
The Role of Network Science in Glioblastoma



Abstract
:Simple Summary
Abstract
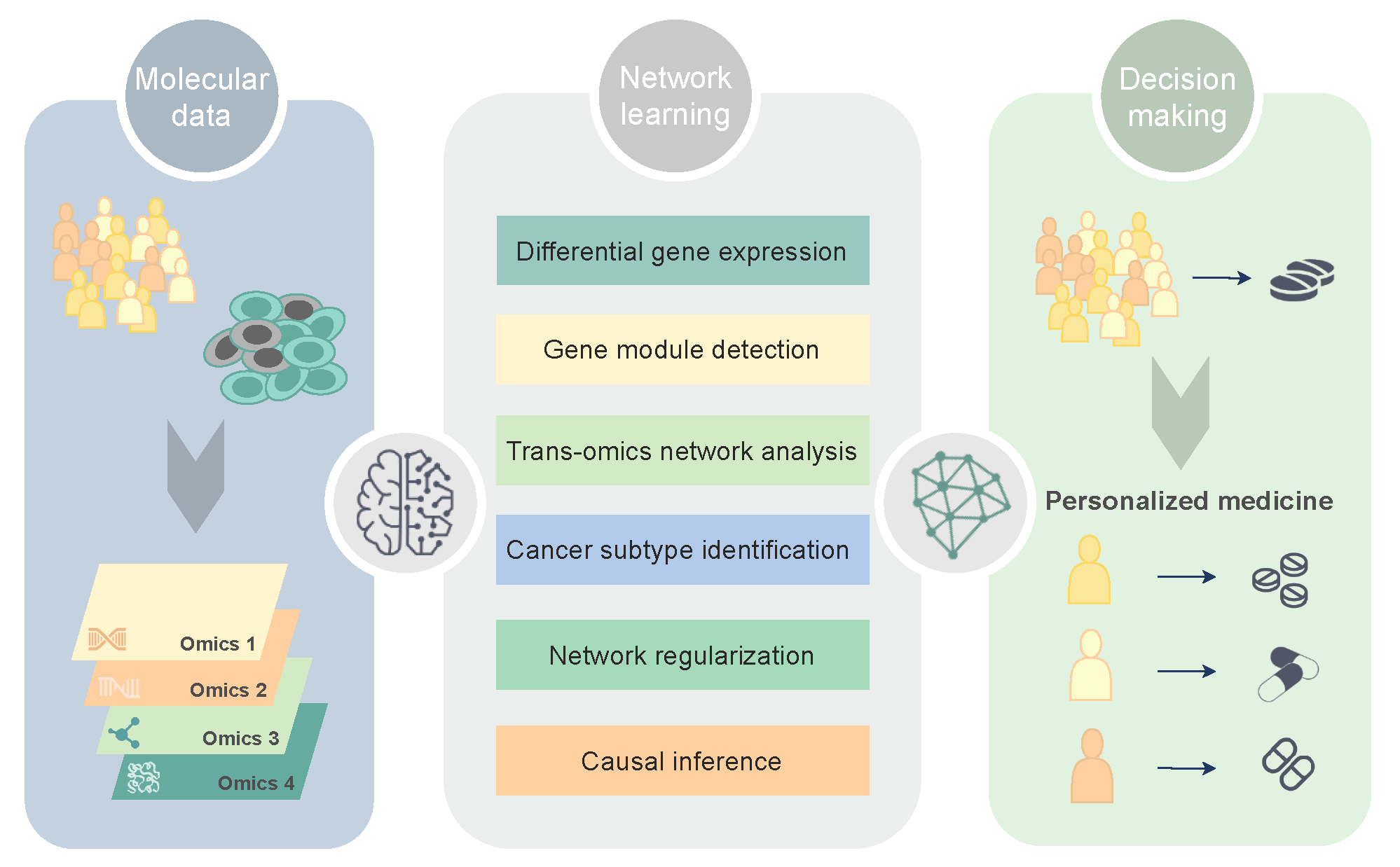
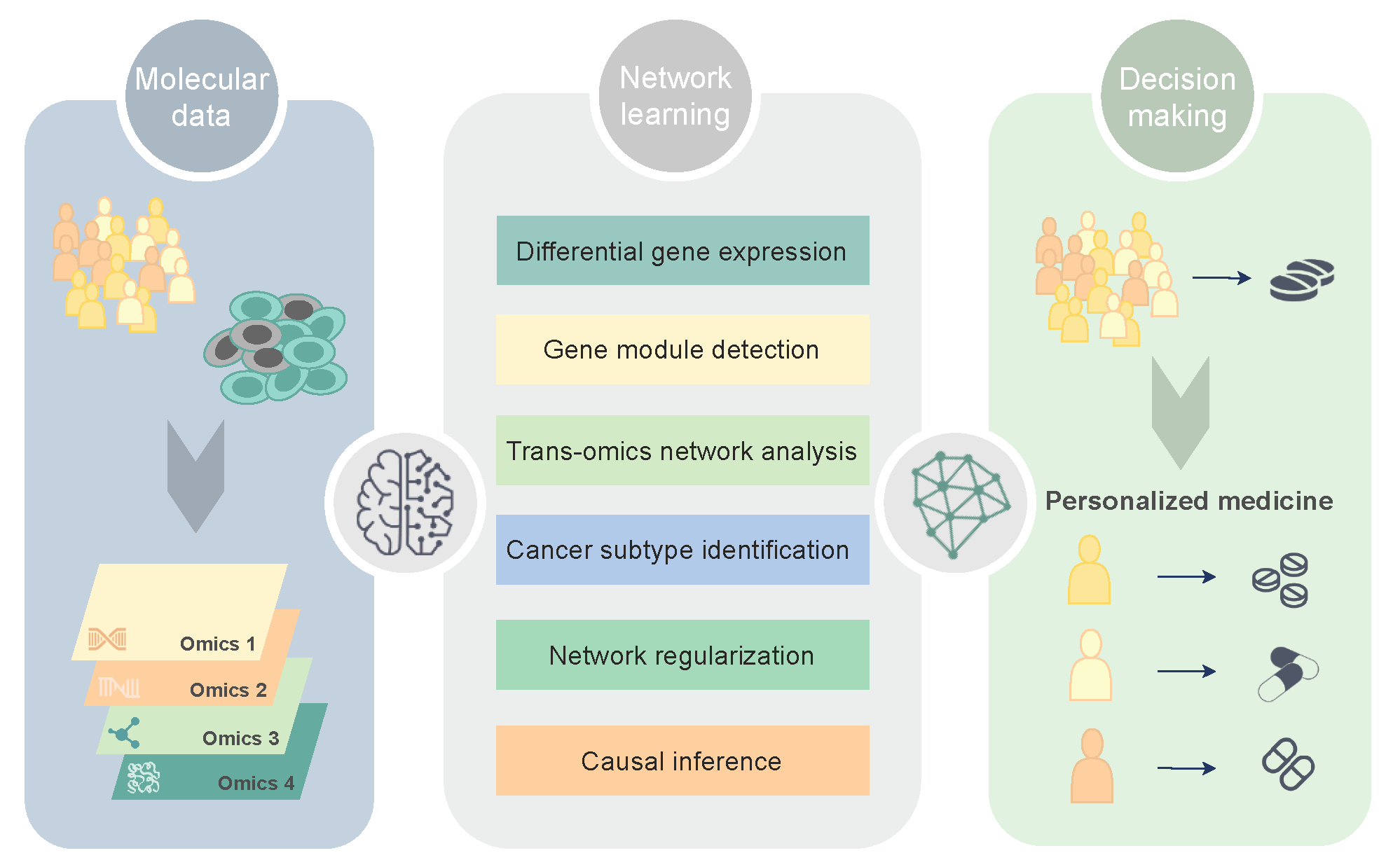
1. Molecular Networks in Precision Oncology
2. Network Discovery in Glioblastoma
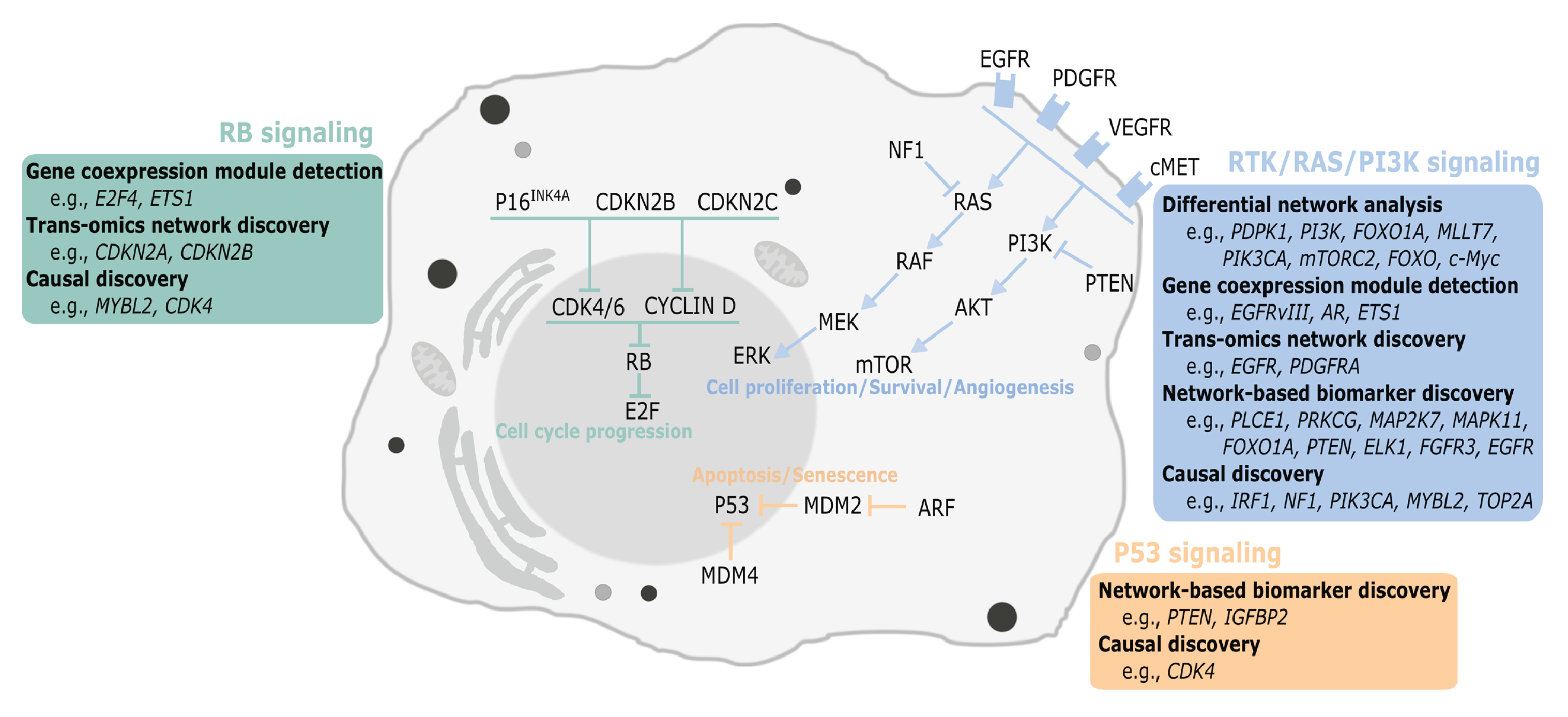
2.1. Differential Network Analysis
2.2. Gene Coexpression Module Detection
2.3. Trans-Omics Network Discovery
2.4. Network-Based Learning
2.4.1. Cancer Subtype Identification
2.4.2. Model-Based Biomarker Discovery
2.5. Causal Discovery
3. Major Challenges and Future Strategies
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Banjo | Bayesian network inference with Java objects |
| BN | Bayesian network |
| CE | Community extraction |
| CLR | Context-likelihood-relatedness |
| CNA | Copy number aberration |
| CSPRV | Cancer subtype prediction using |
| DAG | Directed acyclic graph |
| DEG | Differential expressed gene |
| DiME | Disease module extraction |
| DINGO | Differential network analysis in genomics |
| EMT | Epithelial-to-mesenchymal transition |
| EPoC | Endogenous perturbation analysis of cancer |
| GBM | Glioblastoma multiforme |
| GBM-BioDP | Glioblastoma Bio Discovery Portal |
| GEO | Gene Expression Omnibus |
| GMM | Gaussian mixture model |
| GO | Gene Ontology |
| GWAS | Genome-wide association study |
| HGG | High-grade glioma |
| IAMB | Incremental associated Markov blanket |
| IDA | Intervention calculus when the DAG is absent |
| iDINGO | Integrative differential network analysis in genomics |
| InTRINSiC | Integrative Modeling of Transcription Regulatory Interactions for |
| Systematic Inference of Susceptibility in Cancer | |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KINC | Knowledge Independent Network Construction |
| LGG | Low-grade glioma |
| TCGA | The Cancer Genome Atlas |
| CCLE | Cancer Cell Line Encyclopedia |
| CGGA | Chinese Glioma Genome Atlas |
| EN | Elastic net |
| lasso | Least absolute shrinkage selection operator |
| lncRNA | Long non-coding RNA |
| MI | Mutual information |
| MSLCRN | Module-specific lncRNA-mRNA causal regulatory networks |
| NGS | Next-generation sequencing |
| PDX | Patient-derived xenograft |
| RNA-seq | RNA sequencing |
| scRNA-seq | Single-cell RNA sequencing |
| SNF | Similarity Network Fusion |
| SNP | Single-nucleotide polymorphism |
| SYGNAL | Systems Genetics Network Analysis |
| TAM | Tumor-associated macrophage |
| TCI | Tumor-specific causal inference |
| TF | Transcriptional factor |
| TRN | Transcriptional regulatory network |
| twiner | Twin networks recovery |
| WGCNA | Weighted gene coexpression network analysis |
| WSNF | Weighted similarity network fusion |
References
- Verhaak, R.; Hoadley, K.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.; Miller, C.; Ding, L.; Golub, T.; Cancer Genome Atlas Research Network; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagogo-Jack, I.; Shaw, A. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.; Horswell, S. Intratumor Heterog. Branched Evol. Reveal. Multiregion Seq. N. Engl. J. Med. 2012, 367, 976. [Google Scholar]
- Cohen, R.; Havlin, S. Complex Networks: Structure, Robustness and Function; Cambridge University Press: Cambridge, UK, 2010. [Google Scholar]
- Inda, M.; Bonavia, R.; Seoane, J. Glioblastoma multiforme: A look inside its heterogeneous nature. Cancers 2014, 6, 226–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.; Touloumis, A.; Collins, V.; Marioni, J.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human gliobastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [Green Version]
- Thakkar, J.; Dolecek, T.; Horbinski, C.; Ostrom, Q.; Lightner, D.; Barnholtz-Sloan, S.; Villano, J. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.; Ohgaki, H.; Wiestler, O.; Kleihues, P.; Ellison, D. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.; van den Bent, M.; Weller, M.; Fisher, B.; Taphoorn, M.; Belanger, K.; Brandes, A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Tirosh, I.; Trombetta, J.; Shalek, A.; Gillespie, S.; Wakimoto, H.; Cahill, D.; Nahed, B.; Curry, W.; Martuza, R.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; Verhaak, R.G. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef] [Green Version]
- Gill, B.J.; Pisapiab, D.J.; Malone, H.R.; Goldstein, H.; Lei, L.; Sonabend, A.; Yun, J.; Samanamud, J.; Sims, J.S.; Banu, M.; et al. MRI-localized biopsies reveal subtype-specific differences in molecular and cellular composition at the margins of glioblastoma. Proc. Natl. Acad. Sci. USA 2014, 111, 1250–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silenc. Benefit Temozolomide Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, M.; Baladandayuthapani, V.; Do, K.A. DINGO: Differential network analysis in genomics. Bioinformatics 2015, 31, 3413–3420. [Google Scholar] [CrossRef] [Green Version]
- Velpula, K.K.; Bhasin, A.; Asuthkar, S.; Tsung, A.J. Combined Targeting of PDK1 and EGFR Triggers Regression of Glioblastoma by Reversing the Warburg Effect. Cancer Res. 2013, 73, 7277–7289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meir, E.G.V.; Hadjipanayis, C.G.; Norden, A.D.; Shu, H.K.; Wen, P.Y.; Olson, J.J. Exciting new advances in neuro-oncology: The avenue to a cure for malignant glioma. CA Cancer J. Clin. 2010, 60, 166–193. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Res. Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Mellinghof, I.K.; Wang, M.Y.; Vivanco, I.; Haas-Kogan, D.A.; Zhu, S.; Dia, E.Q.; Lu, K.V.; Yoshimoto, K.; Huang, J.H.; Chute, D.J.; et al. Molecular Determinants of the Response of Glioblastomas to EGFR Kinase Inhibitors. N. Engl. J. Med. 2005, 353, 2012–2024. [Google Scholar] [CrossRef] [Green Version]
- Wen, P.Y.; Lee, E.Q.; Reardon, D.A.; Ligon, K.L.; Yung, W.A. Current clinical development of PI3K pathway inhibitors in glioblastoma. Neuro-Oncology 2012, 14, 819–829. [Google Scholar] [CrossRef] [Green Version]
- Masui, K.; Tanaka, K.; Akhavan, D.; Babic, I.; Gini, B.; Matsutani, T.; Iwanami, A.; Liu, F.; Villa, G.R.; Gu, Y.; et al. mTOR Complex 2 Controls Glycolytic Metabolism in Glioblastoma through FoxO Acetylation and Upregulation of c-Myc. Cell Metab. 2013, 18, 726–739. [Google Scholar] [CrossRef] [Green Version]
- Tateishi, K.; Iafrate, A.J.; Ho, Q.; Curry, W.T.; Batchelor, T.T.; Flaherty, K.T.; Onozato, M.L.; Lelic, N.; Sundaram, S.; Cahill, D.P.; et al. Myc-driven glycolysis is a therapeutic target in glioblastoma. Clin. Cancer Res. 2016, 22, 4452–4465. [Google Scholar] [CrossRef] [Green Version]
- Ning, J.F.; Stanciu, M.; Humphrey, M.R.; Gorham, J.; Wakimoto, H.; Nishihara, R.; Lees, J.; Zou, L.; Martuza, R.L.; Wakimoto, H.; et al. Myc targeted CDK18 promotes ATR and homologous recombination to mediate PARP inhibitor resistance in glioblastoma. Nat. Commun. 2019, 10, 2910. [Google Scholar] [CrossRef] [PubMed]
- Moncini, S.; Salvi, A.; Zuccotti, P.; Viero, G.; Quattrone, A.; Barlati, S.; Petro, G.D.; Venturin, M.; Riva, P. The Role of miR-103 and miR-107 in Regulation of CDK5R1 Expression and in Cellular Migration. PLoS ONE 2011, 6, e20038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, L.H.; Delalle, I.; Caviness, V.S., Jr.; Chae, T.; Harlow, E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 1994, 371, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Catania, A.; Urban, S.; Yan, E.; Hao, C.; Barron, G.; Allalunis-Turner, J. Expression and localization of cyclin- dependent kinase 5 in apoptotic human glioma cells. Neuro-Oncology 2001, 3, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Tian, B.; Gearing, M.; Hunter, S.; Ye, K.; Mao, Z. Cdk5-mediated regulation of the PIKE-A-Akt pathway and glioblastoma cell invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 7570–7575. [Google Scholar] [CrossRef] [Green Version]
- Yushan, R.; Wenjie, C.; Suning, H.; Yiwu, D.; Tengfei, Z.; Madushi, W.M.; Feifei, L.; Changwen, Z.; Xin, W.; Roodrajeetsing, G.; et al. Insights into the clinical value of cyclin-dependent kinase 5 in glioma: A retrospective study. World J. Surg. Oncol. 2015, 13. [Google Scholar] [CrossRef] [Green Version]
- Dorand, R.D.; Nthale, J.; Myers, J.T.; Barkauskas, D.S.; Avril, S.; Chirieleison, S.M.; Pareek, T.K.; Abbott, D.W.; Stearns, D.S.; Letterio, J.J.; et al. Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science 2016, 353, 399–403. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Tucker-Burden, C.; Kaissi, E.; Newsam, A.; Duggireddy, H.; Chau, M.; Zhang, C.; Diwedi, B.; Rupji, M.; Seby, S.; et al. CDK5 Inhibition Resolves PKA/cAMP-Independent Activation of CREB1 Signaling in Glioma Stem Cells. Cell Rep. 2018, 23, 1651–1664. [Google Scholar] [CrossRef] [Green Version]
- Sang, Y.; Li, Y.; Zhang, Y.; Alvarez, A.A.; Yu, B.; Zhang, W.; Hu, B.; Cheng, S.Y.; Feng, H. CDK5-dependent phosphorylation and nuclear translocation of TRIM59 promotes macroH2A1 ubiquitination and tumorigenicity. Nat. Commun. 2019, 10, 4013. [Google Scholar] [CrossRef] [Green Version]
- Madhavan, S.; Zenklusen, J.; Kotliarov, Y.; Sahni, H.; Fine, H.; Buetow, K. Rembrandt: Helping personalized medicine become a reality through integrative translational research. Mol. Cancer Res. 2009, 7, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Yaschenko, E. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2007, 36, D13–D21. [Google Scholar] [CrossRef] [Green Version]
- Celiku, O.; Johnson, S.; Zhao, S.; Camphausen, K.; Shankavaram, U. Visualizing molecular profiles of glioblastoma with GBM-BioDP. PLoS ONE 2014, 9, e101239. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Garraway, L.A. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, K.; Wang, Q.; Li, G.; Zeng, F.; Zhang, Y.; Wu, F.; Chai, R.; Wang, Z.; Zhang, C.; et al. Chinese Glioma Genome Atlas (CGGA): A Comprehensive Resource with Functional Genomic Data for Chinese Glioma Paties. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Tennant, D.; Zhu, Z.; Heath, J.; Yao, X.; He, S. DiME: A Scalable Disease Module Identification Algorithm with Application to Glioma Progression. PLoS ONE 2014, 9, e86693. [Google Scholar] [CrossRef] [PubMed]
- Ficklin, S.; Dunwoodie, L.; Poehlman, W.; Watson, C.; Roche, K.; Feltus, F. Discovering Condition-Specific Gene Co-Expression Patterns Using Gaussian Mixture Models: A Cancer Case Study. Sci. Rep. 2017, 7, 8617. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Le, T.; Liu, L.; Wang, R.; Sun, B.; Li, J. Identifying Cancer Subtypes from miRNA-TFmRNA Regulatory Networks and Expression Data. PLoS ONE 2016, 11, e0152792. [Google Scholar]
- Guo, Y.; Qi, Y.; Li, Z.; Shang, X. Improvement of cancer subtype prediction by incorporating transcriptome expression data and heterogeneous biological networks. BMC Med. Gnomics 2018, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Veríssimo, A.; Vinga, S.; Carrasquinha, E.; Lopes, M. Network Centrality Metrics for Elastic-Net Regularized Models. Bioconductor R Package Version 3.11. 2018. Available online: https://www.bioconductor.org/packages/release/bioc/html/glmSparseNet.html (accessed on 1 February 2021).
- Verissimo, A.; Carrasquinha, E.; Lopes, M.; Oliveira, A.; Sagot, M.F.; Vinga, S. Sparse network-based regularization for the analysis of patientomics high-dimensional survival data. bioRxiv 2018. [Google Scholar] [CrossRef] [Green Version]
- Le, T.; Hoang, T.; Li, J.; Liu, L.; Hu, S. ParallelPC: An R package for efficient constraint based causal exploration. arXiv 2015, arXiv:1510.03042v1. [Google Scholar]
- Zhang, J.; Le, T.; Liu, L.; Li, J. Inferring and analyzing module-specific lncRNA–mRNA causal regulatory networks in human cancer. Briefings Bioinform. 2018, 20, 1403–1419. [Google Scholar] [CrossRef]
- Faith, J.; Hayete, B.; Thaden, J.; Mogno, I.; Wierzbowski, J.; Cottarel, G.; Kasif, S.; Collins, J.; Gardner, T. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007, 5, e8. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shi, N.; Regev, A.; He, S.; Hemann, M. Integrated regulatory models for inference of subtype-specific susceptibilities in glioblastoma. Mol. Syst. Biol. 2020, 16, e9506. [Google Scholar] [CrossRef]
- Scutari, M. Learning Bayesian Networks with the bnlearn R Package. J. Stat. Softw. 2010, 35, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Hartemink, A. Banjo: Bayesian Network Inference with Java Objects; Version 2.2.0; Duke University: Durham, NC, USA, 2005. [Google Scholar]
- Horvath, S.; Zhang, B.; Carlson, M.; Lu, K.; Zhu, S.; Felciano, R.; Laurance, M.; Zhao, W.; Qi, S.; Chen, Z.; et al. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc. Natl. Acad. Sci. USA 2006, 103, 17402–17407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Horvath, S. A General Framework for Weighted Gene Co-Expression Network Analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, C.; Anacker, J.; Gerngras, S.; Kühnel, S.; Said, H.M.; Patel, R.; Kämmerer, U.; Vordermark, D.; Roosen, K.; Vince, G.H. Expression analysis of the autosomal recessive primary microcephaly genes MCPH1 (microcephalin) and MCPH5 (ASPM, abnormal spindle-like, microcephaly associated) in human malignant gliomas. Oncol. Rep. 2008, 20, 301–308. [Google Scholar] [CrossRef] [Green Version]
- Bikeye, S.N.N.; Colin, C.; Marie, Y.; Vampouille, R.; Ravassard, P.; Rousseau, A.; Boisselier, B.; Idbaih, A.; Calvo, C.F.; Leuraud, P.; et al. ASPM-associated stem cell proliferation is involved in malignant progression of gliomas and constitutes an attractive therapeutic target. Cancer Cell Int. 2010, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Visnyei, K.; Onodera, H.; Damoiseaux, R.; Saigusa, K.; Petrosyan, S.; Vries, D.D.; Ferrari, D.; Saxe, J.; Panosyan, E.H.; Masterman-Smith, M.; et al. A molecular screening approach to identify and characterize inhibitors of glioblastoma stem cells. Mol. Cancer Ther. 2011, 10, 1818–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Huang, L.; Yang, Y.; Chen, S.; Sun, J.; Ma, C.; Xie, J.; Song, Y.; Yang, J. ASPM promotes glioblastoma growth by regulating G1 restriction point progression and Wnt-β-catenin signaling. Aging 2020, 12, 224–241. [Google Scholar] [CrossRef]
- Tso, C.L.; Freije, W.A.; Day, A.; Chen, Z.; Merriman, B.; Perlina, A.; Lee, Y.; Dia, E.Q.; Yoshimoto, K.; Mischel, P.S.; et al. Distinct transcription profiles of primary and secondary glioblastoma subgroups. Cancer Res. 2006, 66, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Marie, S.K.N.; Okamoto, O.K.; Uno, M.; Hasegawa, A.P.G.; Oba-Shinjo, S.M.; Cohen, T.; Camargo, A.A.; Kosoy, A.; Carlotti, C.G., Jr.; Toledo, S.; et al. Maternal embryonic leucine zipper kinase transcript abundance correlates with malignancy grade in human astrocytomas. Int. J. Cancer 2008, 122, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Rahane, C.S.; Kutzner, A.; Heese, K. A cancer tissue-specific FAM72 expression profile defines a novel glioblastoma multiform (GBM) gene-mutation signature. J. Neuro-Oncol. 2019, 144, 57–70. [Google Scholar] [CrossRef]
- Zou, Y.F.; Meng, L.B.; He, Z.K.; Hu, C.H.; Shan, M.J.; Wang, D.Y.; Yu, X. Screening and authentication of molecular markers in malignant glioblastoma based on gene expression profiles. Oncol. Lett. 2019, 18, 4593–4604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Zhang, J.; Liu, Z.; Zhao, P. Identification of biomarkers for the transition from low-grade glioma to secondary glioblastoma by an integrated bioinformatic analysis. Am. J. Transl. Res. 2020, 12, 1222–1238. [Google Scholar] [PubMed]
- Chen, T.; Liu, Y.; Chen, L.; Luo, J.; Zhang, C.; Shen, X. Identification of the potential biomarkers in patients with glioma: A weighted gene co-expression network analysis. Carcinogenesis 2020, 41, 743–750. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Xu, P.; Wang, B.; Luo, J.; Fu, R.; Huang, K.; Dai, L.; Lu, J.; Cao, G.; Peng, H.; et al. Identification of a Specific Gene Module for Predicting Prognosis in Glioblastoma Patients. Front. Oncol. 2019, 9, 812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Long, S.; Hu, J.; Wang, Z.; Geng, C.; Ou, S. Systematic identification of lncRNA-based prognostic biomarkers for glioblastoma. Aging 2019, 11, 9405–9423. [Google Scholar] [CrossRef]
- Liang, R.; Zhi, Y.; Zheng, G.; Zhang, B.; Zhu, H.; Wang, M. Analysis of long non-coding RNAs in glioblastoma for prognosis prediction using weighted gene co-expression network analysis, Cox regression, and L1-LASSO penalization. OncoTargets Ther. 2019, 12, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Yang, J.; Liu, J.; Yang, X.; Liao, J.; Yuan, F.; Xu, Y.; Chen, B.L.Q. Identification of glioblastoma gene prognosis modules based on weighted gene co-expression network analysis. BMC Med. Genom. 2018, 11, 96. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Pan, C.; Xu, C.; Sun, Y.; Geng, Y.; Kong, L.; Xiao, X.; Zhao, Z.; Zhou, W.; Huang, L.; et al. Identification of survival-associated key genes and long non-coding RNAs in glioblastoma multiforme by weighted gene co-expression network analysis. Int. J. Mol. Med. 2018, 43, 1709–1722. [Google Scholar] [CrossRef] [PubMed]
- Upton, A.; Arvanitis, T. Using Evolutional Properties of Gene Networks in Understanding Survival Prognosis of Glioblastoma. IEEE J. Biomed. Health Inform. 2014, 18, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Ivliev, A.; ’t Hoen, P.; Sergeeva, M. Coexpression Network Analysis Identifies Transcriptional Modules Related to Proastrocytic Differentiation and Sprouty Signaling in Glioma. Cancer Res. 2010, 70, 10060–10070. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Levina, E.; Zhu, J. Community extraction for social networks. Proc. Natl. Acad. Sci. USA 2011, 108, 7321–7326. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Li, Z.; Wang, M. Expression and prognostic role of E2F transcription factors in high-grade glioma. CNS Neurosci. Ther. 2020, 26, 741–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Jiang, Y.; Wei, W.; Cong, P.; Ding, Y.; Xiang, L.; Wu, K. Androgen receptor signaling regulates growth of glioblastoma multiforme in men. Tumor Biol. 2015, 36, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Fang, D.; Xu, H.; Wang, Q.; Xia, H. The androgen receptor expression and association with patient’s survival in different cancers. Genomics 2020, 112, 1926–1940. [Google Scholar] [CrossRef] [PubMed]
- Werner, C.K.; Nna, U.J.; Sun, H.; Wilder-Romans, K.; Dresser, J.; Kothari, A.U.; Zhou, W.; Yao, Y.; Rao, A.; Stallard, S.; et al. Expression of the Androgen Receptor Governs Radiation Resistance in a Subset of Glioblastomas Vulnerable to Antiandrogen Therapy. Mol. Cancer Ther. 2020, 19, 2163–2174. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.F.; Huang, X.F.; Chang, J.T.; Huang, Y.C.; Weng, J.C.; Tsai, N.M. Cedrol suppresses glioblastoma progression by triggering DNA damage and blocking nuclear translocation of the androgen receptor. Cancer Lett. 2020, 495, 180–190. [Google Scholar] [CrossRef]
- Orozco, M.; Valdez, R.; Ramos, L.; Cabeza, M.; Segovia, J.; Romano, M. Dutasteride combined with androgen receptor antagonists inhibit glioblastoma U87 cell metabolism, proliferation, and invasion capacity: Androgen regulation. Steroids 2020, 164, 108733. [Google Scholar] [CrossRef]
- Chen, T.C.; Chuang, J.Y.; Ko, C.Y.; Kao, T.J.; Yang, P.Y.; Yu, C.H.; Liu, M.S.; Hu, S.L.; Tsai, Y.T.; Chan, H.; et al. AR ubiquitination induced by the curcumin analog suppresses growth of temozolomide-resistant glioblastoma through disrupting GPX4-Mediated redox homeostasis. Redox Biol. 2020, 30, 101413. [Google Scholar] [CrossRef]
- Nakada, M.; Yamashita, J.; Okada, Y.; Sato, H. Ets-1 positively regulates expression of Urokinase-type Plasminogen Activator (uPA) and invasiveness of astrocytic tumors. J. Neuropathol. Exp. Neurol. 1999, 58, 329–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valter, M.M.; Hügel, A.; Huang, H.J.; Cavenee, W.K.; Wiestler, O.D.; Pietsch, T.; Wernert, N. Expression of the Ets-1 transcription factor in human astrocytomas is associated with fms-like tyrosine kinase-1 (Flt-1)/vascular endothelial growth factor receptor-1 synthesis and neoangiogenesis. Cancer Res. 1999, 59, 5608–5614. [Google Scholar]
- Shukla, A.A.; Jain, M.; Chauhan, S.S. Ets-1/Elk-1 is a critical mediator of dipeptidyl-peptidase III transcription in human glioblastoma cells. FEBS J. 2010, 277, 1861–1875. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, Q.L.; Sun, W.; Chandrasekharan, P.; Cheng, H.S.; Ying, Z.; Lakshmanan, M.; Raju, A.; Tenen, D.G.; Cheng, S.Y.; et al. Non-canonical NF-kB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat. Cell Biol. 2015, 17, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.J.; Rube, H.T.; Xavier-Magalhães, A.; Costa, B.M.; Mancini, A.; Song, J.S.; Costello, J.F. Understanding TERT Promoter Mutations: A Common Path to Immortality. Mol. Cancer Res. 2016, 14, 315–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancini, A.; Xavier-Magalhães, A.; Woods, W.S.; Nguyen, K.T.; Amen, A.M.; Hayes, J.L.; Fellmann, C.; Gapinske, M.; McKinney, A.M.; Hong, C.; et al. Disruption of the β1L Isoform of GABP Reverses Glioblastoma Replicative Immortality in a TERT Promoter Mutation-Dependent Manner. Cancer Cell 2018, 34, 513–528. [Google Scholar] [CrossRef] [Green Version]
- Dunwoodie, L.; Poehlman, W.; Ficklin, S.; Feltus, F. Discovery and validation of a glioblastoma co-expressed gene module. Oncotarget 2018, 9, 10995–11008. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ning, W.; Qiu, Z.; Li, S.; Zhang, H.; Yu, C. Tumor-associated macrophages based signaling pathway analysis and hub genes identification in glioma. Medicine 2020, 99, e23840. [Google Scholar] [CrossRef] [PubMed]
- Class, C.; Ha, M.; Baladandayuthapani, V.; Do, K.A. iDINGO—Integrative differential network analysis in genomics with Shiny application. Bioinformatics 2015, 34, 1243–1245. [Google Scholar] [CrossRef] [Green Version]
- Jörnsten, R.; Abenius, T.; Kling, T.; Schmidt, L.; Johansson, E.; Nordling, T.; Nordlander, B.; Sander, C.; Gennemark, P.; Funa, K.; et al. Network modeling of the transcriptional effects of copy number aberrations in glioblastoma. Mol. Syst. Biol. 2011, 7, 486. [Google Scholar] [CrossRef]
- Hansen, L.J.; Sun, R.; Yang, R.; Singh, S.X.; Chen, L.H.; Pirozzi, C.J.; Moure, C.J.; Hemphill, C.; Carpenter, A.B.; Healy, P.; et al. MTAP loss promotes stemness in glioblastoma and confers unique susceptibility to purine starvation. Cancer Res. 2019, 79, 3383–3394. [Google Scholar] [CrossRef]
- De Menezes, W.; Silva, V.A.O.; Gomes, I.N.F.; Rosa, M.N.; Spina, M.L.C.; Carloni, A.C.; Alves, A.L.V.; Melendez, M.; Almeida, G.C.; da Silva, L.S.; et al. Loss of 5’-Methylthioadenosine Phosphorylase (MTAP) is Frequent in High-Grade Gliomas; Nevertheless, it is Not Associated with Higher Tumor Aggressiveness. Cells 2020, 9, 492. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Liu, J.; Liao, Y.; Jin, C.; Zhang, Z.; Zhao, J.; Liu, K.; Huang, H.; Cao, H.; Cheng, Q. Identification of SEC61G as a novel prognostic marker for predicting survival and response to therapies in patients with glioblastoma. Med. Sci. Monit. 2019, 25, 3624–3635. [Google Scholar] [CrossRef]
- Korkolopoulou, P.; Levidou, G.; El-Habr, E.A.; Adamopoulos, C.; Fragkou, P.; Boviatsis, E.; Themistocleous, M.S.; Petraki, K.; Vrettakos, G.; Sakalidou, M.; et al. Sox11 expression in astrocytic gliomas: Correlation with nestin/c-Met/IDH1-R132H expression phenotypes, p-Stat-3 and survival. Br. J. Cancer 2013, 108, 2142–2152. [Google Scholar] [CrossRef] [Green Version]
- Camacho-Urkaray, E.; Santos-Juanes, J.; Gutiérrez-Corres, F.B.; García, B.; Quirós, L.M.; Guerra-Merino, I.; Aguirre, J.J.; Fernández-Vega, I. Establishing cut-off points with clinical relevance for bcl-2, cyclin D1, p16, p21, p27, p53, Sox11 and WT1 expression in glioblastoma—A short report. Cell. Oncol. 2018, 41, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, H.; Lv, S.; Yang, H. High CD133 Expression Is Associated with Worse Prognosis in Patients with Glioblastoma. Mol. Neurobiol. 2016, 53, 2354–2360. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Ergun, A.; Shukla, S.; Campos, B.; Hanna, J.; Ghosh, P.; Quayle, S.; Rai, K.; Colla, S.; Ying, H.; et al. microRNA regulatory network inference identifies miR-34a as a novel regulator of TGF-beta signaling in glioblastoma. Cancer Discov. 2012, 2, 736–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silber, J.; Jacobsen, A.; Ozawa, T.; Harinath, G.; Pedraza, A.; Sander, C.; Holland, E.; Huse, J. miR-34a Repression in Proneural Malignant Gliomas Upregulates Expression of Its Target PDGFRA and Promotes Tumorigenesis. PLoS ONE 2012, 7, e33844. [Google Scholar] [CrossRef]
- Xiong, Y.; Wang, Q. STC1 regulates glioblastoma migration and invasion via the TGF-β/SMAD4 signaling pathway. Mol. Med. Rep. 2019, 20, 3055–3064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Mezlini, A.; Demir, F.; Fiume, M.; Tu, Z.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 2014, 11, 333. [Google Scholar] [CrossRef]
- Smilde, A.; Kiers, H.; Bijlsma, S.; Rubingh, C.; van Erk, M. Matrix correlations for high-dimensional data: The modified RV-coefficient. Bioinformatics 2009, 25, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Vinga, S. Structured sparsity regularization for analyzing high-dimensional omics data. Briefings Bioinform. 2021, 22, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B 2005, 67, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Tibshirani, R.; Saunders, M.; Rosset, S.; Zhu, J.; Knight, K. Sparsity and smoothness via the fused lasso. J. R. Stat. Soc. Ser. B 2005, 67, 91–108. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Hastie, T. The adaptive lasso and its oracle properties. J. Am. Stat. Assoc. 2006, 101, 1418–1429. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Lin, Y. Model selection and estimation in regression with grouped variables. J. R. Stat. Soc. Ser. B 2006, 68, 49–67. [Google Scholar] [CrossRef]
- Li, C.; Li, H. Network-constrained regularization and variable selection for analysis of genomic data. Bioinformatics 2008, 24, 1175–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradisis, S.; Ruvkun, G. Caenorhabditis elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3 kinase to the DAF-16 transcription factor. Genes Dev. 1998, 12, 2488–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, A Putative Protein Tyrosine Phosphatase Gene Mutated Hum. Brain, Breast, Prostate Cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef]
- Carracedo, A.; Pandolfi, P. The PTEN-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [Green Version]
- Swartling, F.; Ferletta, M.; Kastemar, M.; Weiss, W.; Westermark, B. Cyclic GMP-dependent protein kinase II inhibits cell proliferation, Sox9 expression and Akt phosphorylation in human glioma cell lines. Oncogene 2009, 28, 3121–3131. [Google Scholar] [CrossRef] [Green Version]
- Dhanasekaran, D.; Kashef, K.; Lee, C.; Xu, H.; Reddy, E. Scaffold proteins of MAP-kinase modules. Oncogene 2007, 26, 3185–3202. [Google Scholar] [CrossRef] [Green Version]
- Demuth, T.; Reavie, L.B.; Rennert, J.L.; Nakada, M.; Nakada, S.; Hoelzinger, D.B.; Beaudry, C.E.; Henrichs, A.N.; Anderson, E.M.; Berens, M.E. MAP-ing glioma invasion: Mitogen-activated protein kinase kinase 3 and p38 drive glioma invasion and progression and predict patient survival. Mol. Cancer Ther. 2007, 6, 1212–1222. [Google Scholar] [CrossRef] [Green Version]
- Wagner, E.F.; Nebreda, Á.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Uht, R.; Amos, S.; Martin, P.; Riggan, A.; Hussaini, I. The protein kinase Cη- isoform induces proliferation in glioblastoma cell lines through an ERK/Elk-1 pathway. Oncogene 2007, 26, 2885–2893. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Wei, P.; Gong, A.; Chiu, W.T.; Lee, H.T.; Colman, H.; Huang, H.; Xue, J.; Liu, M.; Wang, Y.; et al. FoxM1 Promotes β-Catenin Nuclear Localization and Controls Wnt Target-Gene Expression and Glioma Tumorigenesis. Cancer Cell 2011, 18, 427–442. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Chen, Y.; Wu, Y.; Wang, Z.; Zhou, A.; Zhang, S.; Lin, K.; Aldape, K.; Majumder, S.; Lu, Z.; et al. Tumour suppressor TRIM33 targets nuclear β-catenin degradation. Nat. Commun. 2015, 6, 6156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fecci, P.E.; Ochiai, H.; Mitchell, D.A.; Grossi, P.M.; Sweeney, A.E.; Archer, G.E.; Cummings, T.; Allison, J.P.; Bigner, D.D.; Sampson, J.H. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4 + T cell compartment without affecting regulatory T-cell function. Clin. Cancer Res. 2007, 13, 2158–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebner, S.; Fischmann, A.; Rascher, G.; Duffner, F.; Grote, E.H.; Kalbacher, H.; Wolburg, H. Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol. 2000, 100, 323–331. [Google Scholar] [CrossRef]
- Wolburg, H.; Wolburg-Buchholz, K.; Kraus, J.; Rascher-Eggstein, G.; Liebner, S.; Hamm, S.; Duffner, F.; Grote, E.H.; Risau, W.; Engelhardt, B. Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol. 2003, 105, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.B.; Veríssimo, A.; Carrasquinha, E.; Vinga, S. On the Role of Hub and Orphan Genes in the Diagnosis of Breast Invasive Carcinoma. In Machine Learning, Optimization, and Data Science; Nicosia, G., Pardalos, P., Umeton, R., Giuffrida, G., Sciacca, V., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 631–642. [Google Scholar]
- Carrasquinha, E.; Veríssimo, A.; Lopes, M.B.; Vinga, S. Network-Based Variable Selection for Survival Outcomes in Oncological Data. In Bioinformatics and Biomedical Engineering; Rojas, I., Valenzuela, O., Rojas, F., Herrera, L.J., Ortuño, F., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 550–561. [Google Scholar]
- Veríssimo, A.; Oliveira, A.; Sagot, M.F.; Vinga, S. DegreeCox—A network-based regularization method for survival analysis. J. R. Stat. Soc. Ser. B 2016, 77, 449. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, M.; Vinga, S. Tracking intratumoral heterogeneity in glioblastoma via regularized classification of single-cell RNA-Seq data. BMC Bioinform. 2020, 21, 59. [Google Scholar] [CrossRef] [Green Version]
- Lopes, M.; Casimiro, S.; Vinga, S. Twiner: Correlation-based regularization for identifying common cancer gene signatures. BMC Bioinform. 2019, 20, 356. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, C.; Martins, M.L.M.; Costa, L.; Vinga, S. TCox: Correlation-Based Regularization Applied to Colorectal Cancer Survival Data. Biomedicines 2020, 8, 488. [Google Scholar] [CrossRef]
- Darmanis, S.; Sloan, S.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [Green Version]
- Nørøxe, D.; Poulsen, H.; Lasses, U. Hallmarks of glioblastoma: A systematic review. ESMO Open 2016, 1, e000144. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.; Verhaak, R.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.; Zheng, S.; Chakravarty, D.; Sanborn, J.; Berman, S.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Y.; Yao, X.; Ping, Y.; Jiang, T.; Liu, Q.; Xu, S.; Huang, J.; Mou, H.; Gong, W.; et al. Annexin 1 released by necrotic Human glioblastoma cells stimulates tumor cell growth through the formyl peptide receptor 1. Am. J. Pathol. 2011, 179, 1504–1512. [Google Scholar] [CrossRef]
- Svenningsen, A.; Löring, S.; Sørensen, A.; Huynh, H.; Hjæresen, S.; Martin, N.; Moeller, J.; Elkjær, M.; Holmskov, U.; Illes, Z.; et al. Macrophage migration inhibitory factor (MIF) modulates trophic signaling through interaction with serine protease HTRA1. Cell. Mol. Life Sci. 2017, 74, 4561–4572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Vellanki, R.; Coyaud, E.; Ignatchenko, V.; Li, L.; Krieger, J.; Taylor, P.; Tong, J.; Pham, N.A.; Liu, G.; et al. CHCHD2 is coamplified with EGFR in NSCLC and regulates mitochondrial function and cell migration. Mol. Cancer Res. 2015, 13, 1119–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, N.; Gibaud, A.; Almeida, A.; Ourliac-Garnier, I.; Debatisse, M.; Malfoy, B. Relationships linking amplification level to gene over-expression in gliomas. PLoS ONE 2010, 5, e14249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; He, S.; Yuan, J.; Mao, X.; Cao, Y.; Zong, J.; Tu, Y.; Zhang, Y. Oncogenic role of SOX9 expression in human malignant glioma. Med. Oncol. 2012, 29, 3484–3490. [Google Scholar] [CrossRef]
- Jiang, J.; Zhou, J.; Luo, P.; Gao, H.; Ma, Y.; Chen, Y.S.; Li, L.; Zou, D.; Zhang, Y.; Jing, Z. Prosaposin promotes the proliferation and tumorigenesis in glioma through toll-like receptor 4 (TLR4)-mediated NF-κB signaling pathway. EBioMedicine 2018, 37, 78–90. [Google Scholar] [CrossRef] [Green Version]
- Gont, A.; Daneshmand, M.; Woulfe, J.; Lorimer, I. PREX1 integrates G protein-coupled receptor and phosphoinositide 3-kinase signaling to promote glioblastoma invasion. Eur. J. Cancer 2016, 61, S171–S172. [Google Scholar] [CrossRef] [Green Version]
- Golan-Gerstl, R.; Cohen, M.; Shilo, A.; Suh, S.S.; Bakàcs, A.; Coppola, L.; Karni, R. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res. 2011, 71, 4464–4472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.; Abdelmohsen, K.; Gorospe, M. Posttranscriptional gene regulation by long noncoding RNA. J. Mol. Biol. 2013, 425, 3723–3730. [Google Scholar] [CrossRef] [Green Version]
- Maathuis, H.; Kalisch, M.; Bühlmann, P. Estimating highdimensional intervention effects from observational data. Ann. Stat. 2009, 37, 3133–3164. [Google Scholar] [CrossRef]
- Le, T.; Hoang, T.; Li, J.; Liu, L.; Liu, H.; Hu, S. A fast PC algorithm for high dimensional causal discovery with multi-core PCs. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016, 16, 1483–1495. [Google Scholar] [CrossRef] [Green Version]
- Spirtes, P.; Glymour, C.; Scheines, R. Causation, Prediction, and Search, 2nd ed.; MIT Press: Cambridge, UK, 2000. [Google Scholar]
- Pearl, J. Causality: Models, Reasoning, and Inference; Cambridge University Press: New York, NY, USA, 2000. [Google Scholar]
- Du, Z.; Fei, T.; Verhaak, R.; Su, Z.; Zhang, Y.; Brown, M.; Chen, Y.; Liu, X. Integrative genomic analyses reveal clinically relevant long noncoding RNAs in human cancer. Nat. Struct. Mol. Biol. 2013, 20, 908–913. [Google Scholar] [CrossRef] [Green Version]
- Plaisier, C.; O’Brien, S.; Bernard, B.; Reynolds, S.; Simon, Z.; Toledo, C.; Ding, Y.; Reiss, D.; Paddison, P.; Baliga, N. Causal Mechanistic Regulatory Network for Glioblastoma Deciphered Using Systems Genetics Network Analysis. Cell Syst. 2016, 3, 172–186. [Google Scholar] [CrossRef] [Green Version]
- Musa, J.; Aynaud, M.M.; Mirabeau, O.; Delattre, O.; Grünewald, T.G. MYBL2 (B-Myb): A central regulator of cell proliferation, cell survival and differentiation involved in tumorigenesis. Cell Death Dis. 2017, 8, e2895. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, J.; Bland, C.; KlinkeII, D. Identifying causal networks linking cancer processes and antitumor immunity using Bayesian network inference and metagene constructs. Biotechnol. Prog. 2016, 32, 470–479. [Google Scholar] [CrossRef] [Green Version]
- Tsamardinos, I.; Aliferis, C.; Statnikov, A. Algorithms for large scale Markov Blanket discovery. In Proceedings of the The 16th International FLAIRS Conference, St. Augustine, FL, USA, 12–14 May 2003; pp. 376–380. [Google Scholar]
- Kunkle, B.; Yoo, C.; Roy, D. Reverse Engineering of Modified Genes by Bayesian Network Analysis Defines Molecular Determinants Critical to the Development of Glioblastoma. PLoS ONE 2013, 8, e64140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odreman, F.; Vindigni, M.; Gonzales, M.L.; Niccolini, B.; Candiano, G.; Zanotti, B.; Skrap, M.; Pizzolitto, S.; Stanta, G.; Vindigni, A. Proteomic studies on low- and high-grade human brain astrocytomas. J. Proteome Res. 2005, 4, 698–708. [Google Scholar] [CrossRef]
- Jung, T.Y.; Choi, Y.D.; Kim, Y.H.; Lee, J.J.; Kim, H.S.; Kim, J.S.; Kim, S.K.; Jung, S.; Cho, D. Immunological characterization of glioblastoma cells for immunotherapy. Anticancer Res. 2013, 33, 2525–2534. [Google Scholar] [PubMed]
- Cardoso, L.C.; da S. Soares, R.; de S. Laurentino, T.; Lerario, A.M.; Marie, S.K.N.; Oba-Shinjo, S.M. CD99 expression in glioblastoma molecular subtypes and role in migration and invasion. Int. J. Mol. Sci. 2019, 20, 1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holden, J.A.; Townsend, J.J. DNA topoisomerase II-alpha as a proliferation marker in astrocytic neoplasms of the central nervous system: Correlation with MIB1 expression and patient survival. Mod. Pathol. 1999, 12, 1094–1100. [Google Scholar] [PubMed]
- Kosti, A.; de Araujo, P.R.; Li, W.Q.; Guardia, G.D.A.; Chiou, J.; Yi, C.; Ray, D.; Meliso, F.; Li, Y.M.; Delambre, T.; et al. The RNA-binding protein SERBP1 functions as a novel oncogenic factor in glioblastoma by bridging cancer metabolism and epigenetic regulation. Genome Biol. 2020, 21, 195. [Google Scholar] [CrossRef]
- Cai, C.; Cooper, G.; Lu, K.; Ma, X.; Xu, S.; Zhao, Z.; Chen, X.; Xue, Y.; Lee, A.; Clark, N.; et al. Systematic discovery of the functional impact of somatic genome alterations in individual tumors through tumor-specific causal inference. PLoS Comput. Biol. 2019, 15, e1007088. [Google Scholar] [CrossRef] [Green Version]
- Howell, A.; Zheng, J.; Haycock, P.; McAleenan, A.; Relton, C.; Martin, R.; Kurian, K. Use of Mendelian Randomization for Identifying Risk Factors for Brain Tumors. Front. Genet. 2018, 9, 525. [Google Scholar] [CrossRef]
- Howell, A.E.; Robinson, J.W.; Wootton, R.E.; McAleenan, A.; Tsavachidis, S.; Ostrom, Q.T.; Kurian, K.M. Testing for causality between systematically identified risk factors and glioma: A Mendelian randomization study. BMC Cancer 2020, 508, 1471–2407. [Google Scholar] [CrossRef]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef]
- Linkous, A.; Balamatsias, D.; Snuderl, M.; Edwards, L.; Miyaguchi, K.; Milner, T.; Reich, B.; Cohen-Gould, L.; Storaska, A.; Nakayama, Y.; et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 2019, 26, 3203–3211.e5. [Google Scholar] [CrossRef] [Green Version]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204.e22. [Google Scholar] [CrossRef]
- Yuki, K.; Cheng, N.; Nakano, M.; Kuo, C.J. Organoid Models of Tumor Immunology. Trends Immunol. 2020, 41, 652–664. [Google Scholar] [CrossRef]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef] [Green Version]
- Siolas, D.; Hannon, G.J. Patient Derived Tumor Xenografts: Transforming clinical samples into mouse models. Cancer Res. 2013, 73, 5315–5319. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient Derived Xenograft Models: An Emerging Platform for Translational Cancer Research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [Green Version]
- Vaubel, R.A.; Tian, S.; Remonde, D.; Schroeder, M.A.; Mladek, A.C.; Kitange, G.J.; Caron, A.; Kollmeyer, T.M.; Grove, R.; Peng, S.; et al. Genomic and phenotypic characterization of a broad panel of patient-derived xenografts reflects the diversity of glioblastoma. Clin. Cancer Res. 2020, 26, 1094–1104. [Google Scholar] [CrossRef]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef]
- Zhao, S.G.; Yu, M.; Spratt, D.E.; Chang, S.L.; Feng, F.Y.; Kim, M.M.; Speers, C.W.; Carlson, B.L.; Mladek, A.C.; Lawrence, T.S.; et al. Xenograft-based, platform-independent gene signatures to predict response to alkylating chemotherapy, radiation, and combination therapy for glioblastoma. Neuro-Oncology 2019, 21, 1141–1149. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Circulating tumour cells in cancer patients: Challenges and perspectives. Trends Mol. Med. 2010, 16, 398–406. [Google Scholar] [CrossRef]
- Kilgour, E.; Rothwell, D.G.; Brady, G.; Dive, C. Liquid Biopsy-Based Biomarkers of Treatment Response and Resistance. Cancer Cell 2020, 37, 485–495. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Liquid biopsy and minimal residual disease—Latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef]
- Best, M.G.; Sol, N.; Zijl, S.; Reijneveld, J.C.; Wesseling, P.; Wurdinger, T. Liquid biopsies in patients with diffuse glioma. Acta Neuropathol. 2015, 129, 849–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.M.; Shah, R.H.; Pentsova, E.I.; Pourmaleki, M.; Briggs, S.; Distefano, N.; Zheng, Y.; Skakodub, A.; Mehta, S.A.; Campos, C.; et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 2019, 565, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.; Lee, I. Single-cell network biology for resolving cellular heterogeneity in human diseases. Exp. Mol. Med. 2020, 52, 1798–1808. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopes, M.B.; Martins, E.P.; Vinga, S.; Costa, B.M. The Role of Network Science in Glioblastoma. Cancers 2021, 13, 1045. https://doi.org/10.3390/cancers13051045
Lopes MB, Martins EP, Vinga S, Costa BM. The Role of Network Science in Glioblastoma. Cancers. 2021; 13(5):1045. https://doi.org/10.3390/cancers13051045
Chicago/Turabian StyleLopes, Marta B., Eduarda P. Martins, Susana Vinga, and Bruno M. Costa. 2021. "The Role of Network Science in Glioblastoma" Cancers 13, no. 5: 1045. https://doi.org/10.3390/cancers13051045
APA StyleLopes, M. B., Martins, E. P., Vinga, S., & Costa, B. M. (2021). The Role of Network Science in Glioblastoma. Cancers, 13(5), 1045. https://doi.org/10.3390/cancers13051045