
CD28 Co-Stimulus Achieves Superior CAR T Cell Effector Function against Solid Tumors Than 4-1BB Co-Stimulus

 ,
,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. CAR Construct Generation
2.3. Gamma-Retroviral and Lentiviral Vector Production, Transduction and CAR T Cell Generation
2.4. Flow Cytometry
2.5. Bioluminescence-Based Cytotoxicity and Cytokine Release Assays
2.6. In Vivo Studies
2.7. Tumor-Infiltrating T Cell (TIL) Isolation
2.8. Statistical Analysis
3. Results
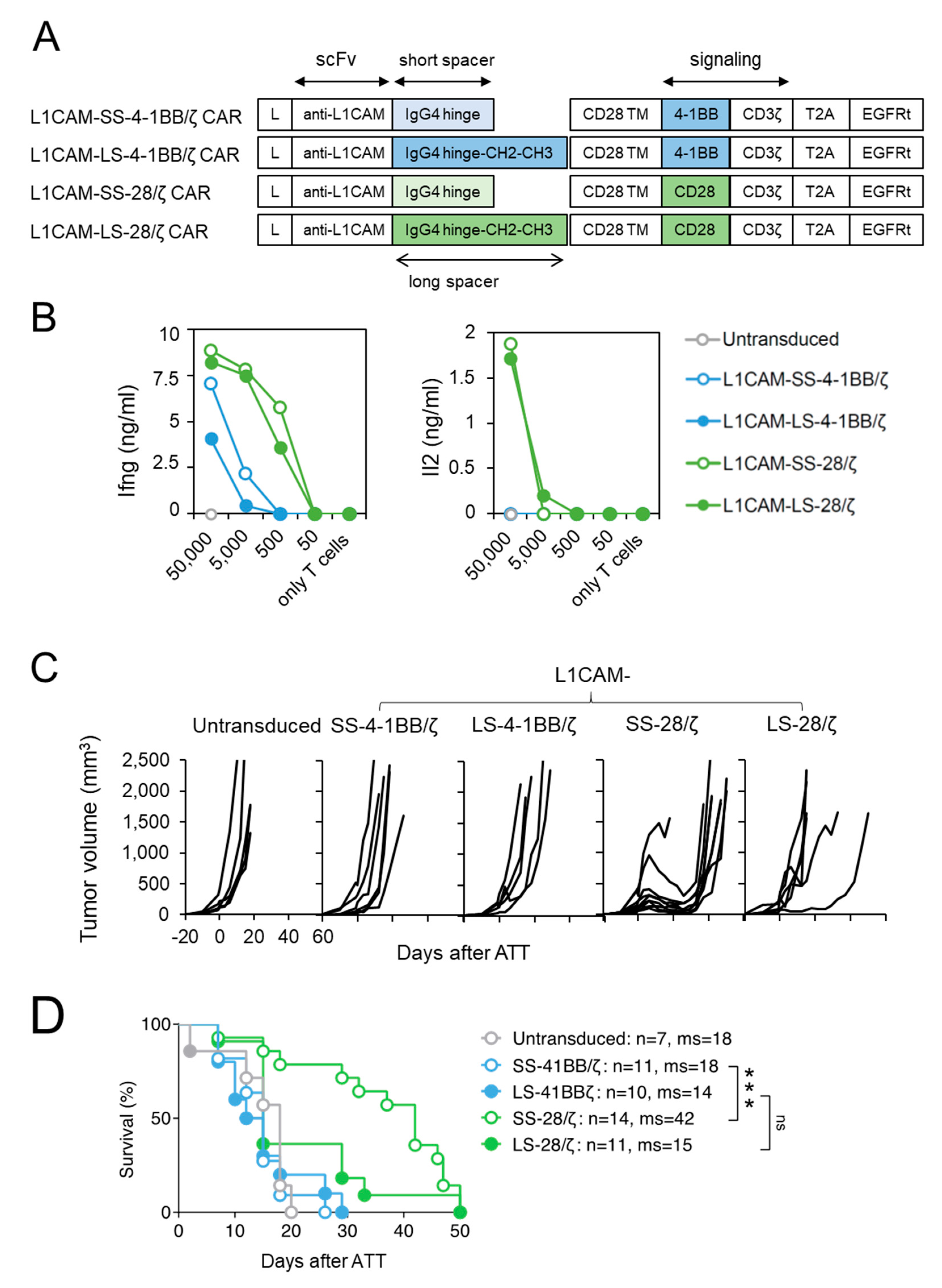
3.1. Short Spacer L1CAM-Specific CAR T Cells with CD28 Signaling Prolonged Survival of SK-N-BE(2) Tumor-Bearing Mice
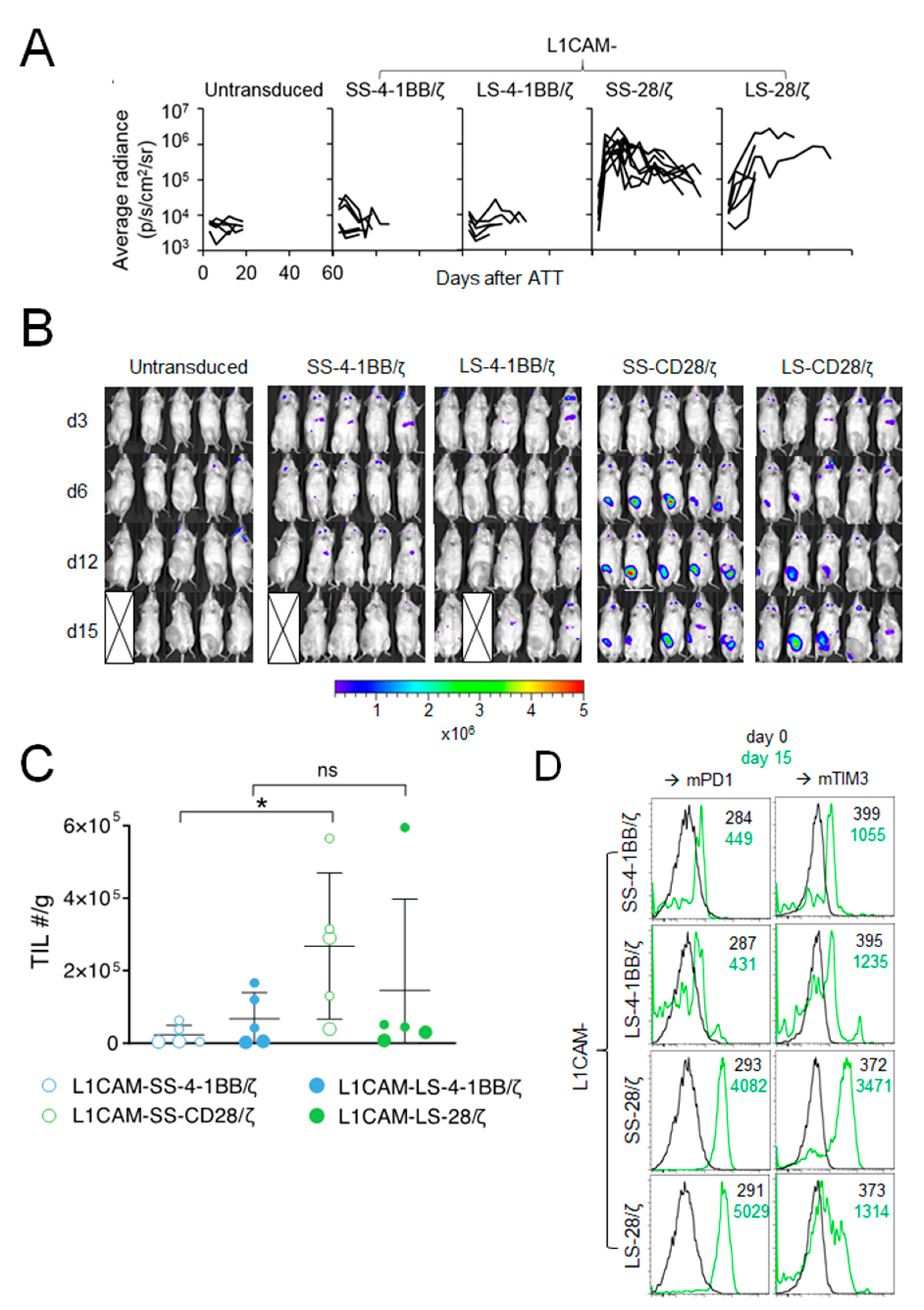
3.2. Anti-Tumor Effect of L1CAM-Specific CAR T Cells Correlates with T Cell Expansion at the Tumor Site
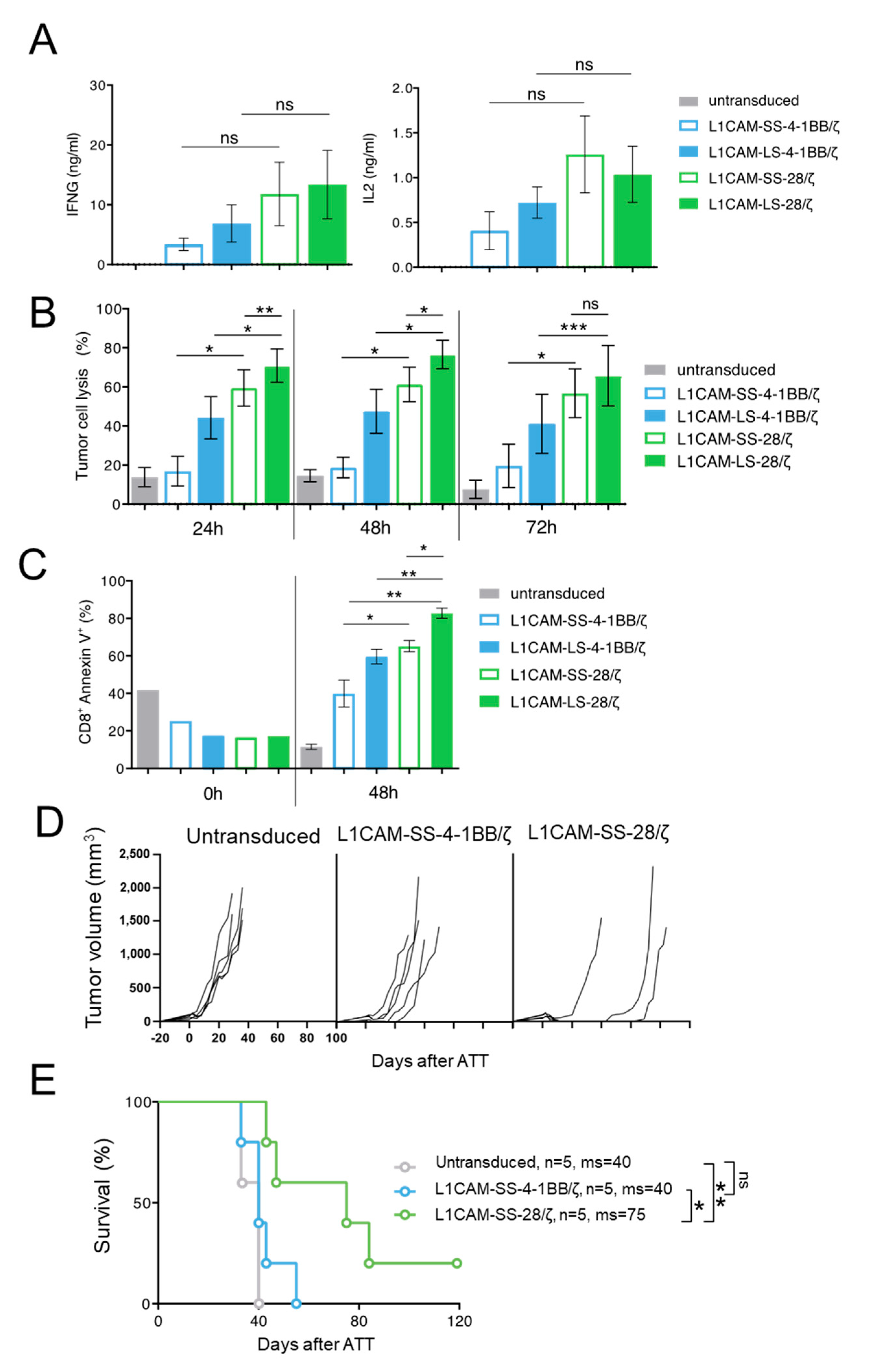
3.3. Superior Function of Human L1CAM-SS-28/ζ CAR T Cells Is Confirmed in a PDX Mouse Model
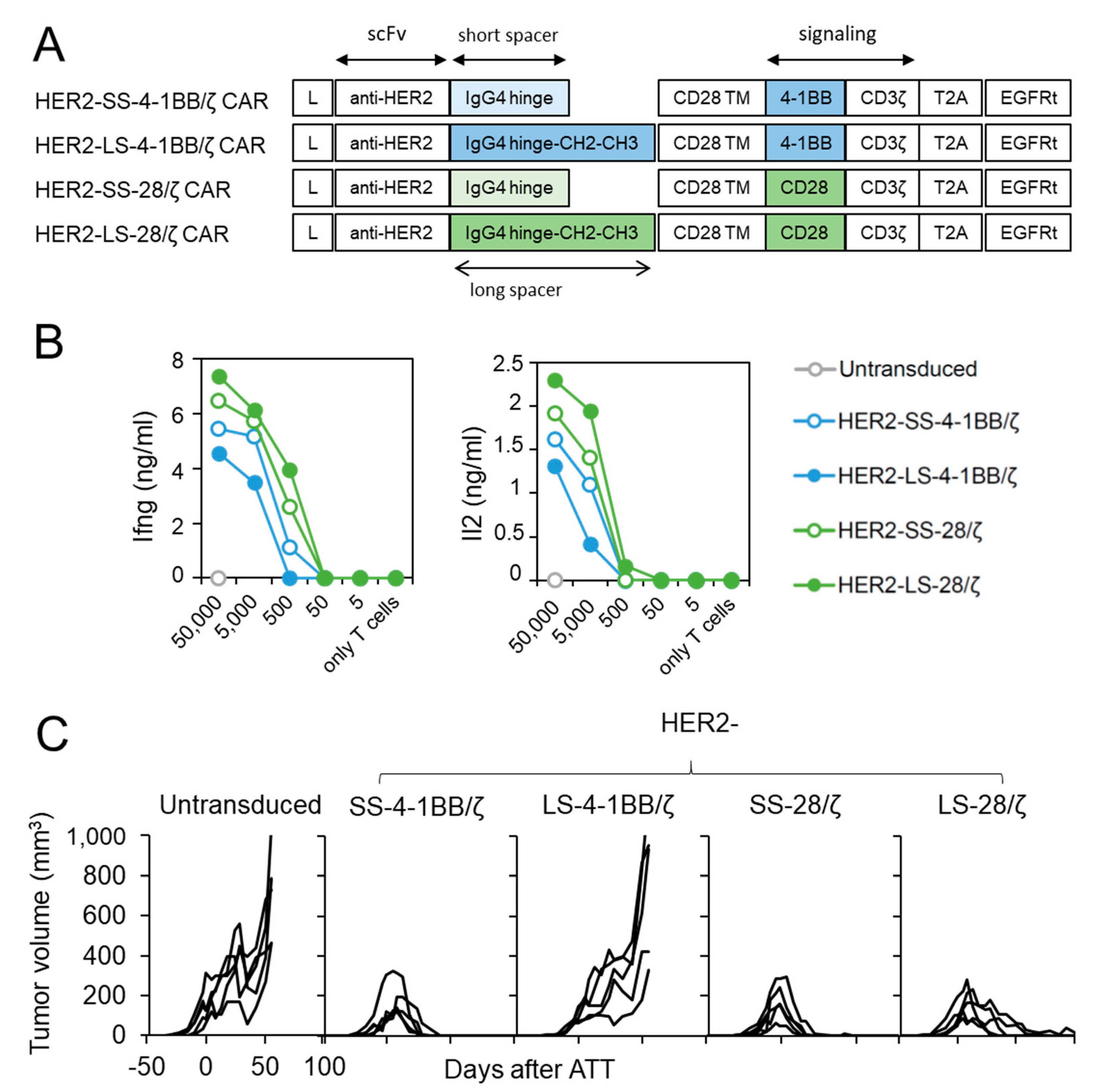
3.4. HER2-Specific CAR T Cells with CD28 Co-Stimulation Possess Higher Effector Function
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maher, J.; Brentjens, R.J.; Gunset, G.; Riviere, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brentjens, R.J.; Santos, E.; Nikhamin, Y.; Yeh, R.; Matsushita, M.; La Perle, K.; Quintas-Cardama, A.; Larson, S.M.; Sadelain, M. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin. Cancer Res. 2007, 13, 5426–5435. [Google Scholar] [CrossRef] [Green Version]
- van der Stegen, S.J.; Hamieh, M.; Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef]
- Kunkele, A.; Taraseviciute, A.; Finn, L.S.; Johnson, A.J.; Berger, C.; Finney, O.; Chang, C.A.; Rolczynski, L.S.; Brown, C.; Mgebroff, S.; et al. Preclinical Assessment of CD171-Directed CAR T-cell Adoptive Therapy for Childhood Neuroblastoma: CE7 Epitope Target Safety and Product Manufacturing Feasibility. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, S.; Naranjo, A.; Serrano, L.M.; Chang, W.C.; Wright, C.L.; Jensen, M.C. Genetic engineering of cytolytic T lymphocytes for adoptive T-cell therapy of neuroblastoma. J. Gene Med. 2004, 6, 704–711. [Google Scholar] [CrossRef]
- Novak-Hofer, I.; Amstutz, H.P.; Morgenthaler, J.J.; Schubiger, P.A. Internalization and degradation of monoclonal antibody chCE7 by human neuroblastoma cells. Int. J. Cancer 1994, 57, 427–432. [Google Scholar] [CrossRef]
- Smith, M.A.; Seibel, N.L.; Altekruse, S.F.; Ries, L.A.; Melbert, D.L.; O’Leary, M.; Smith, F.O.; Reaman, G.H. Outcomes for children and adolescents with cancer: Challenges for the twenty-first century. J. Clin. Oncol. 2010, 28, 2625–2634. [Google Scholar] [CrossRef]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, N.; Bajgain, P.; Sukumaran, S.; Ansari, S.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Leen, A.M.; Vera, J.F. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology 2016, 5, e1253656. [Google Scholar] [CrossRef] [Green Version]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.; Koyanagi, M.; Okada, H.; Takanashi, T.; Wong, Y.W.; Williams, A.F.; Okumura, K.; Yagita, H. CD48 is a counter-receptor for mouse CD2 and is involved in T cell activation. J. Exp. Med. 1992, 176, 1241–1249. [Google Scholar] [CrossRef] [Green Version]
- Johnston, S.C.; Dustin, M.L.; Hibbs, M.L.; Springer, T.A. On the species specificity of the interaction of LFA-1 with intercellular adhesion molecules. J. Immunol. 1990, 145, 1181–1187. [Google Scholar] [PubMed]
- Kammertoens, T.; Friese, C.; Arina, A.; Idel, C.; Briesemeister, D.; Rothe, M.; Ivanov, A.; Szymborska, A.; Patone, G.; Kunz, S.; et al. Tumour ischaemia by interferon-gamma resembles physiological blood vessel regression. Nature 2017, 545, 98–102. [Google Scholar] [CrossRef]
- Textor, A.; Listopad, J.J.; Wuhrmann, L.L.; Perez, C.; Kruschinski, A.; Chmielewski, M.; Abken, H.; Blankenstein, T.; Charo, J. Efficacy of CAR T-cell therapy in large tumors relies upon stromal targeting by IFNgamma. Cancer Res. 2014, 74, 6796–6805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunkele, A.; Johnson, A.J.; Rolczynski, L.S.; Chang, C.A.; Hoglund, V.; Kelly-Spratt, K.S.; Jensen, M.C. Functional Tuning of CARs Reveals Signaling Threshold above Which CD8+ CTL Antitumor Potency Is Attenuated due to Cell Fas-FasL-Dependent AICD. Cancer Immunol. Res. 2015, 3, 368–379. [Google Scholar] [CrossRef] [Green Version]
- Hombach, A.; Hombach, A.A.; Abken, H. Adoptive immunotherapy with genetically engineered T cells: Modification of the IgG1 Fc ‘spacer’ domain in the extracellular moiety of chimeric antigen receptors avoids ‘off-target’ activation and unintended initiation of an innate immune response. Gene 2010, 17, 1206–1213. [Google Scholar] [CrossRef] [Green Version]
- Hudecek, M.; Sommermeyer, D.; Kosasih, P.L.; Silva-Benedict, A.; Liu, L.; Rader, C.; Jensen, M.C.; Riddell, S.R. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol. Res. 2015, 3, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chang, W.C.; Wong, C.W.; Colcher, D.; Sherman, M.; Ostberg, J.R.; Forman, S.J.; Riddell, S.R.; Jensen, M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 2011, 118, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Engels, B.; Cam, H.; Schuler, T.; Indraccolo, S.; Gladow, M.; Baum, C.; Blankenstein, T.; Uckert, W. Retroviral vectors for high-level transgene expression in T lymphocytes. Hum. Gene 2003, 14, 1155–1168. [Google Scholar] [CrossRef]
- Textor, A.; Schmidt, K.; Kloetzel, P.M.; Weissbrich, B.; Perez, C.; Charo, J.; Anders, K.; Sidney, J.; Sette, A.; Schumacher, T.N.; et al. Preventing tumor escape by targeting a post-proteasomal trimming independent epitope. J. Exp. Med. 2016, 213, 2333–2348. [Google Scholar] [CrossRef] [PubMed]
- Ausubel, L.J.; Hall, C.; Sharma, A.; Shakeley, R.; Lopez, P.; Quezada, V.; Couture, S.; Laderman, K.; McMahon, R.; Huang, P.; et al. Production of CGMP-Grade Lentiviral Vectors. Bioprocess Int. 2012, 10, 32–43. [Google Scholar] [PubMed]
- Charo, J.; Perez, C.; Buschow, C.; Jukica, A.; Czeh, M.; Blankenstein, T. Visualizing the dynamic of adoptively transferred T cells during the rejection of large established tumors. Eur. J. Immunol. 2011, 41, 3187–3197. [Google Scholar] [CrossRef] [PubMed]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [Green Version]
- Priceman, S.J.; Gerdts, E.A.; Tilakawardane, D.; Kennewick, K.T.; Murad, J.P.; Park, A.K.; Jeang, B.; Yamaguchi, Y.; Yang, X.; Urak, R.; et al. Co-stimulatory signaling determines tumor antigen sensitivity and persistence of CAR T cells targeting PSCA+ metastatic prostate cancer. Oncoimmunology 2018, 7, e1380764. [Google Scholar] [CrossRef]
- Savoldo, B.; Ramos, C.A.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Investig. 2011, 121, 1822–1826. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Karrison, T.; Rowley, D.A.; Schreiber, H. IFN-gamma- and TNF-dependent bystander eradication of antigen-loss variants in established mouse cancers. J. Clin. Investig. 2008, 118, 1398–1404. [Google Scholar] [CrossRef] [Green Version]
- Anders, K.; Buschow, C.; Herrmann, A.; Milojkovic, A.; Loddenkemper, C.; Kammertoens, T.; Daniel, P.; Yu, H.; Charo, J.; Blankenstein, T. Oncogene-targeting T cells reject large tumors while oncogene inactivation selects escape variants in mouse models of cancer. Cancer Cell 2011, 20, 755–767. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2)-Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Krenciute, G.; Krebs, S.; Torres, D.; Wu, M.F.; Liu, H.; Dotti, G.; Li, X.N.; Lesniak, M.S.; Balyasnikova, I.V.; Gottschalk, S. Characterization and Functional Analysis of scFv-based Chimeric Antigen Receptors to Redirect T Cells to IL13Ralpha2-positive Glioma. Mol. Ther. 2016, 24, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Emtage, P.C.; Lo, A.S.; Gomes, E.M.; Liu, D.L.; Gonzalo-Daganzo, R.M.; Junghans, R.P. Second-generation anti-carcinoembryonic antigen designer T cells resist activation-induced cell death, proliferate on tumor contact, secrete cytokines, and exhibit superior antitumor activity in vivo: A preclinical evaluation. Clin. Cancer Res. 2008, 14, 8112–8122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, A.S.; Xu, C.; Murakami, A.; Marasco, W.A. Regression of established renal cell carcinoma in nude mice using lentivirus-transduced human T cells expressing a human anti-CAIX chimeric antigen receptor. Mol. Ther. Oncolytics 2014, 1, 14003. [Google Scholar] [CrossRef] [PubMed]
- Arch, R.H.; Thompson, C.B. 4-1BB and Ox40 are members of a tumor necrosis factor (TNF)-nerve growth factor receptor subfamily that bind TNF receptor-associated factors and activate nuclear factor kappaB. Mol. Cell. Biol. 1998, 18, 558–565. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jager, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Simon, S.; Labarriere, N. PD-1 expression on tumor-specific T cells: Friend or foe for immunotherapy? Oncoimmunology 2017, 7, e1364828. [Google Scholar] [CrossRef]
- Avery, L.; Filderman, J.; Szymczak-Workman, A.L.; Kane, L.P. Tim-3 co-stimulation promotes short-lived effector T cells, restricts memory precursors, and is dispensable for T cell exhaustion. Proc. Natl. Acad. Sci. USA 2018, 115, 2455–2460. [Google Scholar] [CrossRef] [Green Version]
- John, L.B.; Devaud, C.; Duong, C.P.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.H.; Darcy, P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef] [Green Version]
- Guedan, S.; Madar, A.; Casado-Medrano, V.; Shaw, C.E.; Wing, A.; Liu, F.; Young, R.M.; June, C.H.; Posey, A.D., Jr. Single residue in CD28-costimulated CAR T cells limits long-term persistence and antitumor durability. J. Clin. Investig. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019, 25, 82–88. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Textor, A.; Grunewald, L.; Anders, K.; Klaus, A.; Schwiebert, S.; Winkler, A.; Stecklum, M.; Rolff, J.; Henssen, A.G.; Höpken, U.E.; et al. CD28 Co-Stimulus Achieves Superior CAR T Cell Effector Function against Solid Tumors Than 4-1BB Co-Stimulus. Cancers 2021, 13, 1050. https://doi.org/10.3390/cancers13051050
Textor A, Grunewald L, Anders K, Klaus A, Schwiebert S, Winkler A, Stecklum M, Rolff J, Henssen AG, Höpken UE, et al. CD28 Co-Stimulus Achieves Superior CAR T Cell Effector Function against Solid Tumors Than 4-1BB Co-Stimulus. Cancers. 2021; 13(5):1050. https://doi.org/10.3390/cancers13051050
Chicago/Turabian StyleTextor, Ana, Laura Grunewald, Kathleen Anders, Anika Klaus, Silke Schwiebert, Annika Winkler, Maria Stecklum, Jana Rolff, Anton G. Henssen, Uta E. Höpken, and et al. 2021. "CD28 Co-Stimulus Achieves Superior CAR T Cell Effector Function against Solid Tumors Than 4-1BB Co-Stimulus" Cancers 13, no. 5: 1050. https://doi.org/10.3390/cancers13051050
APA StyleTextor, A., Grunewald, L., Anders, K., Klaus, A., Schwiebert, S., Winkler, A., Stecklum, M., Rolff, J., Henssen, A. G., Höpken, U. E., Eggert, A., Schulte, J. H., Jensen, M. C., Blankenstein, T., & Künkele, A. (2021). CD28 Co-Stimulus Achieves Superior CAR T Cell Effector Function against Solid Tumors Than 4-1BB Co-Stimulus. Cancers, 13(5), 1050. https://doi.org/10.3390/cancers13051050