
Preclinical Models in Prostate Cancer: Resistance to AR Targeting Therapies in Prostate Cancer



Abstract
Simple Summary
Abstract
1. Introduction
2. Overview of Preclinical PCa Models
2.1. Prostate Cancer Derived Cell Lines
2.1.1. Cell Lines
2.1.2. Xenografts and 3D models
2.2. Genetically-Engineered Mouse Models (GEMM)
2.3. Patient-Derived Models
2.4. The Ideal Preclinical Models
3. Preclinical Models to Study AR Resistance Mechanisms
3.1. Reactivation of AR Signaling Output
3.1.1. AR Overexpression
3.1.2. AR Mutation
3.1.3. AR Splice Variants
3.1.4. Glucocorticoid Receptor Takeover
3.1.5. AR Coactivators
3.1.6. AR Outlaw Activation by Cytokines and Growth Factors
3.2. Mechanisms That Bypass AR Signaling
3.2.1. Neuroendocrine Transdifferentiation
3.2.2. Stemness/Stem Cells
4. Challenges and Therapeutic Opportunities
4.1. The Problem of Cross-Resistance
4.1.1. To AR-Targeting Therapies
4.1.2. To Chemotherapeutics
4.2. Heterogeneity
4.3. Targeted Therapies
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sehgal, P.D.; Bauman, T.M.; Nicholson, T.M.; Vellky, J.E.; Ricke, E.A.; Tang, W.; Xu, W.; Huang, W.; Ricke, W.A. Tissue-specific quantification and localization of androgen and estrogen receptors in prostate cancer. Hum. Pathol. 2019, 89, 99–108. [Google Scholar] [CrossRef] [PubMed]
- EAU Guidelines. Edn. Presented at the EAU Annual Congress Amsterdam 2020. Available online: https://uroweb.org/guideline/prostate-cancer/ (accessed on 25 December 2020).
- Tan, M.H.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Yi, P.; Hamilton, R.A.; Shen, H.; Chen, M.; Foulds, C.E.; Mancini, M.A.; Ludtke, S.J.; Wang, Z.; O'Malley, B.W. Structural Insights of Transcriptionally Active, Full-Length Androgen Receptor Coactivator Complexes. Mol. Cell 2020, 79, 812–823.e4. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, K.E.; Arden, K.C.; Cavenee, W.K. Multiple G1 regulatory elements control the androgen-dependent proliferation of prostatic carcinoma cells. J. Biol. Chem. 1998, 273, 20213–20222. [Google Scholar] [CrossRef]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Hoefer, J.; Akbor, M.; Handle, F.; Ofer, P.; Puhr, M.; Parson, W.; Culig, Z.; Klocker, H.; Heidegger, I. Critical role of androgen receptor level in prostate cancer cell resistance to new generation antiandrogen enzalutamide. Oncotarget 2016, 7, 59781–59794. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; You, Z. In vitro and in vivo model systems used in prostate cancer research. J. Biol. Methods 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Horoszewicz, J.S.; Leong, S.S.; Chu, T.M.; Wajsman, Z.L.; Friedman, M.; Papsidero, L.; Kim, U.; Chai, L.S.; Kakati, S.; Arya, S.K.; et al. The LNCaP cell line—A new model for studies on human prostatic carcinoma. Prog Clin. Biol. Res. 1980, 37, 115–132. [Google Scholar]
- Thalmann, G.N.; Anezinis, P.E.; Chang, S.M.; Zhau, H.E.; Kim, E.E.; Hopwood, V.L.; Pathak, S.; von Eschenbach, A.C.; Chung, L.W. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 1994, 54, 2577–2581. [Google Scholar] [PubMed]
- Handle, F.; Prekovic, S.; Helsen, C.; Van den Broeck, T.; Smeets, E.; Moris, L.; Eerlings, R.; Kharraz, S.E.; Urbanucci, A.; Mills, I.G.; et al. Drivers of AR indifferent anti-androgen resistance in prostate cancer cells. Sci. Rep. 2019, 9, 13786. [Google Scholar] [CrossRef] [PubMed]
- Korenchuk, S.; Lehr, J.E.; MClean, L.; Lee, Y.G.; Whitney, S.; Vessella, R.; Lin, D.L.; Pienta, K.J. VCaP, a cell-based model system of human prostate cancer. In Vivo 2001, 15, 163–168. [Google Scholar] [PubMed]
- Lee, Y.G.; Korenchuk, S.; Lehr, J.; Whitney, S.; Vessela, R.; Pienta, K.J. Establishment and characterization of a new human prostatic cancer cell line: DuCaP. In Vivo 2001, 15, 157–162. [Google Scholar] [PubMed]
- Klein, K.A.; Reiter, R.E.; Redula, J.; Moradi, H.; Zhu, X.L.; Brothman, A.R.; Lamb, D.J.; Marcelli, M.; Belldegrun, A.; Witte, O.N.; et al. Progression of metastatic human prostate cancer to androgen independence in immunodeficient SCID mice. Nat. Med. 1997, 3, 402–408. [Google Scholar] [CrossRef]
- Stone, K.R.; Mickey, D.D.; Wunderli, H.; Mickey, G.H.; Paulson, D.F. Isolation of a human prostate carcinoma cell line (DU 145). Int. J. Cancer 1978, 21, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Kaighn, M.E.; Narayan, K.S.; Ohnuki, Y.; Lechner, J.F.; Jones, L.W. Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Investig. Urol. 1979, 17, 16–23. [Google Scholar]
- Sheng, X.; Li, Z.; Wang, D.L.; Li, W.B.; Luo, Z.; Chen, K.H.; Cao, J.J.; Yu, C.; Liu, W.J. Isolation and enrichment of PC-3 prostate cancer stem-like cells using MACS and serum-free medium. Oncol. Lett. 2013, 5, 787–792. [Google Scholar] [CrossRef]
- Tai, S.; Sun, Y.; Squires, J.M.; Zhang, H.; Oh, W.K.; Liang, C.Z.; Huang, J. PC3 is a cell line characteristic of prostatic small cell carcinoma. Prostate 2011, 71, 1668–1679. [Google Scholar] [CrossRef]
- Sramkoski, R.M.; Pretlow, T.G., 2nd; Giaconia, J.M.; Pretlow, T.P.; Schwartz, S.; Sy, M.S.; Marengo, S.R.; Rhim, J.S.; Zhang, D.; Jacobberger, J.W. A new human prostate carcinoma cell line, 22Rv1. In Vitro Cell Dev. Biol. Anim. 1999, 35, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Navone, N.M.; Olive, M.; Ozen, M.; Davis, R.; Troncoso, P.; Tu, S.M.; Johnston, D.; Pollack, A.; Pathak, S.; von Eschenbach, A.C.; et al. Establishment of two human prostate cancer cell lines derived from a single bone metastasis. Clin. Cancer Res. 1997, 3, 2493–2500. [Google Scholar] [PubMed]
- Dependency Map (DepMap). Available online: https://depmap.org/portal/ (accessed on 25 December 2020).
- Cancer Cell Line Encyclopedia. Available online: https://portals.broadinstitute.org/ccle (accessed on 25 December 2020).
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef]
- Tang, S.; Metaferia, N.Y.; Nogueira, M.F.; Gelbard, M.K.; Abou Alaiwi, S.; Seo, J.-H.; Hwang, J.H.; Strathdee, C.A.; Baca, S.C.; Li, J.; et al. A genome-scale CRISPR screen reveals PRMT1 as a critical regulator of androgen receptor signaling in prostate cancer. BioRxiv 2020. [Google Scholar] [CrossRef]
- Pan, Y.; Kytola, S.; Farnebo, F.; Wang, N.; Lui, W.O.; Nupponen, N.; Isola, J.; Visakorpi, T.; Bergerheim, U.S.; Larsson, C. Characterization of chromosomal abnormalities in prostate cancer cell lines by spectral karyotyping. Cytogenet. Cell Genet. 1999, 87, 225–232. [Google Scholar] [CrossRef]
- Nguyen, H.M.; Vessella, R.L.; Morrissey, C.; Brown, L.G.; Coleman, I.M.; Higano, C.S.; Mostaghel, E.A.; Zhang, X.; True, L.D.; Lam, H.M.; et al. LuCaP Prostate Cancer Patient-Derived Xenografts Reflect the Molecular Heterogeneity of Advanced Disease and Serve as Models for Evaluating Cancer Therapeutics. Prostate 2017, 77, 654–671. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.M.; Nguyen, H.M.; Labrecque, M.P.; Brown, L.G.; Coleman, I.M.; Gulati, R.; Lakely, B.; Sondheim, D.; Chatterjee, P.; Marck, B.T.; et al. Durable Response of Enzalutamide-resistant Prostate Cancer to Supraphysiological Testosterone Is Associated with a Multifaceted Growth Suppression and Impaired DNA Damage Response Transcriptomic Program in Patient-derived Xenografts. Eur. Urol. 2020, 77, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Risbridger, G.P.; Toivanen, R.; Taylor, R.A. Preclinical Models of Prostate Cancer: Patient-Derived Xenografts, Organoids, and Other Explant Models. Cold Spring Harb. Perspect. Med. 2018, 8, a030536. [Google Scholar] [CrossRef] [PubMed]
- Neuwirt, H.; Bouchal, J.; Kharaishvili, G.; Ploner, C.; Johrer, K.; Pitterl, F.; Weber, A.; Klocker, H.; Eder, I.E. Cancer-associated fibroblasts promote prostate tumor growth and progression through upregulation of cholesterol and steroid biosynthesis. Cell Commun. Signal. 2020, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Eder, T.; Weber, A.; Neuwirt, H.; Grunbacher, G.; Ploner, C.; Klocker, H.; Sampson, N.; Eder, I.E. Cancer-Associated Fibroblasts Modify the Response of Prostate Cancer Cells to Androgen and Anti-Androgens in Three-Dimensional Spheroid Culture. Int. J. Mol. Sci. 2016, 17, 1458. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef] [PubMed]
- Arriaga, J.M.; Abate-Shen, C. Genetically Engineered Mouse Models of Prostate Cancer in the Postgenomic Era. Cold Spring Harb. Perspect. Med. 2019, 9, a030528. [Google Scholar] [CrossRef]
- Arriaga, J.M.; Panja, S.; Alshalalfa, M.; Zhao, J.; Zou, M.; Giacobbe, A.; Madubata, C.J.; Kim, J.Y.; Rodriguez, A.; Coleman, I.; et al. A MYC and RAS co-activation signature in localized prostate cancer drives bone metastasis and castration resistance. Nat. Cancer 2020, 1, 1082–1096. [Google Scholar] [CrossRef]
- El Kharraz, S.; Christine, H.; Vanessa, D.; Florian, H.; Claude, L.; Martin, V.R.; Adriaan, H.; Claes, O.; Matti, P.; Nina, A.; et al. Dimerization of the ligand-binding domain is crucial for proper functioning of the androgen receptor. Endocr. Abstr. 2020. [Google Scholar] [CrossRef]
- Kerkhofs, S.; Denayer, S.; Haelens, A.; Claessens, F. Androgen receptor knockout and knock-in mouse models. J. Mol. Endocrinol. 2009, 42, 11–17. [Google Scholar] [CrossRef]
- Greenberg, N.M.; DeMayo, F.; Finegold, M.J.; Medina, D.; Tilley, W.D.; Aspinall, J.O.; Cunha, G.R.; Donjacour, A.A.; Matusik, R.J.; Rosen, J.M. Prostate cancer in a transgenic mouse. Proc. Natl. Acad. Sci. USA 1995, 92, 3439–3443. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, A.; Jacobi, A.; Lawrence, M.G.; Risbridger, G.P.; Frydenberg, M.; Williams, E.D.; Vela, I.; Hutmacher, D.W.; Bray, L.J.; Taubenberger, A. Cancer-associated fibroblasts of the prostate promote a compliant and more invasive phenotype in benign prostate epithelial cells. Mater. Today Bio 2020, 8, 100073. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Puca, L.; Bareja, R.; Prandi, D.; Shaw, R.; Benelli, M.; Karthaus, W.R.; Hess, J.; Sigouros, M.; Donoghue, A.; Kossai, M.; et al. Patient derived organoids to model rare prostate cancer phenotypes. Nat. Commun. 2018, 9, 2404. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Weinstein, H.N.W.; Allegakoen, P.; Wadsworth, M.H.; Xie, J.; Yang, H.; Feng, F.Y.; Carroll, P.R.; Wang, B.; Cooperberg, M.R.; et al. Single-cell analysis of human primary prostate cancer reveals the heterogeneity of tumor-associated epithelial cell states. BioRxiv 2020. [Google Scholar] [CrossRef]
- Centenera, M.M.; Hickey, T.E.; Jindal, S.; Ryan, N.K.; Ravindranathan, P.; Mohammed, H.; Robinson, J.L.; Schiewer, M.J.; Ma, S.; Kapur, P.; et al. A patient-derived explant (PDE) model of hormone-dependent cancer. Mol. Oncol. 2018, 12, 1608–1622. [Google Scholar] [CrossRef] [PubMed]
- Handle, F.; Puhr, M.; Schaefer, G.; Lorito, N.; Hoefer, J.; Gruber, M.; Guggenberger, F.; Santer, F.R.; Marques, R.B.; van Weerden, W.M.; et al. The STAT3 Inhibitor Galiellalactone Reduces IL6-Mediated AR Activity in Benign and Malignant Prostate Models. Mol. Cancer Ther. 2018, 17, 2722–2731. [Google Scholar] [CrossRef]
- Visakorpi, T.; Hyytinen, E.; Koivisto, P.; Tanner, M.; Keinanen, R.; Palmberg, C.; Palotie, A.; Tammela, T.; Isola, J.; Kallioniemi, O.P. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995, 9, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chan, S.C.; Brand, L.J.; Hwang, T.H.; Silverstein, K.A.T.; Dehm, S.M. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013, 73, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ning, S.; Lou, W.; Yang, J.C.; Armstrong, C.M.; Lombard, A.P.; D'Abronzo, L.S.; Evans, C.P.; Gao, A.C.; Liu, C. Cross-Resistance Among Next-Generation Antiandrogen Drugs Through the AKR1C3/AR-V7 Axis in Advanced Prostate Cancer. Mol. Cancer Ther. 2020, 19, 1708–1718. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z.; Hoffmann, J.; Erdel, M.; Eder, I.E.; Hobisch, A.; Hittmair, A.; Bartsch, G.; Utermann, G.; Schneider, M.R.; Parczyk, K.; et al. Switch from antagonist to agonist of the androgen receptor bicalutamide is associated with prostate tumour progression in a new model system. Br. J. Cancer 1999, 81, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Kregel, S.; Chen, J.L.; Tom, W.; Krishnan, V.; Kach, J.; Brechka, H.; Fessenden, T.B.; Isikbay, M.; Paner, G.P.; Szmulewitz, R.Z.; et al. Acquired resistance to the second-generation androgen receptor antagonist enzalutamide in castration-resistant prostate cancer. Oncotarget 2016, 7, 26259–26274. [Google Scholar] [CrossRef]
- Li, Q.; Deng, Q.; Chao, H.P.; Liu, X.; Lu, Y.; Lin, K.; Liu, B.; Tang, G.W.; Zhang, D.; Tracz, A.; et al. Linking prostate cancer cell AR heterogeneity to distinct castration and enzalutamide responses. Nat. Commun. 2018, 9, 3600. [Google Scholar] [CrossRef]
- Vlachostergios, P.J.; Puca, L.; Beltran, H. Emerging Variants of Castration-Resistant Prostate Cancer. Curr. Oncol. Rep. 2017, 19, 32. [Google Scholar] [CrossRef]
- Chatterjee, P.; Schweizer, M.T.; Lucas, J.M.; Coleman, I.; Nyquist, M.D.; Frank, S.B.; Tharakan, R.; Mostaghel, E.; Luo, J.; Pritchard, C.C.; et al. Supraphysiological androgens suppress prostate cancer growth through androgen receptor-mediated DNA damage. J. Clin. Investig. 2019, 129, 4245–4260. [Google Scholar] [CrossRef]
- Teply, B.A.; Wang, H.; Luber, B.; Sullivan, R.; Rifkind, I.; Bruns, A.; Spitz, A.; DeCarli, M.; Sinibaldi, V.; Pratz, C.F.; et al. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: An open-label, phase 2, multicohort study. Lancet Oncol. 2018, 19, 76–86. [Google Scholar] [CrossRef]
- Veldscholte, J.; Ris-Stalpers, C.; Kuiper, G.G.J.M.; Jenster, G.; Berrevoets, C.; Claassen, E.; van Rooij, H.C.J.; Trapman, J.; Brinkmann, A.O.; Mulder, E. A mutation in the ligand binding domain of the androgen receptor of human INCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem. Biophys. Res. Commun. 1990, 173, 534–540. [Google Scholar] [CrossRef]
- Moilanen, A.-M.; Riikonen, R.; Oksala, R.; Ravanti, L.; Aho, E.; Wohlfahrt, G.; Nykänen, P.S.; Törmäkangas, O.P.; Palvimo, J.J.; Kallio, P.J. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Prekovic, S.; van Royen, M.E.; Voet, A.R.D.; Geverts, B.; Houtman, R.; Melchers, D.; Zhang, K.Y.J.; van den Broeck, T.; Smeets, E.; Spans, L.; et al. The Effect of F877L and T878A Mutations on Androgen Receptor Response to Enzalutamide. Mol. Cancer Ther. 2016, 15, 1702–1712. [Google Scholar] [CrossRef]
- Prekovic, S.; van den Broeck, T.; Moris, L.; Smeets, E.; Claessens, F.; Joniau, S.; Helsen, C.; Attard, G. Treatment-induced changes in the androgen receptor axis: Liquid biopsies as diagnostic/prognostic tools for prostate cancer. Mol. Cell Endocrinol. 2018, 462, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Conteduca, V.; Wetterskog, D.; Sharabiani, M.T.A.; Grande, E.; Fernandez-Perez, M.P.; Jayaram, A.; Salvi, S.; Castellano, D.; Romanel, A.; Lolli, C.; et al. Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: A multi-institution correlative biomarker study. Ann. Oncol. 2017, 28, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Halabi, S.; Luo, J.; Nanus, D.M.; Giannakakou, P.; Szmulewitz, R.Z.; Danila, D.C.; Healy, P.; Anand, M.; Rothwell, C.J.; et al. Prospective Multicenter Validation of Androgen Receptor Splice Variant 7 and Hormone Therapy Resistance in High-Risk Castration-Resistant Prostate Cancer: The PROPHECY Study. J. Clin. Oncol. 2019, 37, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Wadosky, K.M.; Koochekpour, S. Androgen receptor splice variants and prostate cancer: From bench to bedside. Oncotarget 2017, 8, 18550–18576. [Google Scholar] [CrossRef] [PubMed]
- Cato, L.; de Tribolet-Hardy, J.; Lee, I.; Rottenberg, J.T.; Coleman, I.; Melchers, D.; Houtman, R.; Xiao, T.; Li, W.; Uo, T.; et al. ARv7 Represses Tumor-Suppressor Genes in Castration-Resistant Prostate Cancer. Cancer Cell 2019, 35, 401–413.e6. [Google Scholar] [CrossRef]
- Hirayama, Y.; Tam, T.; Jian, K.; Andersen, R.J.; Sadar, M.D. Combination therapy with androgen receptor N-terminal domain antagonist EPI-7170 and enzalutamide yields synergistic activity in AR-V7-positive prostate cancer. Mol. Oncol. 2020, 14, 2455–2470. [Google Scholar] [CrossRef]
- Liu, C.; Yang, J.C.; Armstrong, C.M.; Lou, W.; Liu, L.; Qiu, X.; Zou, B.; Lombard, A.P.; D'Abronzo, L.S.; Evans, C.P.; et al. AKR1C3 Promotes AR-V7 Protein Stabilization and Confers Resistance to AR-Targeted Therapies in Advanced Prostate Cancer. Mol. Cancer Ther. 2019, 18, 1875–1886. [Google Scholar] [CrossRef]
- Kafka, M.; Mayr, F.; Temml, V.; Moller, G.; Adamski, J.; Hofer, J.; Schwaiger, S.; Heidegger, I.; Matuszczak, B.; Schuster, D.; et al. Dual Inhibitory Action of a Novel AKR1C3 Inhibitor on Both Full-Length AR and the Variant AR-V7 in Enzalutamide Resistant Metastatic Castration Resistant Prostate Cancer. Cancers 2020, 12, 2092. [Google Scholar] [CrossRef]
- Yemelyanov, A.; Bhalla, P.; Yang, X.; Ugolkov, A.; Iwadate, K.; Karseladze, A.; Budunova, I. Differential targeting of androgen and glucocorticoid receptors induces ER stress and apoptosis in prostate cancer cells: A novel therapeutic modality. Cell Cycle 2012, 11, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef]
- Puhr, M.; Hoefer, J.; Eigentler, A.; Ploner, C.; Handle, F.; Schaefer, G.; Kroon, J.; Leo, A.; Heidegger, I.; Eder, I.; et al. The Glucocorticoid Receptor Is a Key Player for Prostate Cancer Cell Survival and a Target for Improved Antiandrogen Therapy. Clin. Cancer Res. 2018, 24, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Cheng, H.; Lin, D.; Liu, L.; Yang, O.; Jia, L.; Fazli, L.; Gleave, M.E.; Wang, Y.; Rennie, P.; et al. The expression of glucocorticoid receptor is negatively regulated by active androgen receptor signaling in prostate tumors. Int. J. Cancer 2015, 136, E27–E38. [Google Scholar] [CrossRef] [PubMed]
- Sahu, B.; Laakso, M.; Pihlajamaa, P.; Ovaska, K.; Sinielnikov, I.; Hautaniemi, S.; Janne, O.A. FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res. 2013, 73, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Claessens, F.; Joniau, S.; Helsen, C. Comparing the rules of engagement of androgen and glucocorticoid receptors. Cell. Mol. Life Sci. 2017, 74, 2217–2228. [Google Scholar] [CrossRef] [PubMed]
- Kluetz, P.G.; Ning, Y.M.; Maher, V.E.; Zhang, L.; Tang, S.; Ghosh, D.; Aziz, R.; Palmby, T.; Pfuma, E.; Zirkelbach, J.F.; et al. Abiraterone acetate in combination with prednisone for the treatment of patients with metastatic castration-resistant prostate cancer: U.S. Food and Drug Administration drug approval summary. Clin. Cancer Res. 2013, 19, 6650–6656. [Google Scholar] [CrossRef]
- Montgomery, B.; Kheoh, T.; Molina, A.; Li, J.; Bellmunt, J.; Tran, N.; Loriot, Y.; Efstathiou, E.; Ryan, C.J.; Scher, H.I.; et al. Impact of baseline corticosteroids on survival and steroid androgens in metastatic castration-resistant prostate cancer: Exploratory analysis from COU-AA-301. Eur. Urol. 2015, 67, 866–873. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Malloy, P.J.; Krishnan, A.V.; Swami, S.; Navone, N.M.; Peehl, D.M.; Feldman, D. Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nat. Med. 2000, 6, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Yemelyanov, A.; Czwornog, J.; Chebotaev, D.; Karseladze, A.; Kulevitch, E.; Yang, X.; Budunova, I. Tumor suppressor activity of glucocorticoid receptor in the prostate. Oncogene 2007, 26, 1885–1896. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.Z.; Lu, J.J.; Liu, Z.D.; Zhang, H.; Wang, S.M.; Xu, H. Dexamethasone suppresses DU145 cell proliferation and cell cycle through inhibition of the extracellular signal-regulated kinase 1/2 pathway and cyclin D1 expression. Asian J. Androl. 2008, 10, 635–641. [Google Scholar] [CrossRef]
- Narayanan, S.; Srinivas, S.; Feldman, D. Androgen-glucocorticoid interactions in the era of novel prostate cancer therapy. Nat. Rev. Urol. 2016, 13, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Wongvipat, J.; Choi, D.; Wang, P.; Lee, Y.S.; Zheng, D.; Watson, P.A.; Gopalan, A.; Sawyers, C.L. GREB1 amplifies androgen receptor output in human prostate cancer and contributes to antiandrogen resistance. eLife 2019, 8, e41913. [Google Scholar] [CrossRef] [PubMed]
- Agoulnik, I.U.; Vaid, A.; Bingman, W.E.; Erdeme, H.; Frolov, A.; Smith, C.L.; Ayala, G.; Ittmann, M.M.; Weigel, N.L. Role of SRC-1 in the promotion of prostate cancer cell growth and tumor progression. Cancer Res. 2005, 65, 7959–7967. [Google Scholar] [CrossRef] [PubMed]
- Luef, B.; Handle, F.; Kharaishvili, G.; Hager, M.; Rainer, J.; Janetschek, G.; Hruby, S.; Englberger, C.; Bouchal, J.; Santer, F.R.; et al. The AR/NCOA1 axis regulates prostate cancer migration by involvement of PRKD1. Endocr. Relat. Cancer 2016, 23, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.K.; Moreira, D.; Won, H.; White, S.W.; Duttagupta, P.; Lucia, M.; Jones, J.; Hsu, J.; Kortylewski, M. Reduced T-cell Numbers and Elevated Levels of Immunomodulatory Cytokines in Metastatic Prostate Cancer Patients De Novo Resistant to Abiraterone and/or Enzalutamide Therapy. Int. J. Mol. Sci. 2019, 20, 1831. [Google Scholar] [CrossRef]
- Hobisch, A.; Ramoner, R.; Fuchs, D.; Godoy-Tundidor, S.; Bartsch, G.; Klocker, H.; Culig, Z. Prostate cancer cells (LNCaP) generated after long-term interleukin 6 (IL-6) treatment express IL-6 and acquire an IL-6 partially resistant phenotype. Clin. Cancer Res. 2001, 7, 2941–2948. [Google Scholar] [PubMed]
- Hobisch, A.; Eder, I.E.; Putz, T.; Horninger, W.; Bartsch, G.; Klocker, H.; Culig, Z. Interleukin-6 regulates prostate-specific protein expression in prostate carcinoma cells by activation of the androgen receptor. Cancer Res. 1998, 58, 4640–4645. [Google Scholar]
- Seaton, A.; Scullin, P.; Maxwell, P.J.; Wilson, C.; Pettigrew, J.; Gallagher, R.; O'Sullivan, J.M.; Johnston, P.G.; Waugh, D.J. Interleukin-8 signaling promotes androgen-independent proliferation of prostate cancer cells via induction of androgen receptor expression and activation. Carcinogenesis 2008, 29, 1148–1156. [Google Scholar] [CrossRef]
- Culig, Z.; Hobisch, A.; Cronauer, M.V.; Radmayr, C.; Trapman, J.; Hittmair, A.; Bartsch, G.; Klocker, H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994, 54, 5474–5478. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.F.; Guan, J.; Qiu, Y.; Kung, H.J. Neuropeptide-induced androgen independence in prostate cancer cells: Roles of nonreceptor tyrosine kinases Etk/Bmx, Src, and focal adhesion kinase. Mol. Cell Biol. 2001, 21, 8385–8397. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, J.; Tachibana, M.; Horiguchi, Y.; Oya, M.; Ohigashi, T.; Asakura, H.; Murai, M. Serum interleukin 6 as a prognostic factor in patients with prostate cancer. Clin. Cancer Res. 2000, 6, 2702–2706. [Google Scholar] [PubMed]
- Culig, Z.; Puhr, M. Interleukin-6 and prostate cancer: Current developments and unsolved questions. Mol. Cell Endocrinol. 2018, 462, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhu, Y.; Lou, W.; Cui, Y.; Evans, C.P.; Gao, A.C. Inhibition of constitutively active Stat3 reverses enzalutamide resistance in LNCaP derivative prostate cancer cells. Prostate 2014, 74, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; de Bono, J.S.; Flechon, A.; Heidenreich, A.; Voog, E.; Davis, N.B.; Qi, M.; Bandekar, R.; Vermeulen, J.T.; Cornfeld, M.; et al. Randomised phase II study of siltuximab (CNTO 328), an anti-IL-6 monoclonal antibody, in combination with mitoxantrone/prednisone versus mitoxantrone/prednisone alone in metastatic castration-resistant prostate cancer. Eur. J. Cancer 2012, 48, 85–93. [Google Scholar] [CrossRef]
- Dorff, T.B.; Goldman, B.; Pinski, J.K.; Mack, P.C.; Lara, P.N., Jr.; van Veldhuizen, P.J., Jr.; Quinn, D.I.; Vogelzang, N.J.; Thompson, I.M., Jr.; Hussain, M.H. Clinical and correlative results of SWOG S0354: A phase II trial of CNTO328 (siltuximab), a monoclonal antibody against interleukin-6, in chemotherapy-pretreated patients with castration-resistant prostate cancer. Clin. Cancer Res. 2010, 16, 3028–3034. [Google Scholar] [CrossRef]
- Chen, T.; Wang, L.H.; Farrar, W.L. Interleukin 6 activates androgen receptor-mediated gene expression through a signal transducer and activator of transcription 3-dependent pathway in LNCaP prostate cancer cells. Cancer Res. 2000, 60, 2132–2135. [Google Scholar] [PubMed]
- Don-Doncow, N.; Marginean, F.; Coleman, I.; Nelson, P.S.; Ehrnstrom, R.; Krzyzanowska, A.; Morrissey, C.; Hellsten, R.; Bjartell, A. Expression of STAT3 in Prostate Cancer Metastases. Eur. Urol. 2017, 71, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Handle, F.; Erb, H.H.; Luef, B.; Hoefer, J.; Dietrich, D.; Parson, W.; Kristiansen, G.; Santer, F.R.; Culig, Z. SOCS3 Modulates the Response to Enzalutamide and Is Regulated by Androgen Receptor Signaling and CpG Methylation in Prostate Cancer Cells. Mol. Cancer Res. 2016, 14, 574–585. [Google Scholar] [CrossRef]
- Thaper, D.; Vahid, S.; Kaur, R.; Kumar, S.; Nouruzi, S.; Bishop, J.L.; Johansson, M.; Zoubeidi, A. Galiellalactone inhibits the STAT3/AR signaling axis and suppresses Enzalutamide-resistant Prostate Cancer. Sci. Rep. 2018, 8, 17307. [Google Scholar] [CrossRef]
- Liu, C.; Lou, W.; Armstrong, C.; Zhu, Y.; Evans, C.P.; Gao, A.C. Niclosamide suppresses cell migration and invasion in enzalutamide resistant prostate cancer cells via Stat3-AR axis inhibition. Prostate 2015, 75, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.C.; Veeramani, S.; Lin, M.F. Neuroendocrine-like prostate cancer cells: Neuroendocrine transdifferentiation of prostate adenocarcinoma cells. Endocr. Relat. Cancer 2007, 14, 531–547. [Google Scholar] [CrossRef]
- Kaur, H.; Samarska, I.; Lu, J.; Faisal, F.; Maughan, B.L.; Murali, S.; Asrani, K.; Alshalalfa, M.; Antonarakis, E.S.; Epstein, J.I.; et al. Neuroendocrine differentiation in usual-type prostatic adenocarcinoma: Molecular characterization and clinical significance. Prostate 2020, 80, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.I.; Amin, M.B.; Beltran, H.; Lotan, T.L.; Mosquera, J.M.; Reuter, V.E.; Robinson, B.D.; Troncoso, P.; Rubin, M.A. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am. J. Surg. Pathol. 2014, 38, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Flesken-Nikitin, A.; Corney, D.C.; Wang, W.; Goodrich, D.W.; Roy-Burman, P.; Nikitin, A.Y. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res. 2006, 66, 7889–7898. [Google Scholar] [CrossRef] [PubMed]
- Faugeroux, V.; Pailler, E.; Oulhen, M.; Deas, O.; Brulle-Soumare, L.; Hervieu, C.; Marty, V.; Alexandrova, K.; Andree, K.C.; Stoecklein, N.H.; et al. Genetic characterization of a unique neuroendocrine transdifferentiation prostate circulating tumor cell-derived eXplant model. Nat. Commun. 2020, 11, 1884. [Google Scholar] [CrossRef] [PubMed]
- Yasumizu, Y.; Rajabi, H.; Jin, C.; Hata, T.; Pitroda, S.; Long, M.D.; Hagiwara, M.; Li, W.; Hu, Q.; Liu, S.; et al. MUC1-C regulates lineage plasticity driving progression to neuroendocrine prostate cancer. Nat. Commun. 2020, 11, 338. [Google Scholar] [CrossRef] [PubMed]
- Ci, X.; Hao, J.; Dong, X.; Xue, H.; Wu, R.; Choi, S.Y.C.; Haegert, A.M.; Collins, C.C.; Liu, X.; Lin, D.; et al. Conditionally Reprogrammed Cells from Patient-Derived Xenograft to Model Neuroendocrine Prostate Cancer Development. Cells 2020, 9, 1398. [Google Scholar] [CrossRef]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [PubMed]
- Friedmann-Morvinski, D.; Verma, I.M. Dedifferentiation and reprogramming: Origins of cancer stem cells. EMBO Rep. 2014, 15, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Banerjee, S.; Ahmad, A.; Li, Y.; Wang, Z.; Sethi, S.; Sarkar, F.H. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PLoS ONE 2010, 5, e12445. [Google Scholar] [CrossRef] [PubMed]
- Rybak, A.P.; Tang, D. SOX2 plays a critical role in EGFR-mediated self-renewal of human prostate cancer stem-like cells. Cell Signal. 2013, 25, 2734–2742. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Liu, X.; Laffin, B.; Chen, X.; Choy, G.; Jeter, C.R.; Calhoun-Davis, T.; Li, H.; Palapattu, G.S.; Pang, S.; et al. The PSA(-/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell 2012, 10, 556–569. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, B.E.; Leong, K.G.; Yue, P.; Li, L.; Jhunjhunwala, S.; Chen, D.; Seo, K.; Modrusan, Z.; Gao, W.Q.; et al. Androgen deprivation causes epithelial-mesenchymal transition in the prostate: Implications for androgen-deprivation therapy. Cancer Res. 2012, 72, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Shankar, E.; Kalayci, F.N.C.; Mukunda, A.; Alassfar, M.; Singh, V.; Chan, E.R.; MacLennan, G.T.; Gupta, S. Androgen Deprivation Induces Transcriptional Reprogramming in Prostate Cancer Cells to Develop Stem Cell-Like Characteristics. Int. J. Mol. Sci. 2020, 21, 9568. [Google Scholar] [CrossRef]
- Shiota, M.; Dejima, T.; Yamamoto, Y.; Takeuchi, A.; Imada, K.; Kashiwagi, E.; Inokuchi, J.; Tatsugami, K.; Kajioka, S.; Uchiumi, T.; et al. Collateral resistance to taxanes in enzalutamide-resistant prostate cancer through aberrant androgen receptor and its variants. Cancer Sci. 2018, 109, 3224–3234. [Google Scholar] [CrossRef] [PubMed]
- van Soest, R.J.; van Royen, M.E.; de Morree, E.S.; Moll, J.M.; Teubel, W.; Wiemer, E.A.; Mathijssen, R.H.; de Wit, R.; van Weerden, W.M. Cross-resistance between taxanes and new hormonal agents abiraterone and enzalutamide may affect drug sequence choices in metastatic castration-resistant prostate cancer. Eur. J. Cancer 2013, 49, 3821–3830. [Google Scholar] [CrossRef]
- Loriot, Y.; Eymard, J.C.; Patrikidou, A.; Ileana, E.; Massard, C.; Albiges, L.; Di Palma, M.; Escudier, B.; Fizazi, K. Prior long response to androgen deprivation predicts response to next-generation androgen receptor axis targeted drugs in castration resistant prostate cancer. Eur. J. Cancer 2015, 51, 1946–1952. [Google Scholar] [CrossRef] [PubMed]
- van Soest, R.J.; de Morree, E.S.; Kweldam, C.F.; de Ridder, C.M.A.; Wiemer, E.A.C.; Mathijssen, R.H.J.; de Wit, R.; van Weerden, W.M. Targeting the Androgen Receptor Confers In Vivo Cross-resistance Between Enzalutamide and Docetaxel, But Not Cabazitaxel, in Castration-resistant Prostate Cancer. Eur. Urol. 2015, 67, 981–985. [Google Scholar] [CrossRef]
- Mezynski, J.; Pezaro, C.; Bianchini, D.; Zivi, A.; Sandhu, S.; Thompson, E.; Hunt, J.; Sheridan, E.; Baikady, B.; Sarvadikar, A.; et al. Antitumour activity of docetaxel following treatment with the CYP17A1 inhibitor abiraterone: Clinical evidence for cross-resistance? Ann. Oncol. 2012, 23, 2943–2947. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, M.T.; Zhou, X.C.; Wang, H.; Bassi, S.; Carducci, M.A.; Eisenberger, M.A.; Antonarakis, E.S. The influence of prior abiraterone treatment on the clinical activity of docetaxel in men with metastatic castration-resistant prostate cancer. Eur. Urol. 2014, 66, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Al Nakouzi, N.; Le Moulec, S.; Albiges, L.; Wang, C.; Beuzeboc, P.; Gross-Goupil, M.; de La Motte Rouge, T.; Guillot, A.; Gajda, D.; Massard, C.; et al. Cabazitaxel Remains Active in Patients Progressing After Docetaxel Followed by Novel Androgen Receptor Pathway Targeted Therapies. Eur. Urol. 2015, 68, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Saad, F.; Winquist, E.; Hubay, S.; Berry, S.; Assi, H.; Levesque, E.; Aucoin, N.; Czaykowski, P.; Lattouf, J.B.; Alloul, K.; et al. Efficacy, quality of life, and safety of cabazitaxel in Canadian metastatic castration-resistant prostate cancer patients treated or not with prior abiraterone. Can. Urol. Assoc. J. 2016, 10, 102–109. [Google Scholar] [CrossRef]
- Bando, Y.; Hinata, N.; Terakawa, T.; Furukawa, J.; Harada, K.I.; Nakano, Y.; Fujisawa, M. Activity of cabazitaxel in patients with metastatic castration-resistant prostate cancer after treatment with single or dual regimens of novel androgen receptor-targeting agents. Med. Oncol. 2017, 34, 163. [Google Scholar] [CrossRef] [PubMed]
- Devlies, W.; Claessens, F. Tracking prostate cancer development at the single-cell level. Nat. Rev. Urol. 2020, 17, 545–546. [Google Scholar] [CrossRef] [PubMed]
- Berglund, E.; Maaskola, J.; Schultz, N.; Friedrich, S.; Marklund, M.; Bergenstrahle, J.; Tarish, F.; Tanoglidi, A.; Vickovic, S.; Larsson, L.; et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat. Commun. 2018, 9, 2419. [Google Scholar] [CrossRef]
- Karthaus, W.R.; Hofree, M.; Choi, D.; Linton, E.L.; Turkekul, M.; Bejnood, A.; Carver, B.; Gopalan, A.; Abida, W.; Laudone, V.; et al. Regenerative potential of prostate luminal cells revealed by single-cell analysis. Science 2020, 368, 497–505. [Google Scholar] [CrossRef]
- Devlies, W.; Eckstein, M.; Cimadamore, A.; Devos, G.; Moris, L.; van den Broeck, T.; Montironi, R.; Joniau, S.; Claessens, F.; Gevaert, T. Clinical Actionability of the Genomic Landscape of Metastatic Castration Resistant Prostate Cancer. Cells 2020, 9, 2494. [Google Scholar] [CrossRef]
- Beltran, H.; Oromendia, C.; Danila, D.C.; Montgomery, B.; Hoimes, C.; Szmulewitz, R.Z.; Vaishampayan, U.; Armstrong, A.J.; Stein, M.; Pinski, J.; et al. A Phase II Trial of the Aurora Kinase A Inhibitor Alisertib for Patients with Castration-resistant and Neuroendocrine Prostate Cancer: Efficacy and Biomarkers. Clin. Cancer Res. 2019, 25, 43–51. [Google Scholar] [CrossRef] [PubMed]
- van Hemelryk, A.; van Weerden, W.M. Novel patient-derived 3D culture models to guide clinical decision-making in prostate cancer. Curr. Opin. Endocr. Metab. Res. 2020, 10, 7–15. [Google Scholar] [CrossRef]
- Jansson, K.H.; Tucker, J.B.; Stahl, L.E.; Simmons, J.K.; Fuller, C.; Beshiri, M.L.; Agarwal, S.; Fang, L.; Hynes, P.G.; Alilin, A.N.; et al. High-throughput screens identify HSP90 inhibitors as potent therapeutics that target inter-related growth and survival pathways in advanced prostate cancer. Sci. Rep. 2018, 8, 17239. [Google Scholar] [CrossRef] [PubMed]
- Maund, S.L.; Nolley, R.; Peehl, D.M. Optimization and comprehensive characterization of a faithful tissue culture model of the benign and malignant human prostate. Lab. Investig. 2014, 94, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Corsello, S.; Nagari, R.; Spangler, R.; Rossen, J.; Kocak, M.; Bryan, J.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.; et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef] [PubMed]


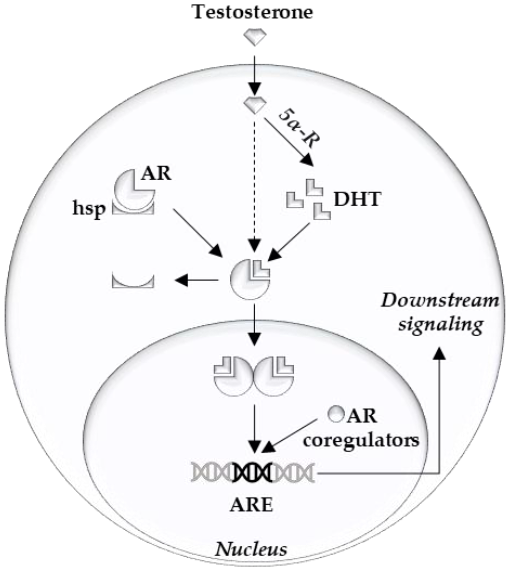{kind=link}
| Cell Line | AR/PSA | Hormone Resistant/Sensitive | Direct/Xenograft (Indirect) | Specific Characteristics |
|---|---|---|---|---|
| Lymph node metastasis | ||||
| LNCaP | + | Sensitive | Direct | T877A AR mutant, PTEN -, androgen dependent |
| LAPC-4 | + | Sensitive | Xenograft | P53 mutant, WT PTEN |
| Bone metastasis | ||||
| VCaP | + | Sensitive | Xenograft | Amplified WT AR, AR splice variants, TMPRSS-ERG fusion |
| MDA PCa 2a/b | + | Sensitive | Direct | AR mutants L702H and T878A |
| PC3 | − | Resistant | Direct | |
| CWR22(Rv1) | + | Resistant | Xenograft | H875Y AR mutant, AR splice variants, WT PTEN, Zinc Finger Duplication |
| Organ metastasis | ||||
| DU-145 (brain) | − | Resistant | Direct | |
| DuCaP (dura) | + | Sensitive | Xenograft | Amplified WT AR |
| GEMM | PCa Inducement | Specific Characteristics |
|---|---|---|
| Localized PCa alterations | ||
| TMPRSS-ERG fusion | + − | Modest phenotype on its own, additive effect with PTEN loss |
| SPOP mutation | + − | Modest phenotype on its own, additive effect with PTEN loss |
| Advanced PCa alterations | ||
| PTEN loss | + | Most frequent GEMM, Clinical stage ranges between models. Additive effects with other alterations |
| Tumor surpressor genes losses (RB/p53) | + − | Modest phenotype on its own, additive effect with PTEN/BRCA mutations/RB/p53 losses |
| DNA repair genes mutation | + | Fewer models available, study of PARP inhibitors. Additive effects with PTEN/RB/p53 losses |
| Oncogenes | + | Clinical stage ranges between Myc levels, additive effects with p53, and PTEN models |
| Other models—mechanistic studies of targets | ||
| Model | Advantages/Disadvantages | |
|---|---|---|
 | PCa cell line experiments | + Easy to culture, high-throughput system − Very homogeneous, not reflective of clinical tumors |
 | Genetically modified PCa cell lines | + Easy validation of de novo resistance mechanisms and alterations from databases − Acquired resistance more difficult to study |
 | 3D cell cultures/organoids | + Study of Cell-Cell interaction − Scaffold material can alter cellular processes |
 | Mouse Xenografts of PCa | + Study of metastasis and treatment response in vivo + Working endocrine system − Limitations similar to injected cell lines |
 | Genetically modified mouse models | + In vivo validation of genetic alterations and underlying pathways − Inter species differences, questionable generalizability to human prostate cancers |
 | Patient derived cell lines | + Good correlation with specific human tumor + Reproducibility of experiments, high throughput − Only representing a certain tumor type, no cell-cell interaction |
 | Patient derived tissue cultures | + Excellent representation of respective type of prostate cancer − Limited life span, cannot be propagated |
 | Patient derived 3D cell cultures/organoids | + Study of Cell-Cell interaction in specific tumor subtypes + Medium to High throughput − Scaffold material can alter cellular processes − Technically challenging |
 | Patient derived Xenografts | + Study of metastasis and treatment of a certain tumor in vivo + Working endocrine system − No immune system, overgrowing murine tissue, tissue collection only at endpoint. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Devlies, W.; Handle, F.; Devos, G.; Joniau, S.; Claessens, F. Preclinical Models in Prostate Cancer: Resistance to AR Targeting Therapies in Prostate Cancer. Cancers 2021, 13, 915. https://doi.org/10.3390/cancers13040915
Devlies W, Handle F, Devos G, Joniau S, Claessens F. Preclinical Models in Prostate Cancer: Resistance to AR Targeting Therapies in Prostate Cancer. Cancers. 2021; 13(4):915. https://doi.org/10.3390/cancers13040915
Chicago/Turabian StyleDevlies, Wout, Florian Handle, Gaëtan Devos, Steven Joniau, and Frank Claessens. 2021. "Preclinical Models in Prostate Cancer: Resistance to AR Targeting Therapies in Prostate Cancer" Cancers 13, no. 4: 915. https://doi.org/10.3390/cancers13040915
APA StyleDevlies, W., Handle, F., Devos, G., Joniau, S., & Claessens, F. (2021). Preclinical Models in Prostate Cancer: Resistance to AR Targeting Therapies in Prostate Cancer. Cancers, 13(4), 915. https://doi.org/10.3390/cancers13040915