
STAMP2 Expression Mediated by Cytokines Attenuates Their Growth-Limiting Effects in Prostate Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. RNA Interference
2.3. Quantitative PCR
2.4. Western Blot Analysis
2.5. Chromatin Immunoprecipitation (ChIP)
2.6. Small-Molecule Inhibitors
2.7. Cell Viability and Colony Formation Assay
2.8. Statistical Analyzes
3. Results
3.1. STAMP2 Is a Direct AR Target Gene in Prostate Cancer Cells
3.2. Temporal Regulation of STAMP2 Expression by Cytokines
3.3. Cytokines and Androgens Synergistically Induce STAMP2 Expression in PCa Cells
3.4. Cytokine-Induced STAMP2 Expression in PCa Cells Is Independent of AR
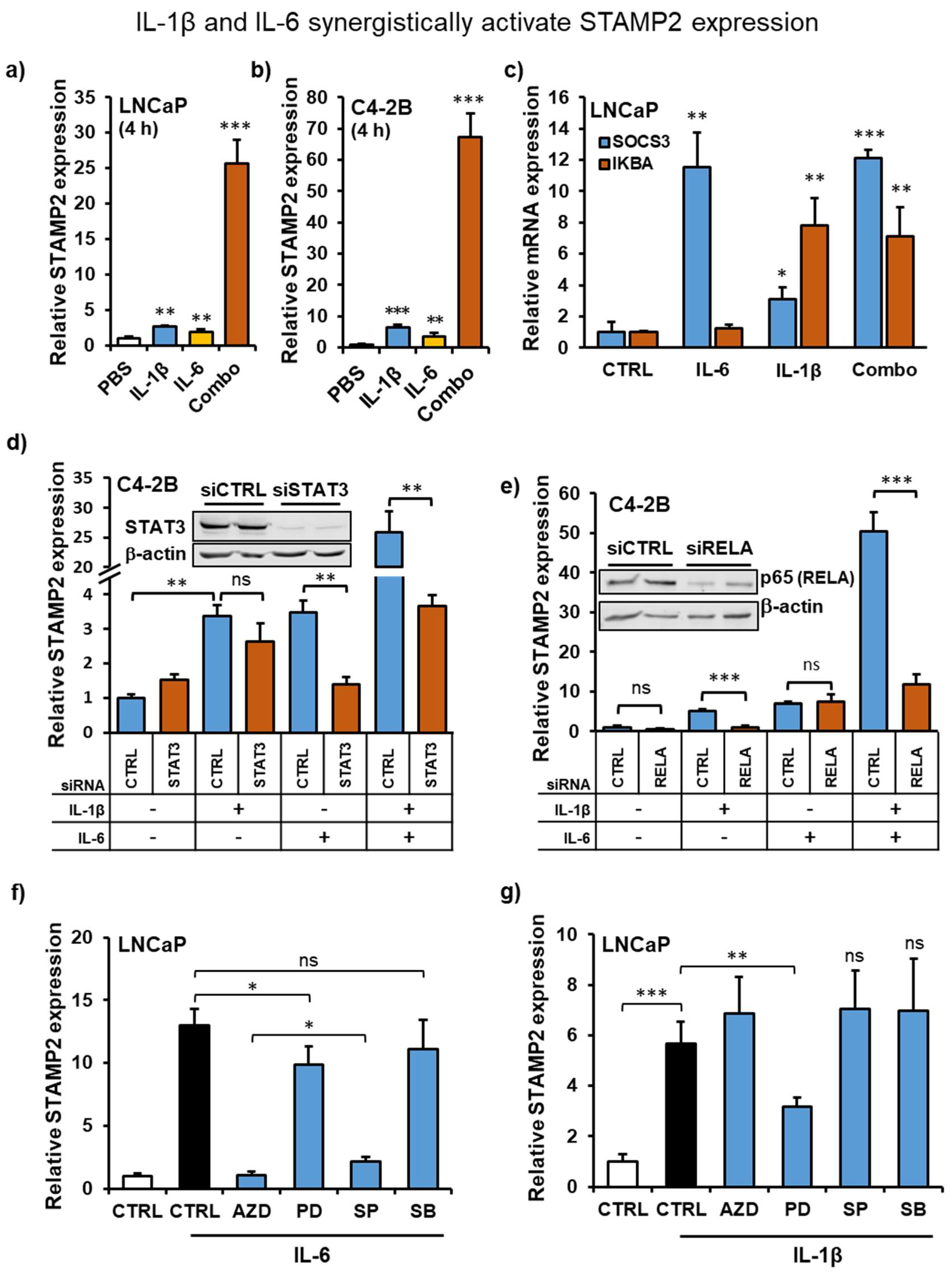
3.5. IL-1β and IL-6 Synergize to Increase STAMP2 Expression
3.6. STAMP2 Expression Attenuates Cytokine-Induced Growth Inhibition in PCa Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Attard, G.; Parker, C.; Eeles, R.A.; Schroder, F.; Tomlins, S.A.; Tannock, I.; Drake, C.G.; de Bono, J.S. Prostate cancer. Lancet 2016, 387, 70–82. [Google Scholar] [CrossRef]
- Sumanasuriya, S.; de Bono, J. Treatment of Advanced Prostate Cancer-A Review of Current Therapies and Future Promise. Cold Spring Harb. Perspect. Med. 2018, 8, a030635. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; Guo, C.; Gurel, B.; de Marzo, A.M.; Sfanos, K.S.; Mani, R.S.; Gil, J.; Drake, C.G.; Alimonti, A. Prostate carcinogenesis: Inflammatory storms. Nat. Rev. Cancer 2020, 20, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Santi, R.; Tamanini, I.; Galli, I.C.; Perletti, G.; Bjerklund Johansen, T.E.; Nesi, G. Current Knowledge of the Potential Links between Inflammation and Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 3833. [Google Scholar] [CrossRef] [Green Version]
- Strasner, A.; Karin, M. Immune Infiltration and Prostate Cancer. Front. Oncol. 2015, 5, 128. [Google Scholar] [CrossRef]
- Sfanos, K.S.; Yegnasubramanian, S.; Nelson, W.G.; de Marzo, A.M. The inflammatory microenvironment and microbiome in prostate cancer development. Nat. Rev. Urol. 2018, 15, 11–24. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, G.T.; Eisenberg, R.; Fleshman, R.L.; Taylor, J.M.; Fu, P.; Resnick, M.I.; Gupta, S. The influence of chronic inflammation in prostatic carcinogenesis: A 5-year followup study. J. Urol. 2006, 176, 1012–1016. [Google Scholar] [CrossRef]
- Hellsten, R.; Lilljebjorn, L.; Johansson, M.; Leandersson, K.; Bjartell, A. The STAT3 inhibitor galiellalactone inhibits the generation of MDSC-like monocytes by prostate cancer cells and decreases immunosuppressive and tumorigenic factors. Prostate 2019, 79, 1611–1621. [Google Scholar] [CrossRef] [Green Version]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [Green Version]
- Bouraoui, Y.; Ricote, M.; Garcia-Tunon, I.; Rodriguez-Berriguete, G.; Touffehi, M.; Rais, N.B.; Fraile, B.; Paniagua, R.; Oueslati, R.; Royuela, M. Pro-inflammatory cytokines and prostate-specific antigen in hyperplasia and human prostate cancer. Cancer Detect. Prev. 2008, 32, 23–32. [Google Scholar] [CrossRef]
- Birnie, R.; Bryce, S.D.; Roome, C.; Dussupt, V.; Droop, A.; Lang, S.H.; Berry, P.A.; Hyde, C.F.; Lewis, J.L.; Stower, M.J.; et al. Gene expression profiling of human prostate cancer stem cells reveals a pro-inflammatory phenotype and the importance of extracellular matrix interactions. Genome Biol. 2008, 9, R83. [Google Scholar] [CrossRef] [Green Version]
- Shariat, S.F.; Kattan, M.W.; Traxel, E.; Andrews, B.; Zhu, K.; Wheeler, T.M.; Slawin, K.M. Association of pre- and postoperative plasma levels of transforming growth factor beta(1) and interleukin 6 and its soluble receptor with prostate cancer progression. Clin. Cancer Res. 2004, 10, 1992–1999. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, J.; Tachibana, M.; Horiguchi, Y.; Oya, M.; Ohigashi, T.; Asakura, H.; Murai, M. Serum interleukin 6 as a prognostic factor in patients with prostate cancer. Clin. Cancer Res. 2000, 6, 2702–2706. [Google Scholar] [PubMed]
- Schroeder, A.; Herrmann, A.; Cherryholmes, G.; Kowolik, C.; Buettner, R.; Pal, S.; Yu, H.; Muller-Newen, G.; Jove, R. Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res. 2014, 74, 1227–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Eiro, N.; Bermudez-Fernandez, S.; Fernandez-Garcia, B.; Atienza, S.; Beridze, N.; Escaf, S.; Vizoso, F.J. Analysis of the expression of interleukins, interferon beta, and nuclear factor-kappa B in prostate cancer and their relationship with biochemical recurrence. J. Immunother. 2014, 37, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Staal, J.; Beyaert, R. Inflammation and NF-kappaB Signaling in Prostate Cancer: Mechanisms and Clinical Implications. Cells 2018, 7, 122. [Google Scholar] [CrossRef] [Green Version]
- Korkmaz, C.G.; Korkmaz, K.S.; Kurys, P.; Elbi, C.; Wang, L.; Klokk, T.I.; Hammarstrom, C.; Troen, G.; Svindland, A.; Hager, G.L.; et al. Molecular cloning and characterization of STAMP2, an androgen-regulated six transmembrane protein that is overexpressed in prostate cancer. Oncogene 2005, 24, 4934–4945. [Google Scholar] [CrossRef] [Green Version]
- Gauss, G.H.; Kleven, M.D.; Sendamarai, A.K.; Fleming, M.D.; Lawrence, C.M. The crystal structure of six-transmembrane epithelial antigen of the prostate 4 (Steap4), a ferri/cuprireductase, suggests a novel interdomain flavin-binding site. J. Biol. Chem. 2013, 288, 20668–20682. [Google Scholar] [CrossRef] [Green Version]
- Kleven, M.D.; Dlakic, M.; Lawrence, C.M. Characterization of a single b-type heme, FAD, and metal binding sites in the transmembrane domain of six-transmembrane epithelial antigen of the prostate (STEAP) family proteins. J. Biol. Chem. 2015, 290, 22558–22569. [Google Scholar] [CrossRef] [Green Version]
- Oosterheert, W.; van Bezouwen, L.S.; Rodenburg, R.N.P.; Granneman, J.; Forster, F.; Mattevi, A.; Gros, P. Cryo-EM structures of human STEAP4 reveal mechanism of iron(III) reduction. Nat. Commun. 2018, 9, 4337. [Google Scholar] [CrossRef]
- Jin, Y.; Wang, L.; Qu, S.; Sheng, X.; Kristian, A.; Maelandsmo, G.M.; Pallmann, N.; Yuca, E.; Tekedereli, I.; Gorgulu, K.; et al. STAMP2 increases oxidative stress and is critical for prostate cancer. EMBO Mol. Med. 2015, 7, 315–331. [Google Scholar] [CrossRef] [Green Version]
- Ten Freyhaus, H.; Calay, E.S.; Yalcin, A.; Vallerie, S.N.; Yang, L.; Calay, Z.Z.; Saatcioglu, F.; Hotamisligil, G.S. Stamp2 controls macrophage inflammation through nicotinamide adenine dinucleotide phosphate homeostasis and protects against atherosclerosis. Cell Metab. 2012, 16, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Xing, X.; Beamer, M.A.; Swindell, W.R.; Sarkar, M.K.; Roberts, L.W.; Voorhees, J.J.; Kahlenberg, J.M.; Harms, P.W.; Johnston, A.; et al. Six-transmembrane epithelial antigens of the prostate comprise a novel inflammatory nexus in patients with pustular skin disorders. J. Allergy Clin. Immunol. 2017, 139, 1217–1227. [Google Scholar] [CrossRef]
- Tanaka, Y.; Matsumoto, I.; Iwanami, K.; Inoue, A.; Minami, R.; Umeda, N.; Kanamori, A.; Ochiai, N.; Miyazawa, K.; Sugihara, M.; et al. Six-transmembrane epithelial antigen of prostate4 (STEAP4) is a tumor necrosis factor alpha-induced protein that regulates IL-6, IL-8, and cell proliferation in synovium from patients with rheumatoid arthritis. Mod. Rheumatol. 2012, 22, 128–136. [Google Scholar] [CrossRef]
- Inoue, A.; Matsumoto, I.; Tanaka, Y.; Umeda, N.; Takai, C.; Kawaguchi, H.; Ebe, H.; Yoshida, H.; Matsumoto, Y.; Segawa, S.; et al. TIARP attenuates autoantibody-mediated arthritis via the suppression of neutrophil migration by reducing CXCL2/CXCR2 and IL-6 expression. Sci. Rep. 2016, 6, 38684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kralisch, S.; Sommer, G.; Weise, S.; Lipfert, J.; Lossner, U.; Kamprad, M.; Schrock, K.; Bluher, M.; Stumvoll, M.; Fasshauer, M. Interleukin-1beta is a positive regulator of TIARP/STAMP2 gene and protein expression in adipocytes in vitro. FEBS Lett. 2009, 583, 1196–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, A.; Matsumoto, I.; Tanaka, Y.; Iwanami, K.; Kanamori, A.; Ochiai, N.; Goto, D.; Ito, S.; Sumida, T. Tumor necrosis factor alpha-induced adipose-related protein expression in experimental arthritis and in rheumatoid arthritis. Arthritis Res. Ther. 2009, 11, R118. [Google Scholar] [CrossRef] [Green Version]
- Wellen, K.E.; Fucho, R.; Gregor, M.F.; Furuhashi, M.; Morgan, C.; Lindstad, T.; Vaillancourt, E.; Gorgun, C.Z.; Saatcioglu, F.; Hotamisligil, G.S. Coordinated regulation of nutrient and inflammatory responses by STAMP2 is essential for metabolic homeostasis. Cell 2007, 129, 537–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klokk, T.I.; Kilander, A.; Xi, Z.; Waehre, H.; Risberg, B.; Danielsen, H.E.; Saatcioglu, F. Kallikrein 4 is a proliferative factor that is overexpressed in prostate cancer. Cancer Res. 2007, 67, 5221–5230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, X.; Nenseth, H.Z.; Qu, S.; Kuzu, O.F.; Frahnow, T.; Simon, L.; Greene, S.; Zeng, Q.; Fazli, L.; Rennie, P.S.; et al. IRE1alpha-XBP1s pathway promotes prostate cancer by activating c-MYC signaling. Nat. Commun. 2019, 10, 323. [Google Scholar] [CrossRef] [Green Version]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [Green Version]
- Cleutjens, K.B.; van der Korput, H.A.; van Eekelen, C.C.; van Rooij, H.C.; Faber, P.W.; Trapman, J. An androgen response element in a far upstream enhancer region is essential for high, androgen-regulated activity of the prostate-specific antigen promoter. Mol. Endocrinol. 1997, 11, 148–161. [Google Scholar] [CrossRef]
- Gonen-Korkmaz, C.; Sevin, G.; Gokce, G.; Arun, M.Z.; Yildirim, G.; Reel, B.; Kaymak, A.; Ogut, D. Analysis of tumor necrosis factor alpha-induced and nuclear factor kappaB-silenced LNCaP prostate cancer cells by RT-qPCR. Exp. Ther. Med. 2014, 8, 1695–1700. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wang, L.H.; Farrar, W.L. Interleukin 6 activates androgen receptor-mediated gene expression through a signal transducer and activator of transcription 3-dependent pathway in LNCaP prostate cancer cells. Cancer Res. 2000, 60, 2132–2135. [Google Scholar] [PubMed]
- Chang, M.A.; Patel, V.; Gwede, M.; Morgado, M.; Tomasevich, K.; Fong, E.L.; Farach-Carson, M.C.; Delk, N.A. IL-1beta induces p62/SQSTM1 and represses androgen receptor expression in prostate cancer cells. J. Cell Biochem. 2014, 115, 2188–2197. [Google Scholar] [CrossRef] [Green Version]
- Oh, K.; Lee, O.Y.; Park, Y.; Seo, M.W.; Lee, D.S. IL-1beta induces IL-6 production and increases invasiveness and estrogen-independent growth in a TG2-dependent manner in human breast cancer cells. BMC Cancer 2016, 16, 724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spooren, A.; Mestdagh, P.; Rondou, P.; Kolmus, K.; Haegeman, G.; Gerlo, S. IL-1beta potently stabilizes IL-6 mRNA in human astrocytes. Biochem. Pharmacol. 2011, 81, 1004–1015. [Google Scholar] [CrossRef] [Green Version]
- Sawada, S.; Chosa, N.; Ishisaki, A.; Naruishi, K. Enhancement of gingival inflammation induced by synergism of IL-1beta and IL-6. Biomed. Res. 2013, 34, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Culig, Z.; Puhr, M. Interleukin-6 and prostate cancer: Current developments and unsolved questions. Mol. Cell Endocrinol. 2018, 462, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Rebe, C.; Ghiringhelli, F. Interleukin-1beta and Cancer. Cancers 2020, 12, 1791. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Bredell, B.X.; Anderson, E.R.; Martin, A.; Mays, C.; Nagao-Kitamoto, H.; Huang, S.; Gyorffy, B.; Greenson, J.K.; Hardiman, K.; et al. Quantitative proteomics identifies STEAP4 as a critical regulator of mitochondrial dysfunction linking inflammation and colon cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9608–E9617. [Google Scholar] [CrossRef] [Green Version]
- Pallmann, N.; Livgard, M.; Tesikova, M.; Zeynep Nenseth, H.; Akkus, E.; Sikkeland, J.; Jin, Y.; Koc, D.; Kuzu, O.F.; Pradhan, M.; et al. Regulation of the unfolded protein response through ATF4 and FAM129A in prostate cancer. Oncogene 2019, 38, 6301–6318. [Google Scholar] [CrossRef]
- Sikkeland, J.; Sheng, X.; Jin, Y.; Saatcioglu, F. STAMPing at the crossroads of normal physiology and disease states. Mol. Cell Endocrinol. 2016, 425, 26–36. [Google Scholar] [CrossRef]
- Loh, C.Y.; Arya, A.; Naema, A.F.; Wong, W.F.; Sethi, G.; Looi, C.Y. Signal Transducer and Activator of Transcription (STATs) Proteins in Cancer and Inflammation: Functions and Therapeutic Implication. Front. Oncol. 2019, 9, 48. [Google Scholar] [CrossRef] [Green Version]
- Bishop, J.L.; Thaper, D.; Zoubeidi, A. The Multifaceted Roles of STAT3 Signaling in the Progression of Prostate Cancer. Cancers 2014, 6, 829–859. [Google Scholar] [CrossRef] [Green Version]
- Giri, D.; Ozen, M.; Ittmann, M. Interleukin-6 is an autocrine growth factor in human prostate cancer. Am. J. Pathol. 2001, 159, 2159–2165. [Google Scholar] [CrossRef] [Green Version]
- Culig, Z.; Bartsch, G.; Hobisch, A. Interleukin-6 regulates androgen receptor activity and prostate cancer cell growth. Mol. Cell Endocrinol. 2002, 197, 231–238. [Google Scholar] [CrossRef]
- Handle, F.; Puhr, M.; Schaefer, G.; Lorito, N.; Hoefer, J.; Gruber, M.; Guggenberger, F.; Santer, F.R.; Marques, R.B.; van Weerden, W.M.; et al. The STAT3 Inhibitor Galiellalactone Reduces IL6-Mediated AR Activity in Benign and Malignant Prostate Models. Mol. Cancer Ther. 2018, 17, 2722–2731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Zhu, Y.; Lou, W.; Cui, Y.; Evans, C.P.; Gao, A.C. Inhibition of constitutively active Stat3 reverses enzalutamide resistance in LNCaP derivative prostate cancer cells. Prostate 2014, 74, 201–209. [Google Scholar] [CrossRef] [Green Version]
- Ueda, T.; Bruchovsky, N.; Sadar, M.D. Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways. J. Biol. Chem. 2002, 277, 7076–7085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traish, A.; Bolanos, J.; Nair, S.; Saad, F.; Morgentaler, A. Do Androgens Modulate the Pathophysiological Pathways of Inflammation? Appraising the Contemporary Evidence. J. Clin. Med. 2018, 7, 549. [Google Scholar] [CrossRef] [Green Version]
- Debelec-Butuner, B.; Alapinar, C.; Varisli, L.; Erbaykent-Tepedelen, B.; Hamid, S.M.; Gonen-Korkmaz, C.; Korkmaz, K.S. Inflammation-mediated abrogation of androgen signaling: An in vitro model of prostate cell inflammation. Mol. Carcinog. 2014, 53, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Malinen, M.; Niskanen, E.A.; Kaikkonen, M.U.; Palvimo, J.J. Crosstalk between androgen and pro-inflammatory signaling remodels androgen receptor and NF-kappaB cistrome to reprogram the prostate cancer cell transcriptome. Nucleic Acids Res. 2017, 45, 619–630. [Google Scholar] [CrossRef] [Green Version]
- Izumi, K.; Fang, L.Y.; Mizokami, A.; Namiki, M.; Li, L.; Lin, W.J.; Chang, C. Targeting the androgen receptor with siRNA promotes prostate cancer metastasis through enhanced macrophage recruitment via CCL2/CCR2-induced STAT3 activation. EMBO Mol. Med. 2013, 5, 1383–1401. [Google Scholar] [CrossRef]
- Liu, Q.; Tong, D.; Liu, G.; Xu, J.; Do, K.; Geary, K.; Zhang, D.; Zhang, J.; Zhang, Y.; Li, Y.; et al. Metformin reverses prostate cancer resistance to enzalutamide by targeting TGF-beta1/STAT3 axis-regulated EMT. Cell Death Dis. 2017, 8, 3007. [Google Scholar] [CrossRef] [PubMed]
- Don-Doncow, N.; Marginean, F.; Coleman, I.; Nelson, P.S.; Ehrnstrom, R.; Krzyzanowska, A.; Morrissey, C.; Hellsten, R.; Bjartell, A. Expression of STAT3 in Prostate Cancer Metastases. Eur. Urol. 2017, 71, 313–316. [Google Scholar] [CrossRef] [Green Version]
- Weber, A.; Wasiliew, P.; Kracht, M. Interleukin-1 (IL-1) pathway. Sci. Signal. 2010, 3, cm1. [Google Scholar] [CrossRef]
- Hobisch, A.; Ramoner, R.; Fuchs, D.; Godoy-Tundidor, S.; Bartsch, G.; Klocker, H.; Culig, Z. Prostate cancer cells (LNCaP) generated after long-term interleukin 6 (IL-6) treatment express IL-6 and acquire an IL-6 partially resistant phenotype. Clin. Cancer Res. 2001, 7, 2941–2948. [Google Scholar] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pihlstrøm, N.; Jin, Y.; Nenseth, Z.; Kuzu, O.F.; Saatcioglu, F. STAMP2 Expression Mediated by Cytokines Attenuates Their Growth-Limiting Effects in Prostate Cancer Cells. Cancers 2021, 13, 1579. https://doi.org/10.3390/cancers13071579
Pihlstrøm N, Jin Y, Nenseth Z, Kuzu OF, Saatcioglu F. STAMP2 Expression Mediated by Cytokines Attenuates Their Growth-Limiting Effects in Prostate Cancer Cells. Cancers. 2021; 13(7):1579. https://doi.org/10.3390/cancers13071579
Chicago/Turabian StylePihlstrøm, Nicklas, Yang Jin, Zeynep Nenseth, Omer F. Kuzu, and Fahri Saatcioglu. 2021. "STAMP2 Expression Mediated by Cytokines Attenuates Their Growth-Limiting Effects in Prostate Cancer Cells" Cancers 13, no. 7: 1579. https://doi.org/10.3390/cancers13071579
APA StylePihlstrøm, N., Jin, Y., Nenseth, Z., Kuzu, O. F., & Saatcioglu, F. (2021). STAMP2 Expression Mediated by Cytokines Attenuates Their Growth-Limiting Effects in Prostate Cancer Cells. Cancers, 13(7), 1579. https://doi.org/10.3390/cancers13071579