemiaNet-Certified Assay Compared to Other Methods for Detection of Minimal Residual Disease in Myelodysplastic Syndrome and Acute Myelogenous Leukemia after Allogeneic Blood Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2018, 24, 2337–2343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valkova, V.; Vydra, J.; Markova, M.; Cerovska, E.; Vrana, M.; Marinov, I.; Cechova, H.; Cetkovsky, P.; Vitek, A.; Salek, C. WT1 Gene Expression in Peripheral Blood Before and After Allogeneic Stem Cell Transplantation Is a Clinically Relevant Prognostic Marker in AML—A Single-Center 14-Year Experience. Clin. Lymphoma Myeloma Leuk. 2020. [Google Scholar] [CrossRef]
- Ossenkoppele, G.J.; Schuurhuis, G.J. MRD in AML: It Is Time to Change the Definition of Remission. Best Pract. Res. Clin. Haematol. 2014, 27, 265–271. [Google Scholar] [CrossRef]
- Chen, X.; Xie, H.; Wood, B.L.; Walter, R.B.; Pagel, J.M.; Becker, P.S.; Sandhu, V.K.; Abkowitz, J.L.; Appelbaum, F.R.; Estey, E.H. Relation of Clinical Response and Minimal Residual Disease and Their Prognostic Impact on Outcome in Acute Myeloid Leukemia. J. Clin. Oncol. 2015, 33, 1258–1264. [Google Scholar] [CrossRef]
- Balsat, M.; Renneville, A.; Thomas, X.; de Botton, S.; Caillot, D.; Marceau, A.; Lemasle, E.; Marolleau, J.-P.; Nibourel, O.; Berthon, C.; et al. Postinduction Minimal Residual Disease Predicts Outcome and Benefit From Allogeneic Stem Cell Transplantation in Acute Myeloid Leukemia With NPM1 Mutation: A Study by the Acute Leukemia French Association Group. J. Clin. Oncol. 2017, 35, 185–193. [Google Scholar] [CrossRef]
- Jourdan, E.; Boissel, N.; Chevret, S.; Delabesse, E.; Renneville, A.; Cornillet, P.; Blanchet, O.; Cayuela, J.-M.; Recher, C.; Raffoux, E.; et al. Prospective Evaluation of Gene Mutations and Minimal Residual Disease in Patients with Core Binding Factor Acute Myeloid Leukemia. Blood 2013, 121, 2213–2223. [Google Scholar] [CrossRef]
- Corbacioglu, A.; Scholl, C.; Schlenk, R.F.; Eiwen, K.; Du, J.; Bullinger, L.; Fröhling, S.; Reimer, P.; Rummel, M.; Derigs, H.-G.; et al. Prognostic Impact of Minimal Residual Disease in CBFB-MYH11—Positive Acute Myeloid Leukemia. J. Clin. Oncol. 2010, 28, 3724–3729. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.A.L.; O’Brien, M.A.; Hills, R.K.; Daly, S.B.; Wheatley, K.; Burnett, A.K. Minimal Residual Disease Monitoring by Quantitative RT-PCR in Core Binding Factor AML Allows Risk Stratification and Predicts Relapse: Results of the United Kingdom MRC AML-15 Trial. Blood 2012, 120, 2826–2835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willekens, C.; Blanchet, O.; Renneville, A.; Cornillet-Lefebvre, P.; Pautas, C.; Guieze, R.; Ifrah, N.; Dombret, H.; Jourdan, E.; Preudhomme, C.; et al. Prospective Long-Term Minimal Residual Disease Monitoring Using RQ-PCR in RUNX1-RUNX1T1-Positive Acute Myeloid Leukemia: Results of the French CBF-2006 Trial. Haematologica 2016, 101, 328–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng-Er, H.; Yu-Chen, Y.; Shu-Rong, C.; Jin-Ren, C.; Jia-Xiang, L.; Lin, Z.; Long-Ju, G.; Zhen-Yi, W. Use of All-Trans Retinoic Acid in the Treatment of Acute Promyelocytic Leukemia. Blood 1988, 72, 567–572, Erratum in 2016, 128, 3017. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.Q.; Shi, X.G.; Tang, W.; Xiong, S.M.; Zhu, J.; Cai, X.; Han, Z.G.; Ni, J.H.; Shi, G.Y.; Jia, P.M.; et al. Use of Arsenic Trioxide (As2O3) in the Treatment of Acute Promyelocytic Leukemia (APL): I. As2O3 Exerts Dose-Dependent Dual Effects on APL Cells. Blood 1997, 89, 3345–3353. [Google Scholar]
- de Thé, H.; Lavau, C.; Marchio, A.; Chomienne, C.; Degos, L.; Dejean, A. The PML-RARalpha Fusion MRNA Generated by the t(15;17) Translocation in Acute Promyelocytic Leukemia Encodes a Functionally Altered RAR. Cell 1991, 66, 675–684. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; Zhu, J.; Puvion, F.; Koken, M.; Honoré, N.; Doubeikovsky, A.; Duprez, E.; Pandolfi, P.P.; Puvion, E.; Freemont, P.; et al. Role of Promyelocytic Leukemia (Pml) Sumolation in Nuclear Body Formation, 11S Proteasome Recruitment, and As2O3-Induced PML or PML/Retinoic Acid Receptor Alpha Degradation. J. Exp. Med. 2001, 193, 1361–1371. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; Jeanne, M.; Benhenda, S.; Nasr, R.; Lei, M.; Peres, L.; Zhou, J.; Zhu, J.; Raught, B.; de Thé, H. Arsenic Degrades PML or PML-RARalpha through a SUMO-Triggered RNF4/Ubiquitin-Mediated Pathway. Nat. Cell Biol. 2008, 10, 547–555. [Google Scholar] [CrossRef]
- Jeanne, M.; Lallemand-Breitenbach, V.; Ferhi, O.; Koken, M.; Le Bras, M.; Duffort, S.; Peres, L.; Berthier, C.; Soilihi, H.; Raught, B.; et al. PML/RARA Oxidation and Arsenic Binding Initiate the Antileukemia Response of As2O3. Cancer Cell 2010, 18, 88–98. [Google Scholar] [CrossRef]
- Ablain, J.; Rice, K.; Soilihi, H.; de Reynies, A.; Minucci, S.; de Thé, H. Activation of a Promyelocytic Leukemia-Tumor Protein 53 Axis Underlies Acute Promyelocytic Leukemia Cure. Nat. Med. 2014, 20, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Ablain, J.; de Thé, H. Retinoic Acid Signaling in Cancer: The Parable of Acute Promyelocytic Leukemia. Int. J. Cancer 2014, 135, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- Schnittger, S.; Schoch, C.; Dugas, M.; Kern, W.; Staib, P.; Wuchter, C.; Löffler, H.; Sauerland, C.M.; Serve, H.; Büchner, T.; et al. Analysis of FLT3 Length Mutations in 1003 Patients with Acute Myeloid Leukemia: Correlation to Cytogenetics, FAB Subtype, and Prognosis in the AMLCG Study and Usefulness as a Marker for the Detection of Minimal Residual Disease. In Proceedings of the 42nd Annual Meeting of the American Society of Hematology, San Francisco, CA, USA, 1–5 December 2000; pp. 59–66. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Piloto, O.; Nguyen, H.B.; Greenberg, K.; Takamiya, K.; Racke, F.; Huso, D.; Small, D. Knock-in of an Internal Tandem Duplication Mutation into Murine FLT3 Confers Myeloproliferative Disease in a Mouse Model. Blood 2008, 111, 3849–3858. [Google Scholar] [CrossRef] [Green Version]
- Stirewalt, D.L.; Pogosova-Agadjanyan, E.L.; Tsuchiya, K.; Joaquin, J.; Meshinchi, S. Copy-Neutral Loss of Heterozygosity Is Prevalent and a Late Event in the Pathogenesis of FLT3/ITD AML. Blood Cancer J. 2014, 4, e208. [Google Scholar] [CrossRef] [Green Version]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.Y.; Hernandez, D.; Rajkhowa, T.; Smith, S.C.; Raman, J.R.; Nguyen, B.; Small, D.; Levis, M. Preclinical Studies of Gilteritinib, a next-Generation FLT3 Inhibitor. Blood 2017, 129, 257–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perl, A.E.; Altman, J.K.; Cortes, J.; Smith, C.; Litzow, M.; Baer, M.R.; Claxton, D.; Erba, H.P.; Gill, S.; Goldberg, S.; et al. Selective Inhibition of FLT3 by Gilteritinib in Relapsed or Refractory Acute Myeloid Leukaemia: A Multicentre, First-in-Human, Open-Label, Phase 1–2 Study. Lancet Oncol. 2017, 18, 1061–1075. [Google Scholar] [CrossRef]
- Usuki, K.; Sakura, T.; Kobayashi, Y.; Miyamoto, T.; Iida, H.; Morita, S.; Bahceci, E.; Kaneko, M.; Kusano, M.; Yamada, S.; et al. Clinical Profile of Gilteritinib in Japanese Patients with Relapsed/Refractory Acute Myeloid Leukemia: An Open-Label Phase 1 Study. Cancer Sci. 2018, 109, 3235–3244. [Google Scholar] [CrossRef]
- McMahon, C.M.; Perl, A.E. Gilteritinib for the Treatment of Relapsed and/or Refractory FLT3-Mutated Acute Myeloid Leukemia. Expert Rev. Clin. Pharmacol. 2019, 12, 841–849. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- Rakheja, D.; Konoplev, S.; Medeiros, L.J.; Chen, W. IDH Mutations in Acute Myeloid Leukemia. Hum. Pathol. 2012, 43, 1541–1551. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in Mutant IDH2 Relapsed or Refractory Acute Myeloid Leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Jonveaux, P.; Quiquandon, I.; Laï, J.L.; Pignon, J.M.; Loucheux-Lefebvre, M.H.; Bauters, F.; Berger, R.; Kerckaert, J.P. P53 Gene Mutations in Acute Myeloid Leukemia with 17p Monosomy. Blood 1991, 78, 1652–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.-M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 Alterations in Acute Myeloid Leukemia with Complex Karyotype Correlate with Specific Copy Number Alterations, Monosomal Karyotype, and Dismal Outcome. Blood 2012, 119, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Bowen, D.; Groves, M.J.; Burnett, A.K.; Patel, Y.; Allen, C.; Green, C.; Gale, R.E.; Hills, R.; Linch, D.C. TP53 Gene Mutation Is Frequent in Patients with Acute Myeloid Leukemia and Complex Karyotype, and Is Associated with Very Poor Prognosis. Leukemia 2009, 23, 203–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christiansen, D.H.; Andersen, M.K.; Pedersen-Bjergaard, J. Mutations with Loss of Heterozygosity of P53 Are Common in Therapy-Related Myelodysplasia and Acute Myeloid Leukemia after Exposure to Alkylating Agents and Significantly Associated with Deletion or Loss of 5q, a Complex Karyotype, and a Poor Prognosis. J. Clin. Oncol. 2001, 19, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, C.; Dicker, F.; Herholz, H.; Schnittger, S.; Kern, W.; Haferlach, T. Mutations of the TP53 Gene in Acute Myeloid Leukemia Are Strongly Associated with a Complex Aberrant Karyotype. Leukemia 2008, 22, 1539–1541. [Google Scholar] [CrossRef]
- Devillier, R.; Mansat-De Mas, V.; Gelsi-Boyer, V.; Demur, C.; Murati, A.; Corre, J.; Prebet, T.; Bertoli, S.; Brecqueville, M.; Arnoulet, C.; et al. Role of ASXL1 and TP53 Mutations in the Molecular Classification and Prognosis of Acute Myeloid Leukemias with Myelodysplasia-Related Changes. Oncotarget 2015, 6, 8388–8396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döhner, H.; Dolnik, A.; Tang, L.; Seymour, J.F.; Minden, M.D.; Stone, R.M.; Del Castillo, T.B.; Al-Ali, H.K.; Santini, V.; Vyas, P.; et al. Cytogenetics and Gene Mutations Influence Survival in Older Patients with Acute Myeloid Leukemia Treated with Azacitidine or Conventional Care. Leukemia 2018, 32, 2546–2557. [Google Scholar] [CrossRef]
- Hunter, A.M.; Sallman, D.A. Current Status and New Treatment Approaches in TP53 Mutated AML. Best Pract. Res. Clin. Haematol. 2019, 32, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Perdrix, A.; Najem, A.; Saussez, S.; Awada, A.; Journe, F.; Ghanem, G.; Krayem, M. PRIMA-1 and PRIMA-1Met (APR-246): From Mutant/Wild Type P53 Reactivation to Unexpected Mechanisms Underlying Their Potent Anti-Tumor Effect in Combinatorial Therapies. Cancers 2017, 9, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.M.R.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1 Reactivates Mutant P53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, JCO2002341. [Google Scholar] [CrossRef] [PubMed]
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; et al. CPX-351 (Cytarabine and Daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients With Newly Diagnosed Secondary Acute Myeloid Leukemia. J. Clin. Oncol. 2018, 36, 2684–2692. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Quek, L.; David, M.D.; Kennedy, A.; Metzner, M.; Amatangelo, M.; Shih, A.; Stoilova, B.; Quivoron, C.; Heiblig, M.; Willekens, C.; et al. Clonal Heterogeneity of Acute Myeloid Leukemia Treated with the IDH2 Inhibitor Enasidenib. Nat. Med. 2018, 24, 1167–1177. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Shih, A.H.; Wang, B.; Nazir, A.; Rustenburg, A.S.; Albanese, S.K.; Patel, M.; Famulare, C.; Correa, F.M.; Takemoto, N.; et al. Acquired Resistance to IDH Inhibition through Trans or Cis Dimer-Interface Mutations. Nature 2018, 559, 125–129. [Google Scholar] [CrossRef]
- DiNardo, C. Enasidenib plus azacitidine significantly improves complete remission and overall response rates versus azacitidine monotherapy in mutant-idh2 newly diagnosed acute myeloid leukemia (nd-aml). Abstr. EHA 2020. [Google Scholar]
- DiNardo, C. Combination of Venetoclax, Azacytidine and Ivosidenib in IDH1 Mutated AML. Abstr. EHA 2020. [Google Scholar]
- Arnone, M.; Konantz, M.; Hanns, P.; Paczulla Stanger, A.M.; Bertels, S.; Godavarthy, P.S.; Christopeit, M.; Lengerke, C. Acute Myeloid Leukemia Stem Cells: The Challenges of Phenotypic Heterogeneity. Cancers 2020, 12, 3742. [Google Scholar] [CrossRef]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A Cell Initiating Human Acute Myeloid Leukaemia after Transplantation into SCID Mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Angelini, D.F.; Ottone, T.; Guerrera, G.; Lavorgna, S.; Cittadini, M.; Buccisano, F.; De Bardi, M.; Gargano, F.; Maurillo, L.; Divona, M.; et al. A Leukemia-Associated CD34/CD123/CD25/CD99+ Immunophenotype Identifies FLT3-Mutated Clones in Acute Myeloid Leukemia. Clin. Cancer Res. 2015, 21, 3977–3985. [Google Scholar] [CrossRef] [Green Version]
- Garg, S.; Reyes-Palomares, A.; He, L.; Bergeron, A.; Lavallée, V.-P.; Lemieux, S.; Gendron, P.; Rohde, C.; Xia, J.; Jagdhane, P.; et al. Hepatic Leukemia Factor Is a Novel Leukemic Stem Cell Regulator in DNMT3A, NPM1, and FLT3-ITD Triple-Mutated AML. Blood 2019, 134, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Paczulla, A.M.; Rothfelder, K.; Raffel, S.; Konantz, M.; Steinbacher, J.; Wang, H.; Tandler, C.; Mbarga, M.; Schaefer, T.; Falcone, M.; et al. Absence of NKG2D Ligands Defines Leukaemia Stem Cells and Mediates Their Immune Evasion. Nature 2019, 572, 254–259. [Google Scholar] [CrossRef]
- Castaigne, S.; Pautas, C.; Terré, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.-N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of Gemtuzumab Ozogamicin on Survival of Adult Patients with De-Novo Acute Myeloid Leukaemia (ALFA-0701): A Randomised, Open-Label, Phase 3 Study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef]
- Larson, R.A.; Sievers, E.L.; Stadtmauer, E.A.; Löwenberg, B.; Estey, E.H.; Dombret, H.; Theobald, M.; Voliotis, D.; Bennett, J.M.; Richie, M.; et al. Final Report of the Efficacy and Safety of Gemtuzumab Ozogamicin (Mylotarg) in Patients with CD33-Positive Acute Myeloid Leukemia in First Recurrence. Cancer 2005, 104, 1442–1452. [Google Scholar] [CrossRef]
- Giles, F.J.; Kantarjian, H.M.; Kornblau, S.M.; Thomas, D.A.; Garcia-Manero, G.; Waddelow, T.A.; David, C.L.; Phan, A.T.; Colburn, D.E.; Rashid, A.; et al. Mylotarg™ (Gemtuzumab Ozogamicin) Therapy Is Associated with Hepatic Venoocclusive Disease in Patients Who Have Not Received Stem Cell Transplantation. Cancer 2001, 92, 406–413. [Google Scholar] [CrossRef]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval Summary: Gemtuzumab Ozogamicin in Relapsed Acute Myeloid Leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar]
- Selby, C.; Yacko, L.R.; Glode, A.E. Gemtuzumab Ozogamicin: Back Again. J. Adv. Pract. Oncol. 2019, 10, 68–82. [Google Scholar]
- Sallman, D.A.; Asch, A.S.; Al Malki, M.M.; Lee, D.J.; Donnellan, W.B.; Marcucci, G.; Kambhampati, S.; Daver, N.G.; Garcia-Manero, G.; Komrokji, R.S.; et al. The First-in-Class Anti-CD47 Antibody Magrolimab (5F9) in Combination with Azacitidine Is Effective in MDS and AML Patients: Ongoing Phase 1b Results. Blood 2019, 134, 569. [Google Scholar] [CrossRef]
- Hutmacher, C.; Volta, L.; Rinaldi, F.; Murer, P.; Myburgh, R.; Manz, M.G.; Neri, D. Development of a Novel Fully-Human Anti-CD123 Antibody to Target Acute Myeloid Leukemia. Leuk. Res. 2019, 84, 106178. [Google Scholar] [CrossRef]
- Ma, H.; Padmanabhan, I.S.; Parmar, S.; Gong, Y. Targeting CLL-1 for Acute Myeloid Leukemia Therapy. J. Hematol. Oncol. 2019, 12, 41. [Google Scholar] [CrossRef]
- Bouvier, A.; Ribourtout, B.; François, S.; Orvain, C.; Paz, D.L.; Beucher, A.; Guérard, A.; Guardiola, P.; Ugo, V.; Blanchet, O.; et al. Donor Cell-Derived Acute Promyelocytic Leukemia after Allogeneic Hematopoietic Stem Cell Transplantation. Eur. J. Haematol. 2018, 101, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Medyouf, H.; Mossner, M.; Jann, J.-C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic Cells in Patients Reprogram Mesenchymal Stromal Cells to Establish a Transplantable Stem Cell Niche Disease Unit. Cell Stem Cell 2014, 14, 824–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schepers, K.; Campbell, T.B.; Passegué, E. Normal and Leukemic Stem Cell Niches: Insights and Therapeutic Opportunities. Cell Stem Cell 2015, 16, 254–267. [Google Scholar] [CrossRef] [Green Version]
- Raaijmakers, M.H.G.P.; Mukherjee, S.; Guo, S.; Zhang, S.; Kobayashi, T.; Schoonmaker, J.A.; Ebert, B.L.; Al-Shahrour, F.; Hasserjian, R.P.; Scadden, E.O.; et al. Bone Progenitor Dysfunction Induces Myelodysplasia and Secondary Leukaemia. Nature 2010, 464, 852–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kode, A.; Manavalan, J.S.; Mosialou, I.; Bhagat, G.; Rathinam, C.V.; Luo, N.; Khiabanian, H.; Lee, A.; Murty, V.V.; Friedman, R.; et al. Leukaemogenesis Induced by an Activating β-Catenin Mutation in Osteoblasts. Nature 2014, 506, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Passaro, D.; Di Tullio, A.; Abarrategi, A.; Rouault-Pierre, K.; Foster, K.; Ariza-McNaughton, L.; Montaner, B.; Chakravarty, P.; Bhaw, L.; Diana, G.; et al. Increased Vascular Permeability in the Bone Marrow Microenvironment Contributes to Disease Progression and Drug Response in Acute Myeloid Leukemia. Cancer Cell 2017, 32, 324–341.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blau, O.; Baldus, C.D.; Hofmann, W.-K.; Thiel, G.; Nolte, F.; Burmeister, T.; Türkmen, S.; Benlasfer, O.; Schümann, E.; Sindram, A.; et al. Mesenchymal Stromal Cells of Myelodysplastic Syndrome and Acute Myeloid Leukemia Patients Have Distinct Genetic Abnormalities Compared with Leukemic Blasts. Blood 2011, 118, 5583–5592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skayneh, H.; Jishi, B.; Hleihel, R.; Hamieh, M.; Darwiche, N.; Bazarbachi, A.; El Sabban, M.; El Hajj, H. A Critical Review of Animal Models Used in Acute Myeloid Leukemia Pathophysiology. Genes 2019, 10, 614. [Google Scholar] [CrossRef] [Green Version]
- Crozatier, M.; Meister, M. Drosophila Haematopoiesis. Cell. Microbiol. 2007, 9, 1117–1126. [Google Scholar] [CrossRef]
- Osman, D.; Gobert, V.; Ponthan, F.; Heidenreich, O.; Haenlin, M.; Waltzer, L. A Drosophila Model Identifies Calpains as Modulators of the Human Leukemogenic Fusion Protein AML1-ETO. Proc. Natl. Acad. Sci. USA 2009, 106, 12043–12048. [Google Scholar] [CrossRef] [Green Version]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The Zebrafish Reference Genome Sequence and Its Relationship to the Human Genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, J.-R.J.; Munson, K.M.; Elagib, K.E.; Goldfarb, A.N.; Sweetser, D.A.; Peterson, R.T. Discovering Chemical Modifiers of Oncogene-Regulated Hematopoietic Differentiation. Nat. Chem. Biol. 2009, 5, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Skipper, H.E.; Schabel, F.M.; Wilcox, W.S. Experimental Evaluation of Potential Anticancer Agents. XXI. Scheduling of Arabinosylcytosine to Take Advantage of Its S-Phase Specificity against Leukemia Cells. Cancer Chemother. Rep. 1967, 51, 125–165. [Google Scholar]
- Skipper, H.E.; Perry, S. Kinetics of Normal and Leukemic Leukocyte Populations and Relevance to Chemotherapy. Cancer Res. 1970, 30, 1883–1897. [Google Scholar]
- Early, E.; Moore, M.A.; Kakizuka, A.; Nason-Burchenal, K.; Martin, P.; Evans, R.M.; Dmitrovsky, E. Transgenic Expression of PML/RARalpha Impairs Myelopoiesis. Proc. Natl. Acad. Sci. USA 1996, 93, 7900–7904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grisolano, J.L.; Wesselschmidt, R.L.; Pelicci, P.G.; Ley, T.J. Altered Myeloid Development and Acute Leukemia in Transgenic Mice Expressing PML-RARalpha under Control of Cathepsin G Regulatory Sequences. Blood 1997, 89, 376–387. [Google Scholar] [CrossRef] [Green Version]
- Feuring-Buske, M.; Gerhard, B.; Cashman, J.; Humphries, R.K.; Eaves, C.J.; Hogge, D.E. Improved Engraftment of Human Acute Myeloid Leukemia Progenitor Cells in Beta 2-Microglobulin-Deficient NOD/SCID Mice and in NOD/SCID Mice Transgenic for Human Growth Factors. Leukemia 2003, 17, 760–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wunderlich, M.; Chou, F.-S.; Link, K.A.; Mizukawa, B.; Perry, R.L.; Carroll, M.; Mulloy, J.C. AML Xenograft Efficiency Is Significantly Improved in NOD/SCID-IL2RG Mice Constitutively Expressing Human SCF, GM-CSF and IL-3. Leukemia 2010, 24, 1785–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinisch, A.; Thomas, D.; Corces, M.R.; Zhang, X.; Gratzinger, D.; Hong, W.-J.; Schallmoser, K.; Strunk, D.; Majeti, R. A Humanized Bone Marrow Ossicle Xenotransplantation Model Enables Improved Engraftment of Healthy and Leukemic Human Hematopoietic Cells. Nat. Med. 2016, 22, 812–821. [Google Scholar] [CrossRef]
- Reinisch, A.; Hernandez, D.C.; Schallmoser, K.; Majeti, R. Generation and Use of a Humanized Bone-Marrow-Ossicle Niche for Hematopoietic Xenotransplantation into Mice. Nat. Protoc. 2017, 12, 2169–2188. [Google Scholar] [CrossRef] [PubMed]
- Fenouille, N.; Bassil, C.F.; Ben-Sahra, I.; Benajiba, L.; Alexe, G.; Ramos, A.; Pikman, Y.; Conway, A.S.; Burgess, M.R.; Li, Q.; et al. The Creatine Kinase Pathway Is a Metabolic Vulnerability in EVI1-Positive Acute Myeloid Leukemia. Nat. Med. 2017, 23, 301–313. [Google Scholar] [CrossRef]
- Benajiba, L.; Alexe, G.; Su, A.; Raffoux, E.; Soulier, J.; Hemann, M.T.; Hermine, O.; Itzykson, R.; Stegmaier, K.; Puissant, A. Creatine Kinase Pathway Inhibition Alters GSK3 and WNT Signaling in EVI1-Positive AML. Leukemia 2019, 33, 800–804. [Google Scholar] [CrossRef]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi Screen Identifies Brd4 as a Therapeutic Target in Acute Myeloid Leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Tzelepis, K.; Koike-Yusa, H.; De Braekeleer, E.; Li, Y.; Metzakopian, E.; Dovey, O.M.; Mupo, A.; Grinkevich, V.; Li, M.; Mazan, M.; et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 2016, 17, 1193–1205. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, T.; Masuda, T.; Canver, M.C.; Seiler, M.; Semba, Y.; Shboul, M.; Al-Raqad, M.; Maeda, M.; Schoonenberg, V.A.C.; Cole, M.A.; et al. Genome-Wide CRISPR-Cas9 Screen Identifies Leukemia-Specific Dependence on a Pre-MRNA Metabolic Pathway Regulated by DCPS. Cancer Cell 2018, 33, 386–400.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Marker | Prognostic Impact | Marker of MRD | Targeted Therapy |
|---|---|---|---|
| PML-RARa | FAVORABLE | Yes | ATRA and Arsenic Trioxide |
| RUNX1-RUNX1T1 | FAVORABLE | Yes | None |
| CBFb-MYH11 | FAVORABLE | Yes | None |
| NPM1 mutant | without FLT3-ITD or with FLT3-ITD low ratio FAVORABLE | Yes | None |
| with FLT3-ITD high ratio INTERMEDIATE | |||
| FLT3 mutant (ITD or TKD) | FLT3-ITD low ratio with NPM1 wild type FLT3-ITD high ratio with NPM1 mutant INTERMEDIATE | No | FLT3 inhibitors (1) Three generational categoriesMIDOSTAURINE (1st) GILTERITINIB (3rd) (2) Two functional categories |
| FLT3-ITD high ratio with NPM1 wild type UNFAVORABLE | |||
| IDH1/IDH2 mutant | None | No | IVOSIDENIB (IDH1 inhibitor) ENASIDENIB (IDH2 inhibitor) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasquer, H.; Tostain, M.; Kaci, N.; Roux, B.; Benajiba, L. Descriptive and Functional Genomics in Acute Myeloid Leukemia (AML): Paving the Road for a Cure. Cancers 2021, 13, 748. https://doi.org/10.3390/cancers13040748
Pasquer H, Tostain M, Kaci N, Roux B, Benajiba L. Descriptive and Functional Genomics in Acute Myeloid Leukemia (AML): Paving the Road for a Cure. Cancers. 2021; 13(4):748. https://doi.org/10.3390/cancers13040748
Chicago/Turabian StylePasquer, Hélène, Maëlys Tostain, Nina Kaci, Blandine Roux, and Lina Benajiba. 2021. "Descriptive and Functional Genomics in Acute Myeloid Leukemia (AML): Paving the Road for a Cure" Cancers 13, no. 4: 748. https://doi.org/10.3390/cancers13040748
APA StylePasquer, H., Tostain, M., Kaci, N., Roux, B., & Benajiba, L. (2021). Descriptive and Functional Genomics in Acute Myeloid Leukemia (AML): Paving the Road for a Cure. Cancers, 13(4), 748. https://doi.org/10.3390/cancers13040748