
Distinct Signatures of Genomic Copy Number Variants Define Subgroups of Merkel Cell Carcinoma Tumors

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
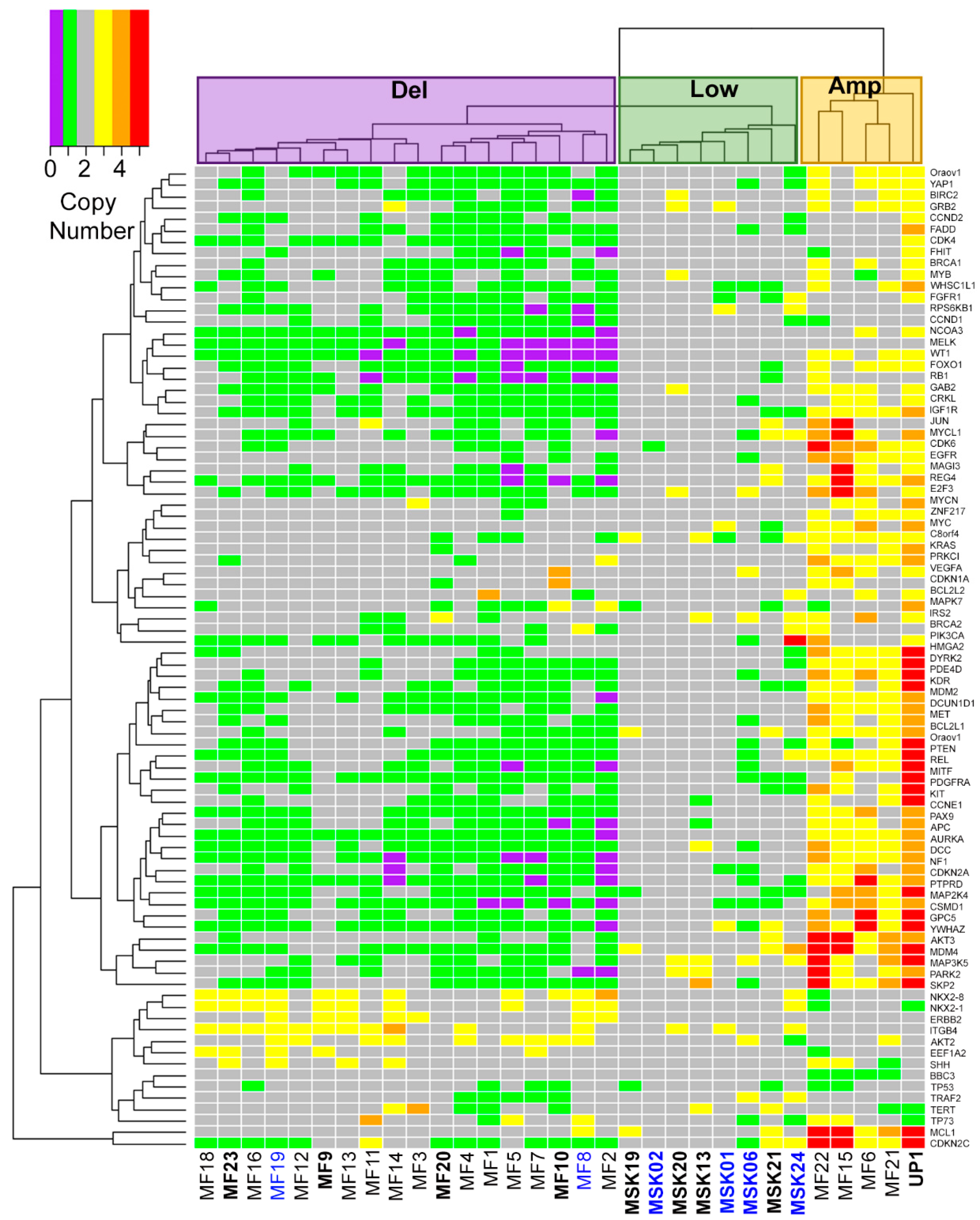
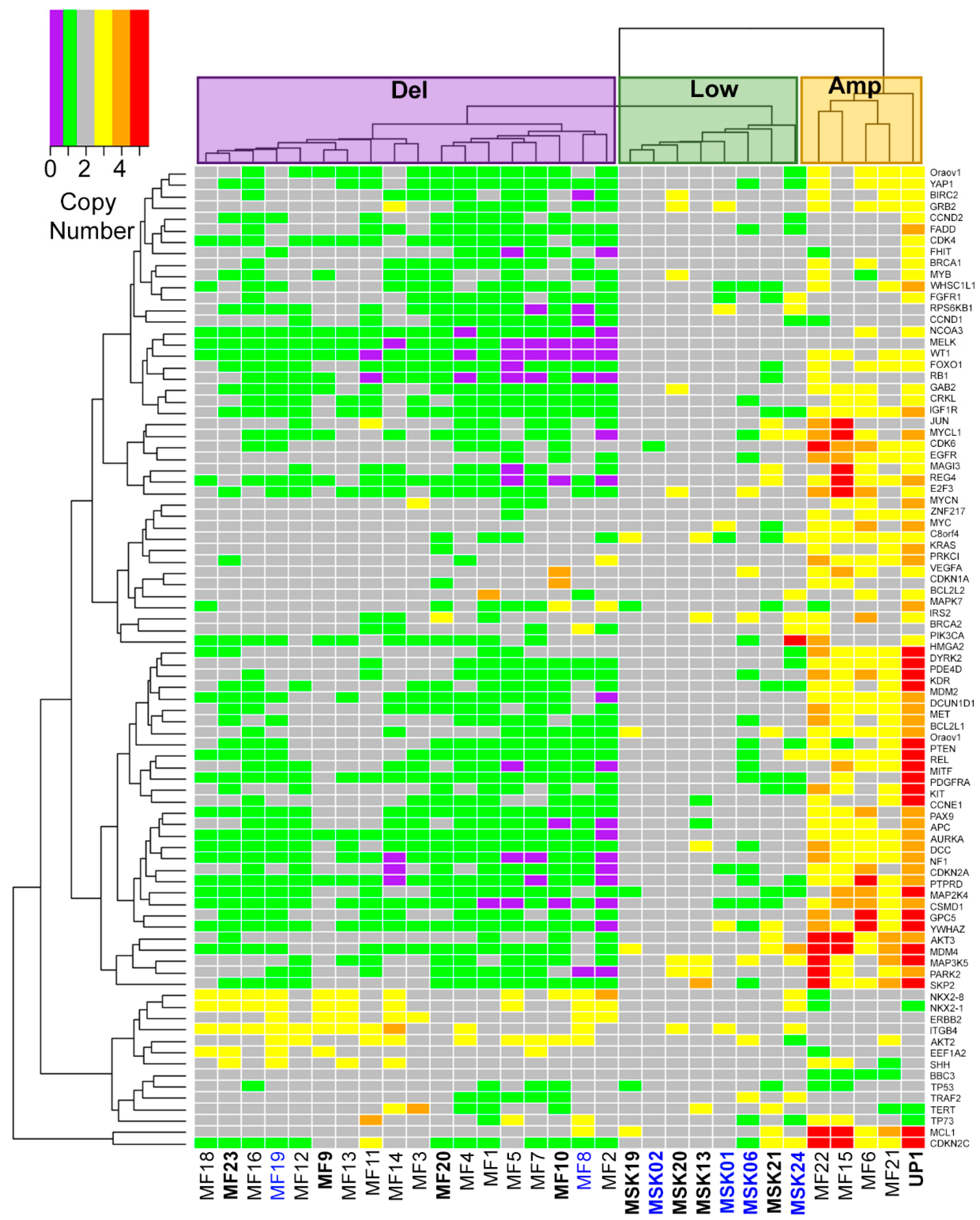
2.1. Three Genomic Structural Variant Signatures Identified in MCC Tumors
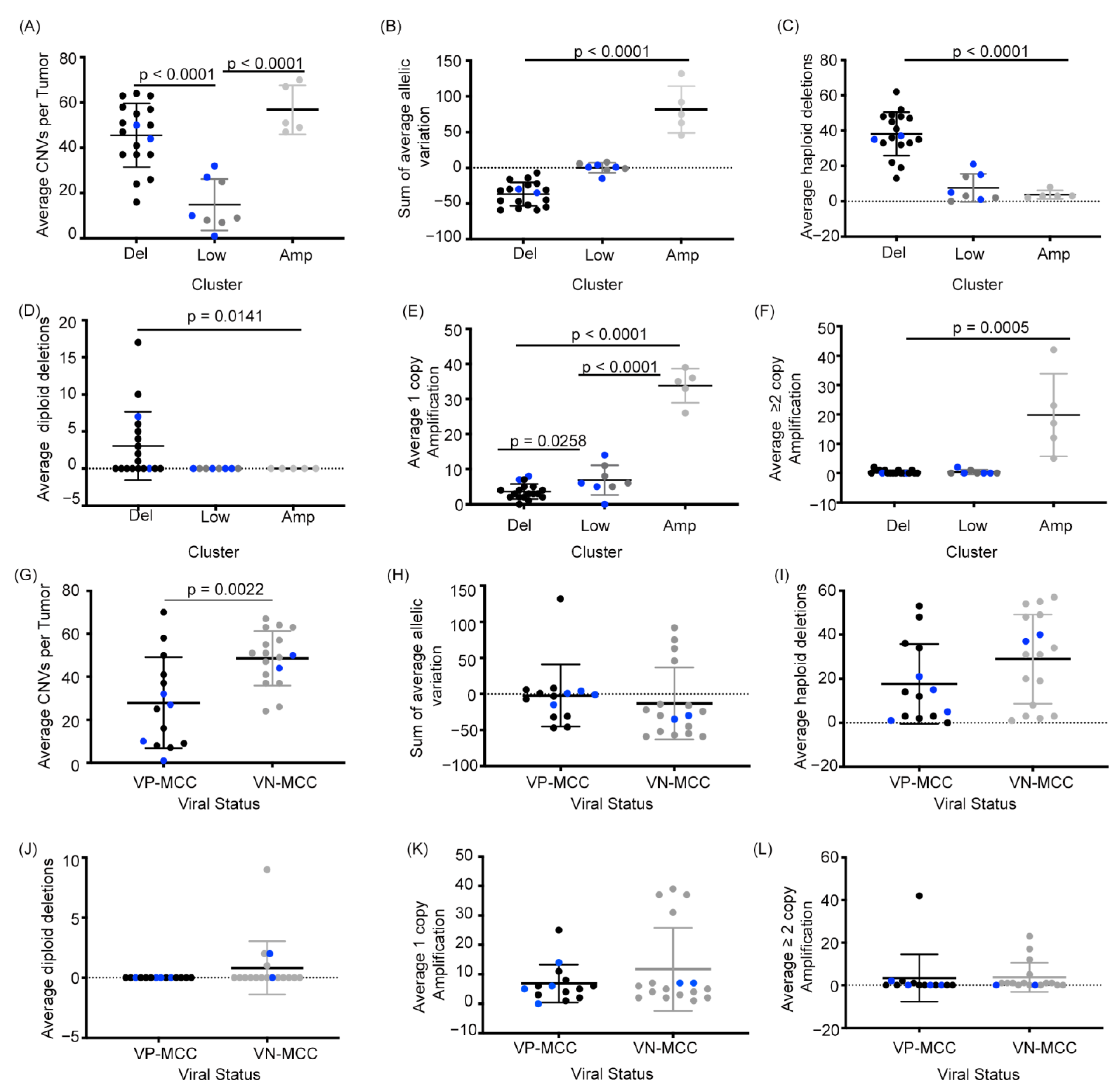
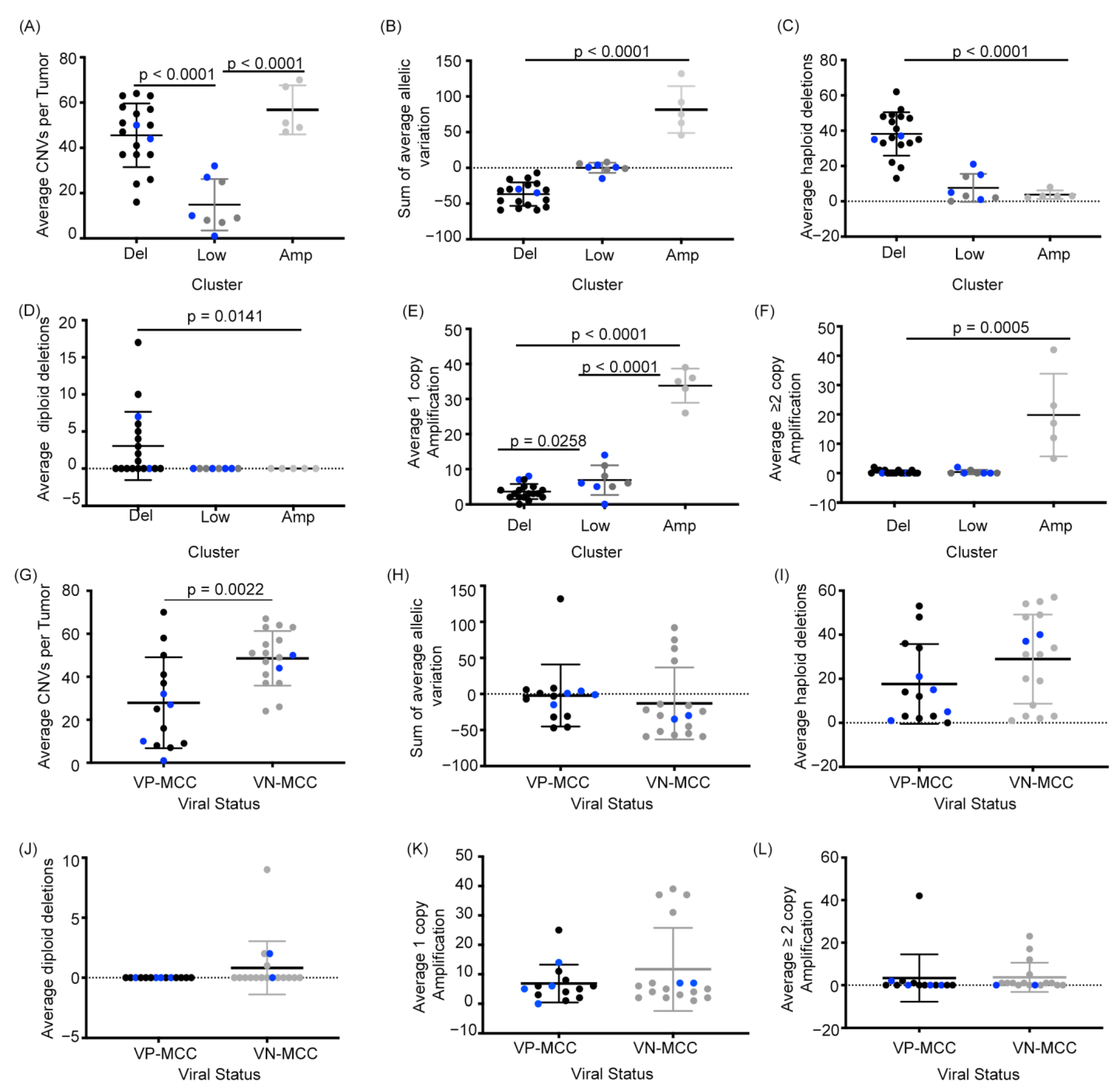
2.2. MCC Structural Variant Signatures Are Characterized by Deletions, Absence of Copy Changes, or Amplifications
2.3. VN-MCC Contains More Structural Variants Than VP-MCC
2.4. MCC Structural Variant Signatures Are Not Predictors of Survival
3. Discussion
4. Materials and Methods
4.1. Inclusion Criteria and Patient Samples
4.2. Genomic DNA Isolation
4.3. Virus Detection
4.4. Cell Lines
4.5. Nanostring Prep and Run
4.6. Nanostring Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Albores-Saavedra, J.; Batich, K.; Chable-Montero, F.; Sagy, N.; Schwartz, A.M.; Henson, D.E. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: A population-based study. J. Cutan. Pathol. 2010, 37, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Harms, P.W.; Harms, K.L.; Moore, P.S.; DeCaprio, J.A.; Nghiem, P.; Wong, M.K.; Brownell, I. International workshop on Merkle cell carcinoma research working. The biology and treatment of Merkel cell carcinoma: Current understanding and research priorities. Nat. Rev. Clin. Oncol. 2018, 15, 763–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, R.A.; Lambert, W.C. The Merkel cell carcinoma: A 50-year retrospect. J. Surg. Oncol. 2005, 89, 5. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, T.L.; Dennis, S.; Kachare, S.D.; Vohra, N.A.; Wong, J.H.; Zervos, E.E. Dramatic increase in the incidence and mortality from Merkel cell carcinoma in the United States. Am. Surg. 2015, 81, 802–806. [Google Scholar] [CrossRef]
- van Veenendaal, L.M.; van Akkooi, A.C.J.; Verhoef, C.; Grunhagen, D.J.; Klop, W.M.C.; Valk, G.D.; Tesselaar, M.E.T. Merkel cell carcinoma: Clinical outcome and prognostic factors in 351 patients. J. Surg. Oncol. 2018, 117, 1768–1775. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, P.W. Update on Merkel cell carcinoma. Clin. Lab. Med. 2017, 37, 485–501. [Google Scholar] [CrossRef]
- Al-Rohil, R.N.; Milton, D.R.; Nagarajan, P.; Curry, J.L.; Feldmeyer, L.; Torres-Cabala, C.A.; Ivan, D.; Prieto, V.G.; Tetzlaff, M.T.; Aung, P.P. Intratumoral and peritumoral lymphovascular invasion detected by D2-40 immunohistochemistry correlates with metastasis in primary cutaneous Merkel cell carcinoma. Hum. Pathol. 2018, 77, 98–107. [Google Scholar] [CrossRef]
- Aung, P.P.; Parra, E.R.; Barua, S.; Sui, D.; Ning, J.; Mino, B.; Ledesma, D.A.; Curry, J.L.; Nagarajan, P.; Torres-Cabala, C.A.; et al. B7-H3 Expression in Merkel cell carcinoma-associated endothelial cells correlates with locally aggressive primary tumor features and increased vascular density. Clin. Cancer Res. 2019, 25, 3455–3467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldmeyer, L.; Hudgens, C.W.; Ray-Lyons, G.; Nagarajan, P.; Aung, P.P.; Curry, J.L.; Torres-Cabala, C.A.; Mino, B.; Rodriguez-Canales, J.; Reuben, A.; et al. Density, distribution, and composition of immune infiltrates correlate with survival in Merkel cell carcinoma. Clin. Cancer Res. 2016, 22, 5553–5563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, P.W.; Patel, R.M.; Verhaegen, M.E.; Giordano, T.J.; Nash, K.T.; Johnson, C.N.; Daignault, S.; Thomas, D.G.; Gudjonsson, J.E.; Elder, J.T.; et al. Distinct gene expression profiles of viral- and nonviral-associated merkel cell carcinoma revealed by transcriptome analysis. J. Investig. Dermatol. 2013, 133, 936–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Harms, P.W.; Palanisamy, N.; Carskadon, S.; Cao, X.; Siddiqui, J.; Patel, R.M.; Zelenka-Wang, S.; Durham, A.B.; Fullen, D.R.; et al. Age and gender associations of virus positivity in Merkel cell carcinoma characterized using a novel RNA in situ hybridization assay. Clin. Cancer Res. 2017, 23, 5622–5630. [Google Scholar] [CrossRef] [Green Version]
- Goh, G.; Walradt, T.; Markarov, V.; Blom, A.; Riaz, N.; Doumani, R.; Stafstrom, K.; Moshiri, A.; Yelistratova, L.; Levinsohn, J.; et al. Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget 2016, 7, 3403–3415. [Google Scholar] [CrossRef] [Green Version]
- Harms, P.W.; Collie, A.M.; Hovelson, D.H.; Cani, A.K.; Verhaegen, M.E.; Patel, R.M.; Fullen, D.R.; Omata, K.; Dlugosz, A.A.; Tomlins, S.A.; et al. Next generation sequencing of Cytokeratin 20-negative Merkel cell carcinoma reveals ultraviolet-signature mutations and recurrent TP53 and RB1 inactivation. Mod. Pathol. 2016, 29, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Q.; Waldeck, K.; Vergara, I.A.; Schroder, J.; Madore, J.; Wilmott, J.S.; Colebatch, A.J.; De Paoli-Iseppi, R.; Li, J.; Lupat, R.; et al. UV-associated mutations underlie the etiology of MCV-negative merkel cell carcinomas. Cancer Res. 2015, 75, 5228–5234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veija, T.; Sarhadi, V.K.; Koljonen, V.; Bohling, T.; Knuutila, S. Hotspot mutations in polyomavirus positive and negative Merkel cell carcinomas. Cancer Genet. 2016, 209, 30–35. [Google Scholar] [CrossRef]
- Kwun, H.J.; Shuda, M.; Feng, H.; Camacho, C.J.; Moore, P.S.; Chang, Y. Merkel cell polyomavirus small T antigen controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase SCFFbw7. Cell Host Microbe 2013, 14, 125–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuda, M.; Kwun, H.J.; Feng, H.; Chang, Y.; Moore, P.S. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J. Clin. Investig. 2011, 121, 3623–3634. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Sada, H.; Muller, M.; Mehta, R.; Toth, R.; Arthur, J.S.C.; Whitehouse, A.; Macdonald, A. The PP4R1 sub-unit of protein phosphatase PP4 is essential for inhibition of NF-kappaB by merkel polyomavirus small tumour antigen. Oncotarget 2017, 8, 25418–25432. [Google Scholar] [CrossRef] [Green Version]
- Van Gele, M.; Leonard, J.H.; Van Roy, N.; Van Limbergen, H.; Van Belle, S.; Cocquyt, V.; Salwen, H.; De Paepe, A.; Speleman, F. Combined karyotyping, CGH and M-FISH analysis allows detailed characterization of unidentified chromosomal rearrangements in Merkel cell carcinoma. Int. J. Cancer 2002, 101, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Starrett, G.J.; Thakuria, M.; Chen, T.; Marcelus, C.; Cheng, J.; Nomburg, J.; Thorner, A.R.; Slevin, M.K.; Powers, W.; Burns, R.T.; et al. Clinical and molecular characterization of virus-positive and virus-negative Merkel cell carcinoma. Genome Med. 2020, 12, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evan, G.I.; Vousden, K.H. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L. Defects in a cell cycle checkpoint may be responsible for the genomic instability of cancer cells. Cell 1992, 71, 543–546. [Google Scholar] [CrossRef]
- Limpose, K.L.; Trego, K.S.; Li, Z.; Leung, S.W.; Sarker, A.H.; Shah, J.A.; Ramalingam, S.S.; Werner, E.M.; Dynan, W.S.; Cooper, P.K.; et al. Overexpression of the base excision repair NTHL1 glycosylase causes genomic instability and early cellular hallmarks of cancer. Nucleic Acids Res. 2018, 46, 4515–4532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garneski, K.M.; DeCaprio, J.A.; Nghiem, P. Does a new polyomavirus contribute to Merkel cell carcinoma? Genome Biol. 2008, 9, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popp, S.; Waltering, S.; Herbst, C.; Moll, I.; Boukamp, P. UV-B-type mutations and chromosomal imbalances indicate common pathways for the development of Merkel and skin squamous cell carcinomas. Int. J. Cancer 2002, 99, 352–360. [Google Scholar] [CrossRef]
- Hartschuh, W.; Schulz, T. Merkel cell hyperplasia in chronic radiation-damaged skin: Its possible relationship to fibroepithelioma of Pinkus. J. Cutan. Pathol. 1997, 24, 477–483. [Google Scholar] [CrossRef]
- Miller, R.W.; Rabkin, C.S. Merkel cell carcinoma and melanoma: Etiological similarities and differences. Cancer Epidemiol. Biomark. Prev. 1999, 8, 153–158. [Google Scholar]
- Agelli, M.; Clegg, L.X. Epidemiology of primary Merkel cell carcinoma in the United States. J. Am. Acad. Dermatol. 2003, 49, 832–841. [Google Scholar] [CrossRef]
- Brenner, B.; Sulkes, A.; Rakowsky, E.; Feinmesser, M.; Yukelson, A.; Bar-Haim, E.; Katz, A.; Idelevich, E.; Neuman, A.; Barhana, M.; et al. Second neoplasms in patients with Merkel cell carcinoma. Cancer 2001, 91, 1358–1362. [Google Scholar] [CrossRef]
- Ratner, D.; Nelson, B.R.; Brown, M.D.; Johnson, T.M. Merkel cell carcinoma. J. Am. Acad. Dermatol. 1993, 29, 143–156. [Google Scholar] [CrossRef]
- Cerroni, L.; Kerl, H. Primary cutaneous neuroendocrine (Merkel cell) carcinoma in association with squamous- and basal-cell carcinoma. Am. J. Dermatopathol. 1997, 19, 610–613. [Google Scholar] [CrossRef] [PubMed]
- Aydin, A.; Kocer, N.E.; Bekerecioglu, M.; Sari, I. Cutaneous undifferentiated small (Merkel) cell carcinoma, that developed synchronously with multiple actinic keratoses, squamous cell carcinomas and basal cell carcinoma. J. Dermatol. 2003, 30, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Pulitzer, M.P.; Brannon, A.R.; Berger, M.F.; Louis, P.; Scott, S.N.; Jungbluth, A.A.; Coit, D.G.; Brownell, I.; Busam, K.J. Cutaneous squamous and neuroendocrine carcinoma: Genetically and immunohistochemically different from Merkel cell carcinoma. Mod. Pathol. 2015, 28, 1023–1032. [Google Scholar] [CrossRef] [Green Version]
- Iacocca, M.V.; Abernethy, J.L.; Stefanato, C.M.; Allan, A.E.; Bhawan, J. Mixed Merkel cell carcinoma and squamous cell carcinoma of the skin. J. Am. Acad. Dermatol. 1998, 39, 882–887. [Google Scholar] [CrossRef]
- Panich, U.; Sittithumcharee, G.; Rathviboon, N.; Jirawatnotai, S. Ultraviolet radiation-induced skin aging: The role of DNA damage and oxidative stress in epidermal stem cell damage mediated skin aging. Stem. Cells Int. 2016, 2016, 7370642. [Google Scholar] [CrossRef] [Green Version]
- Morales-Sanchez, A.; Fuentes-Panana, E.M. Human viruses and cancer. Viruses 2014, 6, 4047–4079. [Google Scholar] [CrossRef] [Green Version]
- Heath, M.; Jaimes, N.; Lemos, B.; Mostaghimi, A.; Wang, L.C.; Penas, P.F.; Nghiem, P. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: The AEIOU features. J. Am. Acad. Dermatol. 2008, 58, 375–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, S.M.; Barton, C.M.; Lee, S.J.; Morton, D.G.; Wallace, D.M.; Lemoine, N.R.; Neoptolemos, J.P. Loss of the retinoblastoma susceptibility gene (RB1) is a frequent and early event in prostatic tumorigenesis. Br. J. Cancer 1994, 70, 1252–1257. [Google Scholar] [CrossRef] [Green Version]
- Phillips, S.M.; Morton, D.G.; Lee, S.J.; Wallace, D.M.; Neoptolemos, J.P. Loss of heterozygosity of the retinoblastoma and adenomatous polyposis susceptibility gene loci and in chromosomes 10p, 10q and 16q in human prostate cancer. Br. J. Urol. 1994, 73, 390–395. [Google Scholar] [CrossRef]
- Bettendorf, O.; Schmidt, H.; Staebler, A.; Grobholz, R.; Heinecke, A.; Boecker, W.; Hertle, L.; Semjonow, A. Chromosomal imbalances, loss of heterozygosity, and immunohistochemical expression of TP53, RB1, and PTEN in intraductal cancer, intraepithelial neoplasia, and invasive adenocarcinoma of the prostate. Genes Chromosom. Cancer 2008, 47, 565–572. [Google Scholar] [CrossRef]
- Dimaras, H.; Khetan, V.; Halliday, W.; Orlic, M.; Prigoda, N.L.; Piovesan, B.; Marrano, P.; Corson, T.W.; Eagle, R.C., Jr.; Squire, J.A.; et al. Loss of RB1 induces non-proliferative retinoma: Increasing genomic instability correlates with progression to retinoblastoma. Hum. Mol. Genet. 2008, 17, 1363–1372. [Google Scholar] [CrossRef] [Green Version]
- Xing, F.; Persaud, Y.; Pratilas, C.A.; Taylor, B.S.; Janakiraman, M.; She, Q.B.; Gallardo, H.; Liu, C.; Merghoub, T.; Hefter, B.; et al. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring (V600E)BRAF. Oncogene 2012, 31, 446–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesbacher, S.; Pfitzer, L.; Wiedorfer, K.; Angermeyer, S.; Borst, A.; Haferkamp, S.; Scholz, C.J.; Wobser, M.; Schrama, D.; Houben, R. RB1 is the crucial target of the Merkel cell polyomavirus Large T antigen in Merkel cell carcinoma cells. Oncotarget 2016, 7, 32956–32968. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Sato, Y.; Watanabe, D.; Ito, H.; Shimonohara, N.; Tsuji, T.; Nakajima, N.; Suzuki, Y.; Matsuo, K.; Nakagawa, H.; et al. Nuclear localization of Merkel cell polyomavirus large T antigen in Merkel cell carcinoma. Virology 2010, 398, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Dresang, L.R.; Guastafierro, A.; Arora, R.; Normolle, D.; Chang, Y.; Moore, P.S. Response of Merkel cell polyomavirus-positive merkel cell carcinoma xenografts to a survivin inhibitor. PLoS ONE 2013, 8, e80543. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Weaver, Z.; Linke, S.P.; Li, C.; Gotay, J.; Wang, X.W.; Harris, C.C.; Ried, T.; Deng, C.X. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol. Cell 1999, 3, 389–395. [Google Scholar] [CrossRef]
- Keyomarsi, K.; Pardee, A.B. Redundant cyclin overexpression and gene amplification in breast cancer cells. Proc. Natl. Acad. Sci. USA 1993, 90, 1112–1116. [Google Scholar] [CrossRef] [Green Version]
- Paulson, K.G.; Lemos, B.D.; Feng, B.; Jaimes, N.; Penas, P.F.; Bi, X.; Maher, E.; Cohen, L.; Leonard, J.H.; Granter, S.R.; et al. Array-CGH reveals recurrent genomic changes in Merkel cell carcinoma including amplification of L-Myc. J. Investig. Dermatol. 2009, 129, 1547–1555. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.W.; Wu, N.; Kim, Y.C.; Cheng, P.F.; Basom, R.; Kim, D.; Dunn, C.T.; Lee, A.Y.; Kim, K.; Lee, C.S.; et al. Genetic requirement for Mycl and efficacy of RNA Pol I inhibition in mouse models of small cell lung cancer. Genes Dev. 2016, 30, 1289–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Q.; Byrum, S.D.; Moreland, L.E.; Mackintosh, S.G.; Kannan, A.; Lin, Z.; Morgan, M.; Stack, B.C., Jr.; Cornelius, L.A.; Tackett, A.J.; et al. A proteomic study of human Merkel cell carcinoma. J. Proteom. Bioinform. 2013, 6, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; McDermott, A.; Shao, L.; Kannan, A.; Morgan, M.; Stack, B.C., Jr.; Moreno, M.; Davis, D.A.; Cornelius, L.A.; Gao, L. Chronic mTOR activation promotes cell survival in Merkel cell carcinoma. Cancer Lett. 2014, 344, 272–281. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.W.; Lee, M.C.; Hsu, N.Y.; Wu, T.C.; Wu, J.Y.; Wang, Y.C.; Cheng, Y.W.; Chen, C.Y.; Lee, H. FHIT loss confers cisplatin resistance in lung cancer via the AKT/NF-kappaB/Slug-mediated PUMA reduction. Oncogene 2015, 34, 3882–3883. [Google Scholar] [CrossRef] [Green Version]
- Semba, S.; Trapasso, F.; Fabbri, M.; McCorkell, K.A.; Volinia, S.; Druck, T.; Iliopoulos, D.; Pekarsky, Y.; Ishii, H.; Garrison, P.N.; et al. Fhit modulation of the Akt-survivin pathway in lung cancer cells: Fhit-tyrosine 114 (Y114) is essential. Oncogene 2006, 25, 2860–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiske, J.; Albring, K.F.; Huber, O. The tumor suppressor Fhit acts as a repressor of beta-catenin transcriptional activity. Proc. Natl. Acad. Sci. USA 2007, 104, 20344–20349. [Google Scholar] [CrossRef] [Green Version]
- Robertson, B.W.; Chellaiah, M.A. Osteopontin induces beta-catenin signaling through activation of Akt in prostate cancer cells. Exp. Cell. Res. 2010, 316, 1–11. [Google Scholar] [CrossRef] [Green Version]
- He, X.C.; Zhang, J.; Tong, W.G.; Tawfik, O.; Ross, J.; Scoville, D.H.; Tian, Q.; Zeng, X.; He, X.; Wiedemann, L.M.; et al. BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling. Nat. Genet. 2004, 36, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Masugi, Y.; Yamazaki, K.; Emoto, K.; Effendi, K.; Tsujikawa, H.; Kitago, M.; Itano, O.; Kitagawa, Y.; Sakamoto, M. Upregulation of integrin beta4 promotes epithelial-mesenchymal transition and is a novel prognostic marker in pancreatic ductal adenocarcinoma. Lab. Investig. 2015, 95, 308–319. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; de la Cruz, C.C.; Sayles, L.C.; Alleyne-Chin, C.; Vaka, D.; Knaak, T.D.; Bigos, M.; Xu, Y.; Hoang, C.D.; Shrager, J.B.; et al. A rare population of CD24(+)ITGB4(+)Notch(hi) cells drives tumor propagation in NSCLC and requires Notch3 for self-renewal. Cancer Cell 2013, 24, 59–74. [Google Scholar] [CrossRef] [Green Version]
- nanoString, Analyzing FFPE Specimens with the nCounter® Copy Number Variation (CNV) Assay. 2019. Available online: http://108.166.79.23/products/CNV, nanoString Tech, https://www.nanostring.com/wp-content/uploads/2020/12/TN_MK1113_FFPE_CNV_R3.pdf (accessed on 16 February 2021).
- Do, H.; Dobrovic, A. Sequence artifacts in DNA from formalin-fixed tissues: Causes and strategies for minimization. Clin. Chem. 2015, 61, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakubek, Y.A.; Chang, K.; Sivakumar, S.; Yu, Y.; Giordano, M.R.; Fowler, J.; Huff, C.D.; Kadara, H.; Vilar, E.; Scheet, P. Large-scale analysis of acquired chromosomal alterations in non-tumor samples from patients with cancer. Nat. Biotechnol. 2020, 38, 90–96. [Google Scholar] [CrossRef]
- Ronan, S.G.; Green, A.D.; Shilkaitis, A.; Huang, T.S.; Das Gupta, T.K. Merkel cell carcinoma: In vitro and in vivo characteristics of a new cell line. J. Am. Acad. Dermatol. 1993, 29, 715–722. [Google Scholar] [CrossRef]
- Rosen, S.T.; Gould, V.E.; Salwen, H.R.; Herst, C.V.; Le Beau, M.M.; Lee, I.; Bauer, K.; Marder, R.J.; Andersen, R.; Kies, M.S.; et al. Establishment and characterization of a neuroendocrine skin carcinoma cell line. Lab. Investig. 1987, 56, 302–312. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
| MCC Sample | Sex | MCPyV Status | Age | Site of MCC | Specimen Code | Cluster | Tissue Source |
|---|---|---|---|---|---|---|---|
| MF1 | Female | Negative | 68 | right upper arm | primary | Del | FFPE |
| MF2 | Male | Negative | 72 | left hand | primary | Del | FFPE |
| MF3 | Female | Negative | 80 | right gluteal | primary | Del | FFPE |
| MF4 | Male | Negative | 64 | abdominal wall | primary | Del | FFPE |
| MF5 | Male | Negative | 89 | left ala of nose | primary | Del | FFPE |
| MF6 | Male | Negative | - | frontal scalp | primary | Amp | FFPE |
| MF7 | Female | Negative | 94 | right scalp | primary | Del | FFPE |
| MF8 | Female | Negative | - | lymph node | metastasis | Del | FFPE |
| MF9 | Male | Positive | 58 | left thigh | primary | Del | FFPE |
| MF10 | Male | Positive | 67 | left index finger | primary | Del | FFPE |
| MF11 | Male | Negative | 72 | left cheek | primary | Del | FFPE |
| MF12 | Male | Positive | - | right neck | primary | Del | FFPE |
| MF13 | Female | Negative | - | right leg | primary | Del | FFPE |
| MF14 | Female | Negative | 100 | right forehead | primary | Del | FFPE |
| MF15 | Male | Negative | 93 | left cheek, nose | primary | Amp | FFPE |
| MF16 | Female | Negative | 74 | left buttock | primary | Del | FFPE |
| MF18 | Female | Negative | - | right forearm | primary | Del | FFPE |
| MF19 | Male | Negative | 77 | right face | metastasis | Del | FFPE |
| MF20 | Male | Positive | 75 | top of head | primary | Del | FFPE |
| MF21 | Male | Negative | 87 | right wrist | primary | Amp | FFPE |
| MF22 | Female | Negative | 88 | forehead | primary | Amp | FFPE |
| MF23 | Male | Positive | 81 | left cheek | primary | Del | FFPE |
| UP1 | Female | Positive | 75 | left brow | - | Amp | FFPE |
| MSK1 | Female | Positive | 80 | lymph nodes | metastasis | Low | Frozen |
| MSK2 | Male | Positive | 73 | pancreas | metastasis | Low | Frozen |
| MSK6 | Male | Positive | 53 | groin | metastasis | Low | Frozen |
| MSK13 | Female | Positive | 62 | skin | primary | Low | Frozen |
| MSK19 | Male | Positive | 59 | skin | primary | Low | Frozen |
| MSK20 | Female | Positive | 63 | skin | primary | Low | Frozen |
| MSK21 | Male | Positive | 87 | skin | primary | Low | Frozen |
| MSK24 | Male | Positive | 82 | lymph nodes | metastasis | Low | Frozen |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hill, N.T.; Kim, D.; Busam, K.J.; Chu, E.Y.; Green, C.; Brownell, I. Distinct Signatures of Genomic Copy Number Variants Define Subgroups of Merkel Cell Carcinoma Tumors. Cancers 2021, 13, 1134. https://doi.org/10.3390/cancers13051134
Hill NT, Kim D, Busam KJ, Chu EY, Green C, Brownell I. Distinct Signatures of Genomic Copy Number Variants Define Subgroups of Merkel Cell Carcinoma Tumors. Cancers. 2021; 13(5):1134. https://doi.org/10.3390/cancers13051134
Chicago/Turabian StyleHill, Natasha T., David Kim, Klaus J. Busam, Emily Y. Chu, Clayton Green, and Isaac Brownell. 2021. "Distinct Signatures of Genomic Copy Number Variants Define Subgroups of Merkel Cell Carcinoma Tumors" Cancers 13, no. 5: 1134. https://doi.org/10.3390/cancers13051134
APA StyleHill, N. T., Kim, D., Busam, K. J., Chu, E. Y., Green, C., & Brownell, I. (2021). Distinct Signatures of Genomic Copy Number Variants Define Subgroups of Merkel Cell Carcinoma Tumors. Cancers, 13(5), 1134. https://doi.org/10.3390/cancers13051134