
Integrin αvβ3 Engagement Regulates Glucose Metabolism and Migration through Focal Adhesion Kinase (FAK) and Protein Arginine Methyltransferase 5 (PRMT5) in Glioblastoma Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
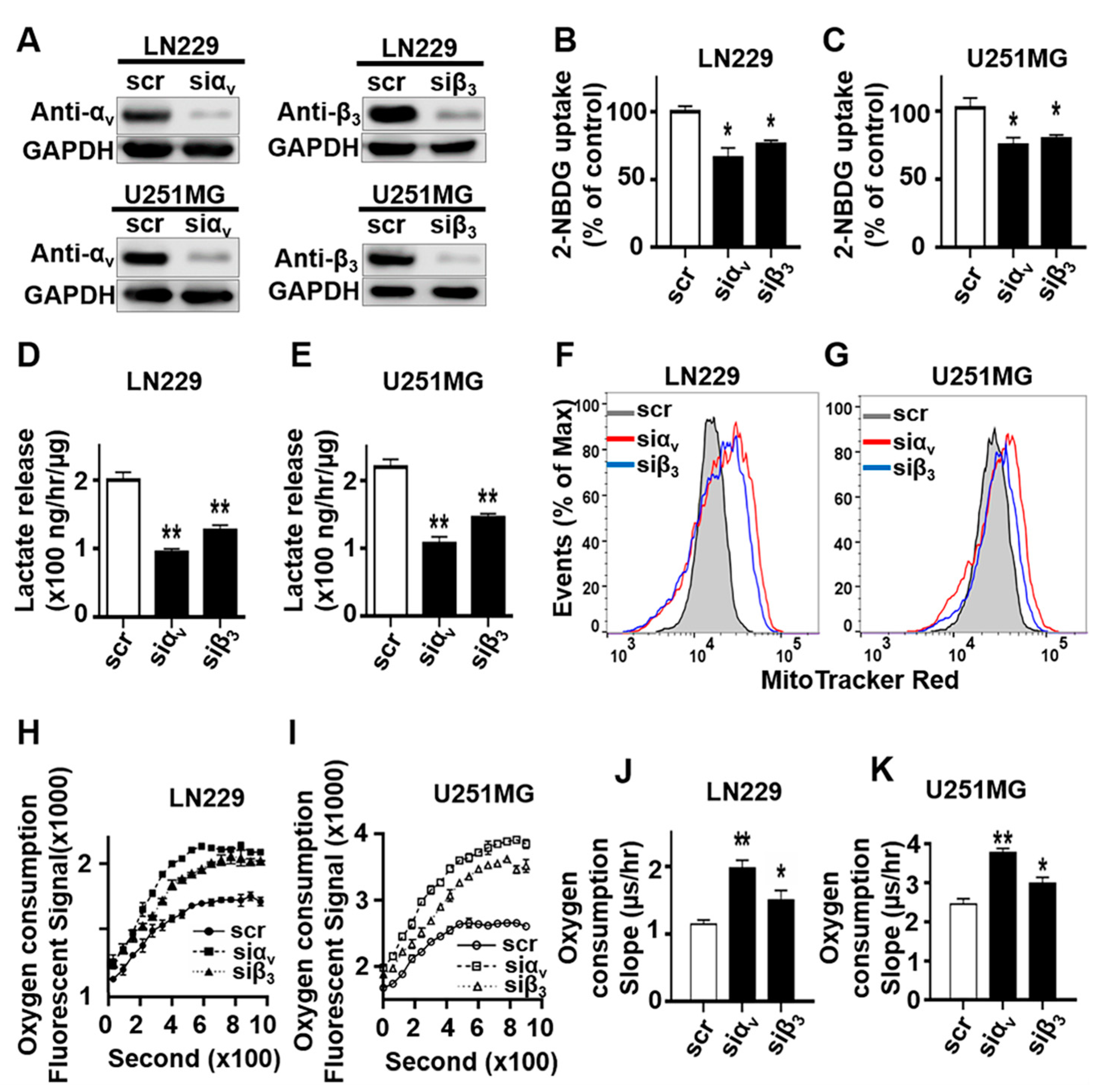
2.1. Integrin αvβ3 Plays an Important Role in Metabolic Reprogramming toward Glycolysis in Glioblastoma (GBM) Cells
2.2. Engagement of Integrin αvβ3 with Osteopontin Is Associated with a Metabolic Shift toward Glycolysis in GBM Cells
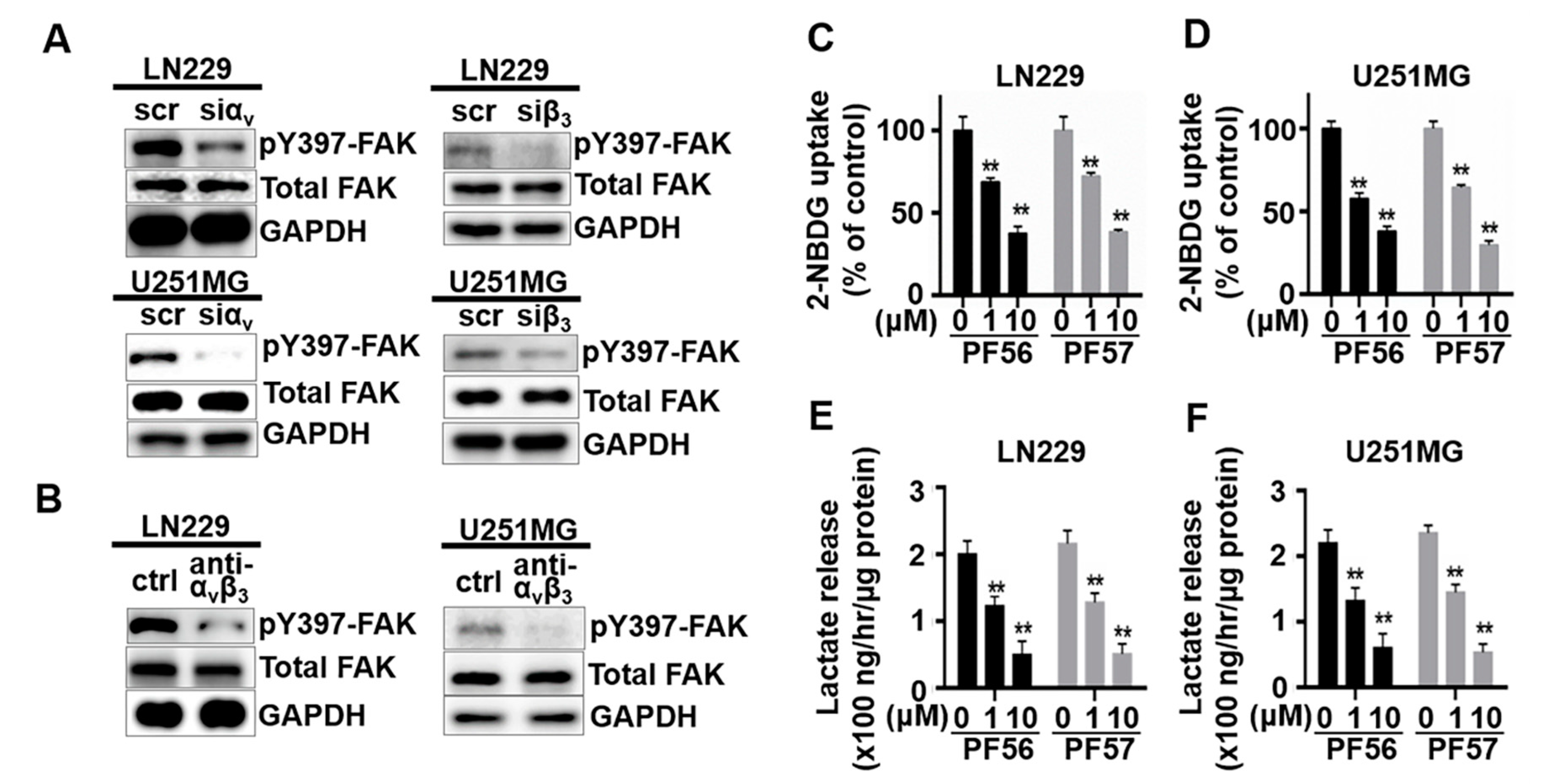
2.3. Blockade of FAK Activation Inhibits Glucose Uptake and Glycolysis but Promotes Mitochondrial Function in GBM Cells
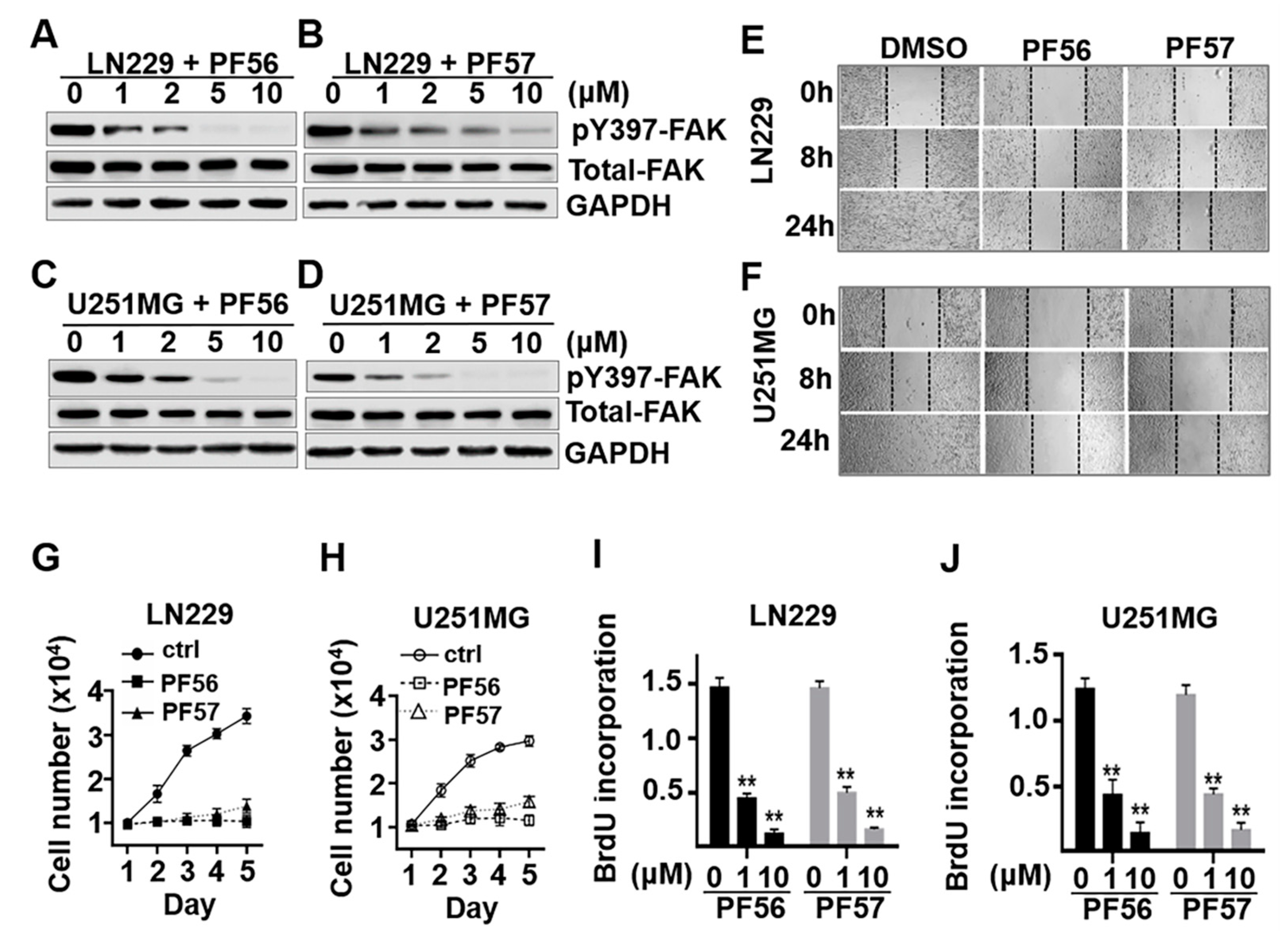
2.4. Blockade of αvβ3 and Osteopontin Engagement through FAK Inhibition Significantly Decreases Cell Migration and Proliferation in GBM Cells
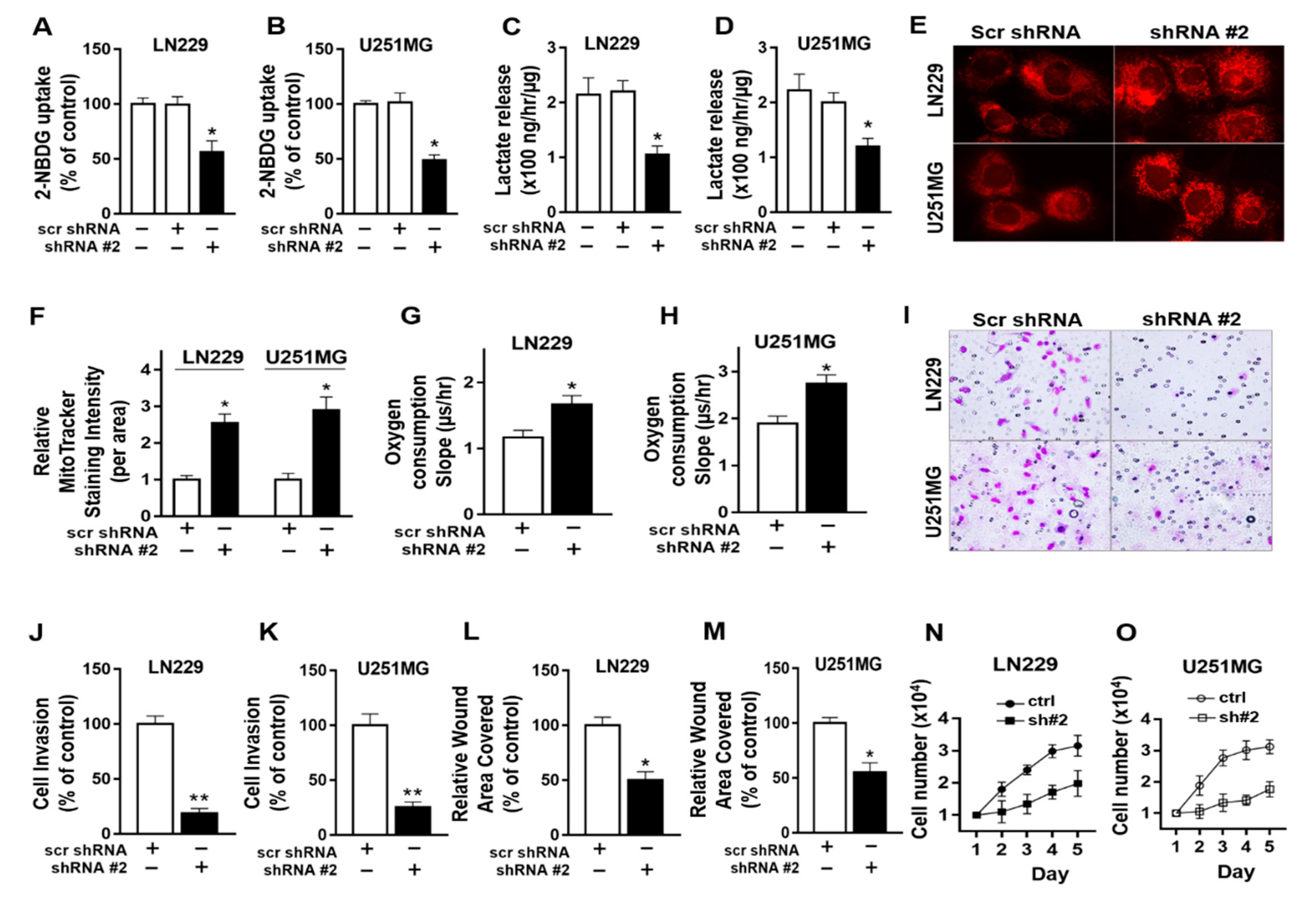
2.5. Protein Arginine Methyltransferase -5 (PRMT-5) Regulates Metabolic Shift towards Glycolysis, Migration and Invasion in GBM Cells
3. Discussion
4. Materials and Methods
4.1. Reagent and Antibodies
4.2. Cell Lines and Cell Culture
4.3. Immunoblotting
4.4. BrdU Cell Proliferation Assay and Cell Count
4.5. Wound Closure and Invasion Assays
4.6. Glucose Uptake Assay
4.7. Mitochondrial Activity Assay
4.8. Measurement of Extracellular Oxygen Consumption Rate
4.9. Lactate Assay
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mittelbronn, M.; Warth, A.; Meyermann, R.; Goodman, S.; Weller, M. Expression of integrins αvβ3 and αvβ5 and their ligands in primary and secondary central nervous system neoplasms. Histol. Histopathol. 2013, 28, 749–758. [Google Scholar]
- Schnell, O.; Krebs, B.; Wagner, E.; Romagna, A.; Beer, A.J.; Grau, S.J.; Thon, N.; Goetz, C.; Kretzschmar, H.A.; Tonn, J.C.; et al. Expression of integrin alphavbeta3 in gliomas correlates with tumor grade and is not restricted to tumor vasculature. Brain Pathol. 2008, 18, 378–386. [Google Scholar] [CrossRef] [Green Version]
- Gladson, C.L.; Cheresh, D.A. Glioblastoma expression of vitronectin and the alpha v beta 3 integrin. Adhesion mechanism for transformed glial cells. J. Clin. Investig. 1991, 88, 1924–1932. [Google Scholar] [CrossRef] [Green Version]
- Brooks, P.C.; Strömblad, S.; Sanders, L.C.; von Schalscha, T.L.; Aimes, R.T.; Stetler-Stevenson, W.G.; Quigley, J.P.; Cheresh, D.A. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell 1996, 85, 683–693. [Google Scholar] [CrossRef] [Green Version]
- Silletti, S.; Kessler, T.; Goldberg, J.; Boger, D.L.; Cheresh, D.A. Disruption of matrix metalloproteinase 2 binding to integrin alpha vbeta 3 by an organic molecule inhibits angiogenesis and tumor growth in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 119–124. [Google Scholar] [CrossRef]
- Bello, L.; Lucini, V.; Carrabba, G.; Giussani, C.; Machluf, M.; Pluderi, M.; Nikas, D.; Zhang, J.; Tomei, G.; Villani, R.M.; et al. Simultaneous inhibition of glioma angiogenesis, cell proliferation, and invasion by a naturally occurring fragment of human metalloproteinase-2. Cancer Res. 2001, 61, 8730–8736. [Google Scholar]
- Fisher, L.W.; Jain, A.; Tayback, M.; Fedarko, N.S. Small integrin binding ligand N-linked glycoprotein gene family expression in different cancers. Clin. Cancer Res. 2004, 10, 8501–8511. [Google Scholar] [CrossRef] [Green Version]
- Bellahcène, A.; Castronovo, V.; Ogbureke, K.U.; Fisher, L.W.; Fedarko, N.S. Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): Multifunctional proteins in cancer. Nat. Rev. Cancer 2008, 8, 212–226. [Google Scholar] [CrossRef] [Green Version]
- Said, H.M.; Hagemann, C.; Staab, A.; Stojic, J.; Kühnel, S.; Vince, G.H.; Flentje, M.; Roosen, K.; Vordermark, D. Expression patterns of the hypoxia-related genes osteopontin, CA9, erythropoietin, VEGF and HIF-1alpha in human glioma in vitro and in vivo. Radiother. Oncol. 2007, 83, 398–405. [Google Scholar] [CrossRef]
- Matusan-Ilijas, K.; Behrem, S.; Jonjic, N.; Zarkovic, K.; Lucin, K. Osteopontin expression correlates with angiogenesis and survival in malignant astrocytoma. Pathol. Oncol. Res. 2008, 14, 293–298. [Google Scholar] [CrossRef]
- Ding, Q.; Stewart, J., Jr.; Prince, C.W.; Chang, P.L.; Trikha, M.; Han, X.; Grammer, J.R.; Gladson, C.L. Promotion of malignant astrocytoma cell migration by osteopontin expressed in the normal brain: Differences in integrin signaling during cell adhesion to osteopontin versus vitronectin. Cancer Res. 2002, 62, 5336–5343. [Google Scholar]
- Burgett, M.E.; Lathia, J.D.; Roth, P.; Nowacki, A.S.; Galileo, D.S.; Pugacheva, E.; Huang, P.; Vasanji, A.; Li, M.; Byzova, T.; et al. Direct contact with perivascular tumor cells enhances integrin αvβ3 signaling and migration of endothelial cells. Oncotarget. 2016, 7, 43852–43867. [Google Scholar] [CrossRef] [Green Version]
- Sreekanthreddy, P.; Srinivasan, H.; Kumar, D.M.; Nijaguna, M.B.; Sridevi, S.; Vrinda, M.; Arivazhagan, A.; Balasubramaniam, A.; Hegde, A.S.; Chandramouli, B.A.; et al. Identification of potential serum biomarkers of glioblastoma: Serum osteopontin levels correlate with poor prognosis. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1409–1422. [Google Scholar] [CrossRef] [Green Version]
- Lamour, V.; Henry, A.; Kroonen, J.; Nokin, M.J.; von Marschall, Z.; Fisher, L.W.; Chau, T.L.; Chariot, A.; Sanson, M.; Delattre, J.Y.; et al. Targeting osteopontin suppresses glioblastoma stem-like cell character and tumorigenicity in vivo. Int. J. Cancer. 2015, 137, 1047–1057. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Keibler, M.A.; Wasylenko, T.M.; Kelleher, J.K.; Iliopoulos, O.; Vander Heiden, M.G.; Stephanopoulos, G. Metabolic requirements for cancer cell proliferation. Cancer Metab. 2016, 4, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhou, Y.; Shingu, T.; Feng, L.; Chen, Z.; Ogasawara, M.; Keating, M.J.; Kondo, S.; Huang, P. Metabolic alterations in highly tumorigenic glioblastoma cells: Preference for hypoxia and high dependency on glycolysis. J. Biol. Chem. 2011, 286, 32843–32853. [Google Scholar] [CrossRef] [Green Version]
- Granchi, C.; Minutolo, F. Anticancer agents that counteract tumor glycolysis. ChemMedChem 2012, 7, 1318–1350. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Grammer, J.R.; Nelson, M.A.; Guan, J.L.; Stewart, J.E., Jr.; Gladson, C.L. p27Kip1 and cyclin D1 are necessary for focal adhesion kinase regulation of cell cycle progression in glioblastoma cells propagated in vitro and in vivo in the scid mouse brain. J. Biol. Chem. 2005, 280, 6802–6815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Li, R.; Zhang, W.; Yang, X.; Wheeler, C.G.; Friedman, G.K.; Province, P.; Ding, Q.; You, Z.; Fathallah-Shaykh, H.M.; et al. Expression of PRMT5 correlates with malignant grade in gliomas and plays a pivotal role in tumor growth in vitro. J. Neurooncol. 2014, 118, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pendergrass, W.; Wolf, N.; Poot, M. Efficacy of MitoTracker Green and CMXrosamine to measure changes in mitochondrial membrane potentials in living cells and tissues. Cytometry A 2004, 61, 162–169. [Google Scholar] [CrossRef]
- Puleston, D. Detection of Mitochondrial Mass, Damage, and Reactive Oxygen Species by Flow Cytometry. Cold Spring Harb. Protoc. 2015, 2015. [Google Scholar] [CrossRef]
- Parsons, J.T. Focal adhesion kinase: The first ten years. J. Cell Sci. 2003, 116, 1409–1416. [Google Scholar] [CrossRef] [Green Version]
- Golubovskaya, V.M.; Huang, G.; Ho, B.; Yemma, M.; Morrison, C.D.; Lee, J.; Eliceiri, B.P.; Cance, W.G. Pharmacologic blockade of FAK autophosphorylation decreases human glioblastoma tumor growth and synergizes with temozolomide. Mol. Cancer Ther. 2013, 12, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Hecker, T.P.; Ding, Q.; Rege, T.A.; Hanks, S.K.; Gladson, C.L. Overexpression of FAK promotes Ras activity through the formation of a FAK/p120RasGAP complex in malignant astrocytoma cells. Oncogene 2004, 23, 3962–3971. [Google Scholar] [CrossRef] [Green Version]
- Haskell, H.; Natarajan, M.; Hecker, T.P.; Ding, Q.; Stewart, J.; Jr Grammer, J.R.; Gladson, C.L. Focal adhesion kinase is expressed in the angiogenic blood vessels of malignant astrocytic tumors in vivo and promotes capillary tube formation of brain microvascular endothelial cells. Clin. Cancer Res. 2003, 9, 2157–2165. [Google Scholar]
- Cai, G.Q.; Zheng, A.; Tang, Q.; White, E.S.; Chou, C.F.; Gladson, C.L.; Olman, M.A.; Ding, Q. Downregulation of FAK-related non-kinase mediates the migratory phenotype of human fibrotic lung fibroblasts. Exp. Cell Res. 2010, 316, 1600–1609. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef]
- Pal, S.; Baiocchi, R.A.; Byrd, J.C.; Grever, M.R.; Jacob, S.T.; Sif, S. Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J. 2007, 26, 3558–3569. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Pal, S.; Sif, S. Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells. Mol. Cell. Biol. 2008, 28, 6262–6277. [Google Scholar] [CrossRef] [Green Version]
- Eckert, D.; Biermann, K.; Nettersheim, D.; Gillis, A.J.; Steger, K.; Jäck, H.M.; Müller, A.M.; Looijenga, L.H.; Schorle, H. Expression of BLIMP1/PRMT5 and concurrent histone H2A/H4 arginine 3 dimethylation in fetal germ cells, CIS/IGCNU and germ cell tumors. BMC Dev. Biol. 2008, 8, 106. [Google Scholar] [CrossRef] [Green Version]
- Powers, M.A.; Fay, M.M.; Factor, R.E.; Welm, A.L.; Ullman, K.S. Protein arginine methyltransferase 5 accelerates tumor growth by arginine methylation of the tumor suppressor programmed cell death 4. Cancer Res. 2011, 71, 5579–5587. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.; Zhao, S.; Liu, T.; Liu, Y.; Liu, Y.; Yang, X. Overexpression of PRMT5 promotes tumor cell growth and is associated with poor disease prognosis in epithelial ovarian cancer. J. Histochem. Cytochem. 2013, 61, 206–217. [Google Scholar] [CrossRef] [Green Version]
- Velpula, K.K.; Bhasin, A.; Asuthkar, S.; Tsung, A.J. Combined targeting of PDK1 and EGFR triggers regression of glioblastoma by reversing the Warburg effect. Cancer Res. 2013, 73, 7277–7289. [Google Scholar] [CrossRef] [Green Version]
- Poteet, E.; Choudhury, G.R.; Winters, A.; Li, W.; Ryou, M.G.; Liu, R.; Tang, L.; Ghorpade, A.; Wen, Y.; Yuan, F.; et al. Reversing the Warburg effect as a treatment for glioblastoma. J. Biol. Chem. 2013, 288, 9153–9164. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.A.; Chao, Y.; Shiah, S.G.; Lin, W.W. Nutrient deprivation induces the Warburg effect through ROS/AMPK-dependent activation of pyruvate dehydrogenase kinase. Biochim. Biophys. Acta 2013, 1833, 1147–1156. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Erfani, S.; Liu, Z.; Jia, C.; Chen, Y.; Xu, B.; Deng, X.; Alfáro, J.E.; Chen, L.; Napier, D.; et al. CD151-α3β1 integrin complexes are prognostic markers of glioblastoma and cooperate with EGFR to drive tumor cell motility and invasion. Oncotarget 2015, 6, 29675–29693. [Google Scholar] [CrossRef] [Green Version]
- Riemenschneider, M.J.; Mueller, W.; Betensky, R.A.; Mohapatra, G.; Louis, D.N. In situ analysis of integrin and growth factor receptor signaling pathways in human glioblastomas suggests overlapping relationships with focal adhesion kinase activation. Am. J. Pathol. 2005, 167, 1379–1387. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.; Machado, J., Jr.; Merlo, A. Loss of focal adhesion kinase (FAK) inhibits epidermal growth factor receptor-dependent migration and induces aggregation of nh(2)-terminal FAK in the nuclei of apoptotic glioblastoma cells. Cancer Res. 2001, 61, 4978–4981. [Google Scholar]
- Tamura, M.; Gu, J.; Takino, T.; Yamada, K.M. Tumor suppressor PTEN inhibition of cell invasion, migration, and growth: Differential involvement of focal adhesion kinase and p130Cas. Cancer Res. 1999, 59, 442–449. [Google Scholar]
- D’Abaco, G.M.; Kaye, A.H. Integrins: Molecular determinants of glioma invasion. J. Clin. Neurosci. 2007, 14, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gao, Q.; Zhou, Y.; Dier, U.; Hempel, N.; Hochwald, S.N. Focal adhesion kinase-promoted tumor glucose metabolism is associated with a shift of mitochondrial respiration to glycolysis. Oncogene 2016, 35, 1926–1942. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Hochwald, S.N. The role of FAK in tumor metabolism and therapy. Pharmacol. Ther. 2014, 142, 154–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Hu, Q.; Xu, J.; Ji, S.; Dai, W.; Liu, W.; Xu, W.; Sun, Q.; Zhang, Z.; Ni, Q.; et al. PRMT5 enhances tumorigenicity and glycolysis in pancreatic cancer via the FBW7/cMyc axis. Cell Commun. Signal. 2019, 17, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barczak, W.; Jin, L.; Carr, S.M.; Munro, S.; Ward, S.; Kanapin, A.; Samsonova, A.; La Thangue, N.B. PRMT5 promotes cancer cell migration and invasion through the E2F pathway. Cell Death Dis. 2020, 11, 572. [Google Scholar] [CrossRef]
- Chittka, A.; Nitarska, J.; Grazini, U.; Richardson, W.D. Transcription factor positive regulatory domain 4 (PRDM4) recruits protein arginine methyltransferase 5 (PRMT5) to mediate histone arginine methylation and control neural stem cell proliferation and differentiation. J. Biol. Chem. 2012, 287, 42995–43006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayasaka, H.; Martin, K.H.; Hershey, E.D.; Parsons, J.T. Disruption of FRNK expression by gene targeting of the intronic promoter within the focal adhesion kinase gene. J. Cell Biochem. 2007, 102, 947–954. [Google Scholar] [CrossRef]
- Ding, Q.; Cai, G.Q.; Hu, M.; Yang, Y.; Zheng, A.; Tang, Q.; Gladson, C.L.; Hayasaka, H.; Wu, H.; You, Z.; et al. FAK-related nonkinase is a multifunctional negative regulator of pulmonary fibrosis. Am. J. Pathol. 2013, 182, 1572–1584. [Google Scholar] [CrossRef] [Green Version]
- Che, P.; Yang, Y.; Han, X.; Hu, M.; Sellers, J.C.; Londono-Joshi, A.I.; Cai, G.Q.; Buchsbaum, D.J.; Christein, J.D.; Tang, Q.; et al. S100A4 promotes pancreatic cancer progression through a dual signaling pathway mediated by Src and focal adhesion kinase. Sci. Rep. 2015, 5, 8453. [Google Scholar] [CrossRef] [Green Version]
- Leira, F.; Louzao, M.C.; Vieites, J.M.; Botana, L.M.; Vieytes, M.R. Fluorescent microplate cell assay to measure uptake and metabolism of glucose in normal human lung fibroblasts. Toxicol. In Vitro 2002, 16, 267–273. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Che, P.; Yu, L.; Friedman, G.K.; Wang, M.; Ke, X.; Wang, H.; Zhang, W.; Nabors, B.; Ding, Q.; Han, X. Integrin αvβ3 Engagement Regulates Glucose Metabolism and Migration through Focal Adhesion Kinase (FAK) and Protein Arginine Methyltransferase 5 (PRMT5) in Glioblastoma Cells. Cancers 2021, 13, 1111. https://doi.org/10.3390/cancers13051111
Che P, Yu L, Friedman GK, Wang M, Ke X, Wang H, Zhang W, Nabors B, Ding Q, Han X. Integrin αvβ3 Engagement Regulates Glucose Metabolism and Migration through Focal Adhesion Kinase (FAK) and Protein Arginine Methyltransferase 5 (PRMT5) in Glioblastoma Cells. Cancers. 2021; 13(5):1111. https://doi.org/10.3390/cancers13051111
Chicago/Turabian StyleChe, Pulin, Lei Yu, Gregory K. Friedman, Meimei Wang, Xiaoxue Ke, Huafeng Wang, Wenbin Zhang, Burt Nabors, Qiang Ding, and Xiaosi Han. 2021. "Integrin αvβ3 Engagement Regulates Glucose Metabolism and Migration through Focal Adhesion Kinase (FAK) and Protein Arginine Methyltransferase 5 (PRMT5) in Glioblastoma Cells" Cancers 13, no. 5: 1111. https://doi.org/10.3390/cancers13051111