
Bifidobacterium Strain-Specific Enhances the Efficacy of Cancer Therapeutics in Tumor-Bearing Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Cell Culture
2.3. Bacteria
2.4. Flow Cytometry Analysis
2.5. Quantitative PCR (qPCR) Analysis of Tumor Tissue
2.6. Mice RNA Sequencing and Data Analysis
2.7. Bacterial RNA Sequencing and Data Analysis
2.8. Statistical Analysis
3. Results
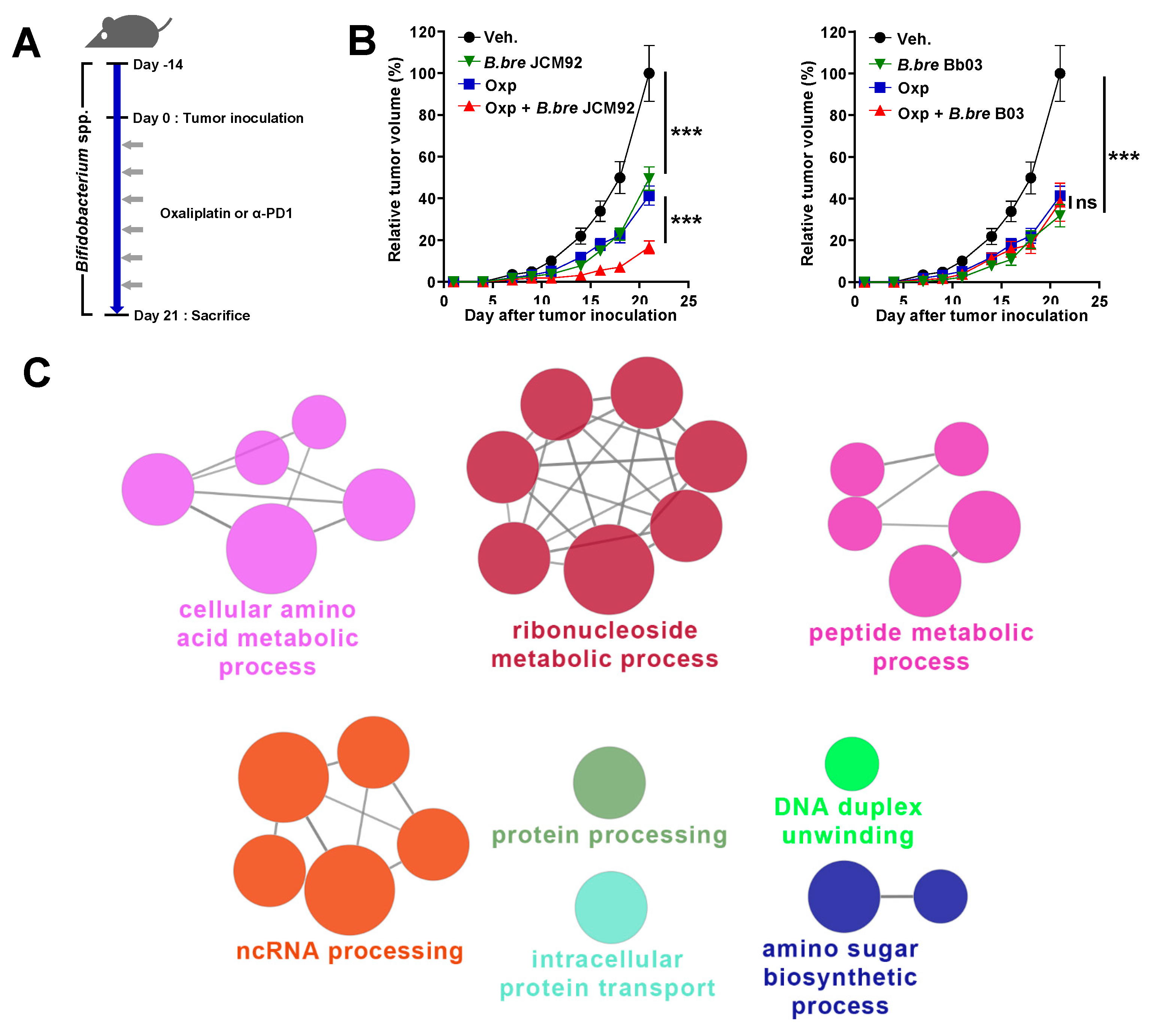
3.1. Mice Supplemented with B. breve Strains Exhibit Anti-Tumor Effects
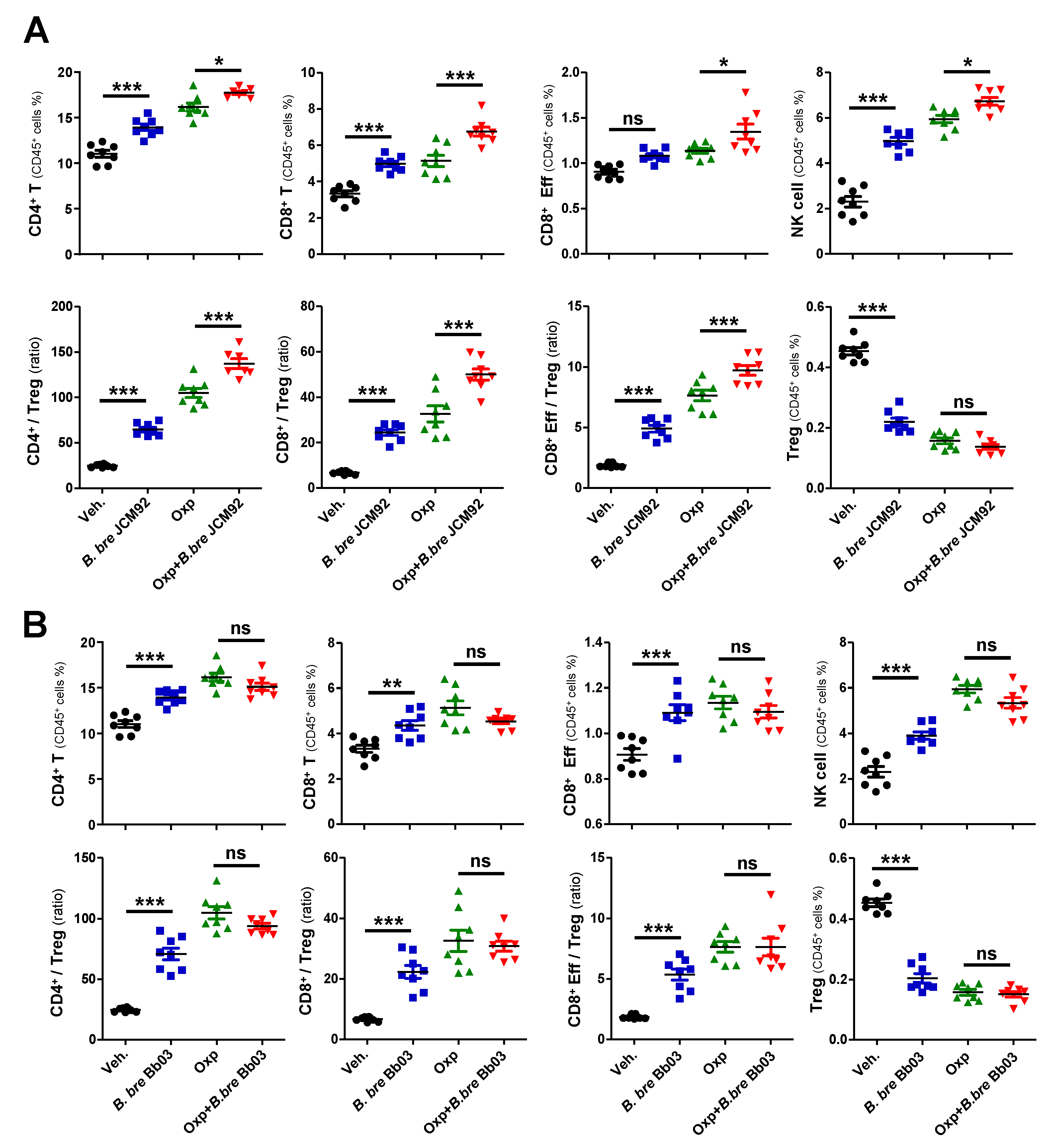
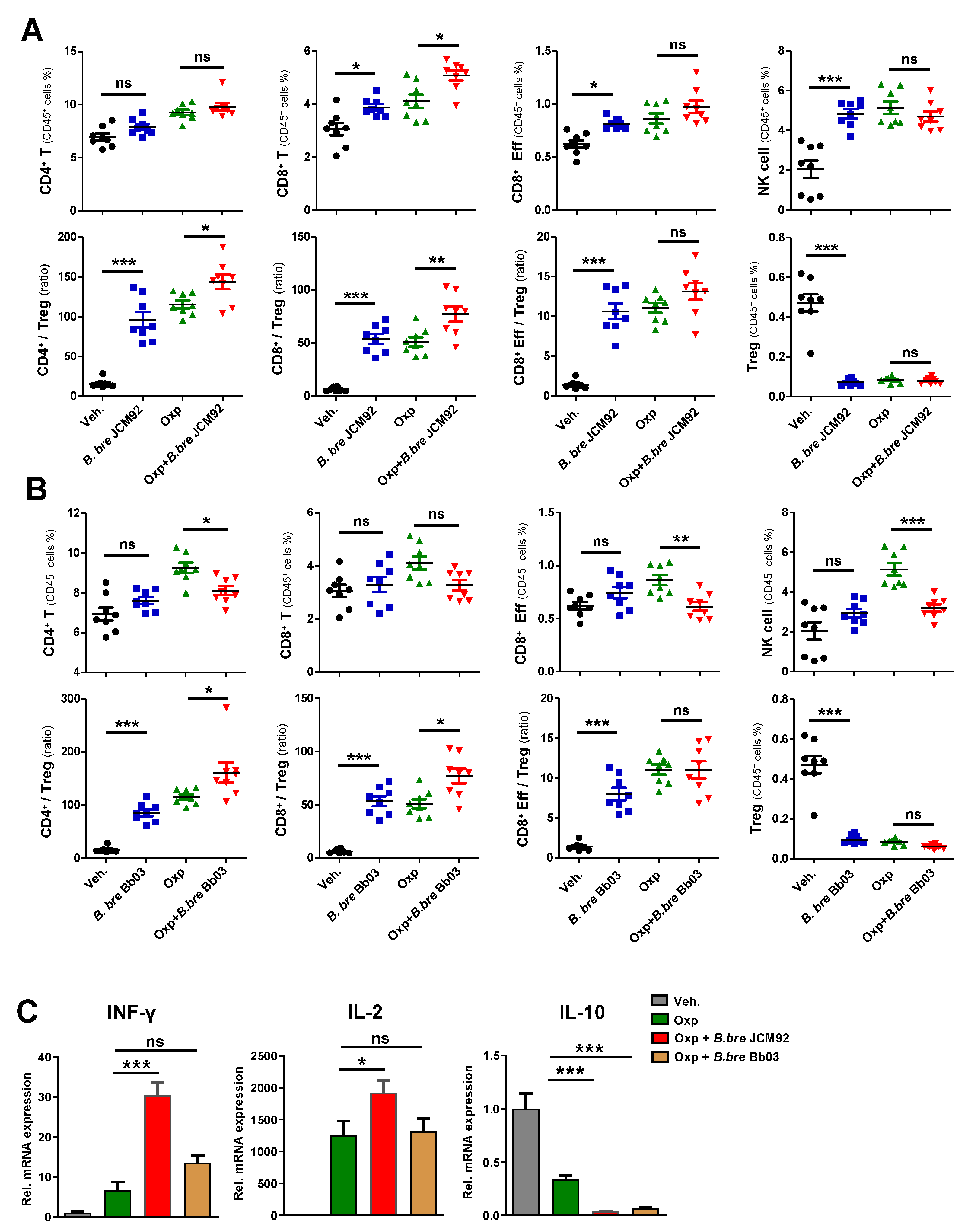
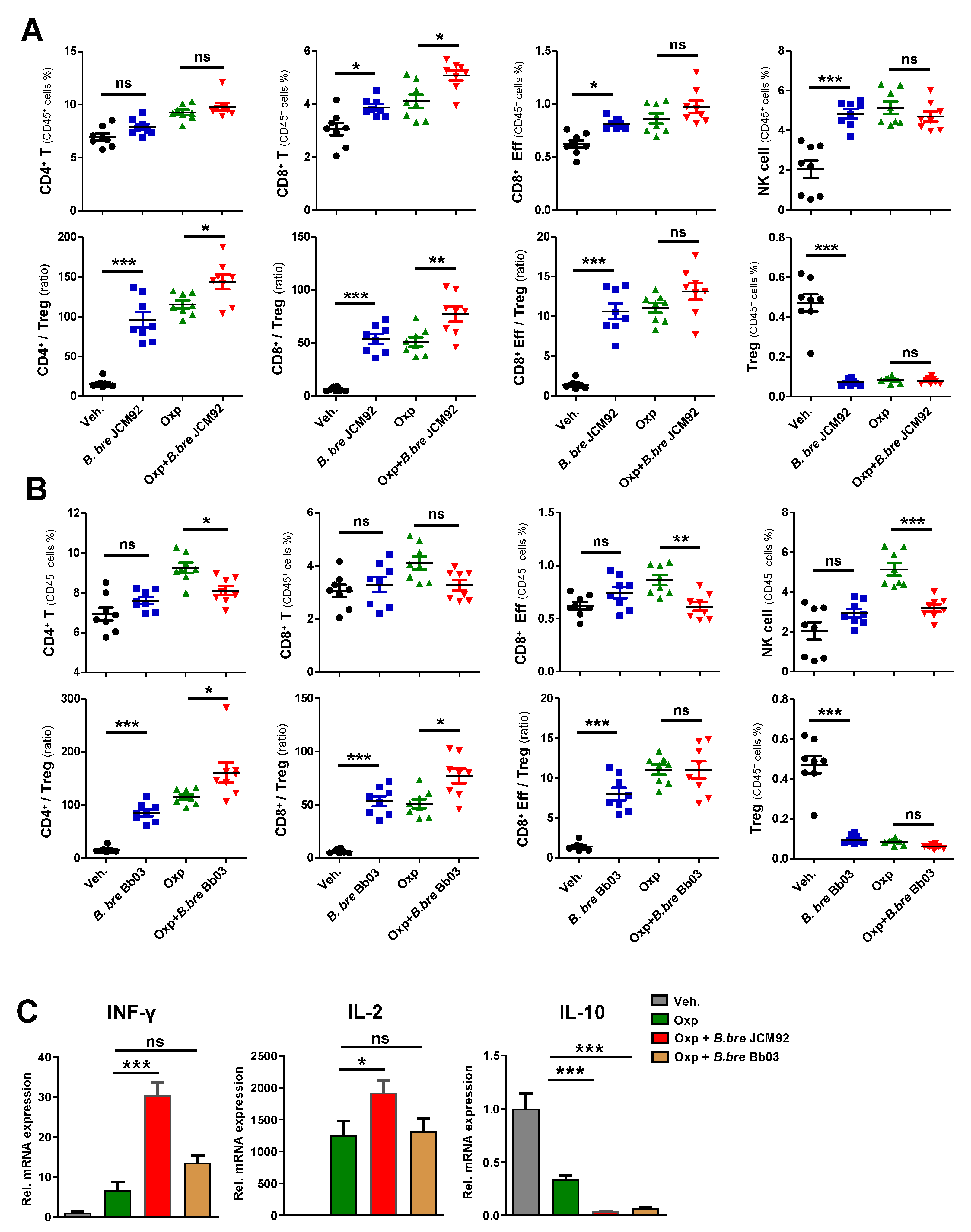
3.2. B. bre JCM92 Boosts the Efficacy of Oxaliplatin by Enhancing Anti-Tumor Immunity
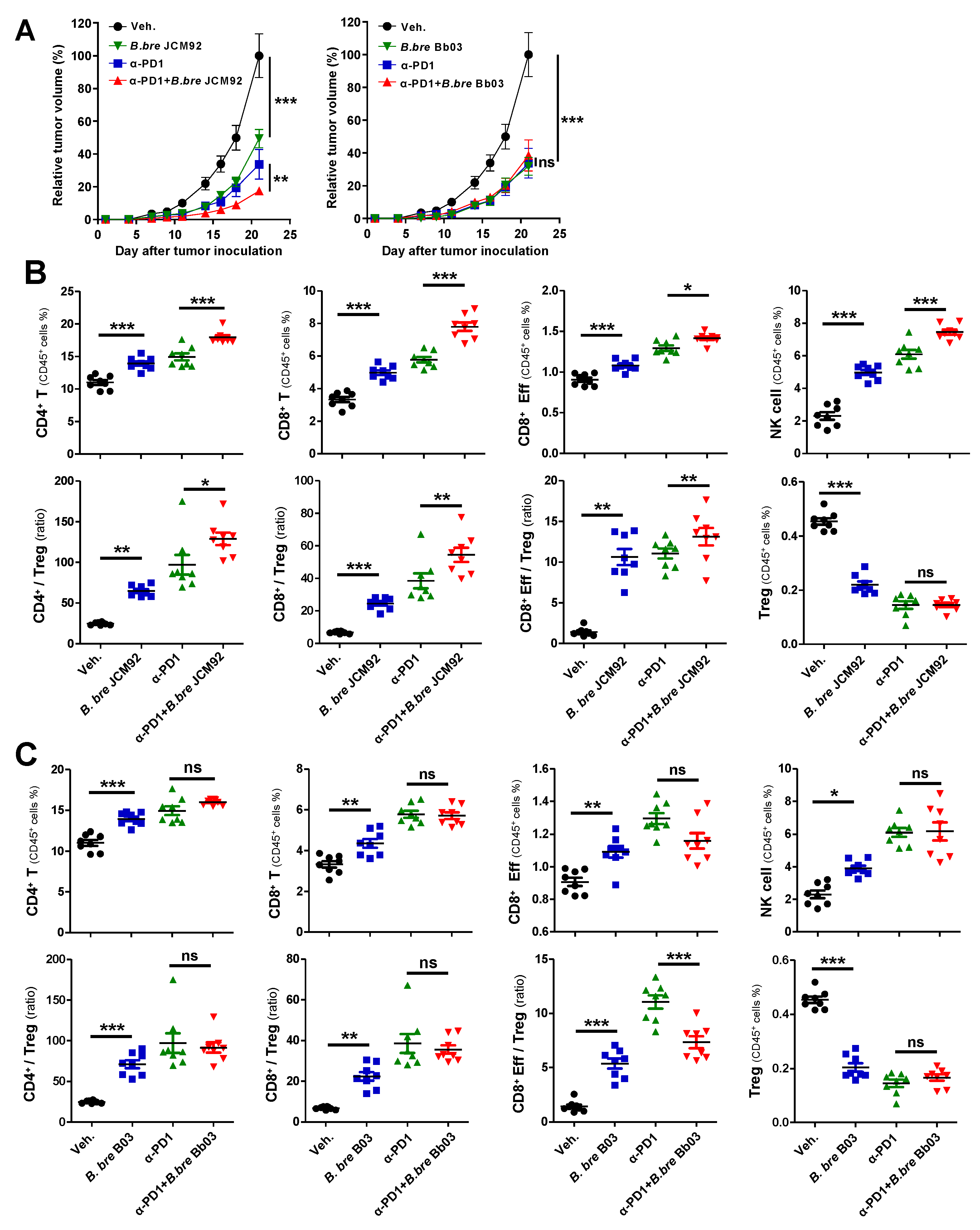
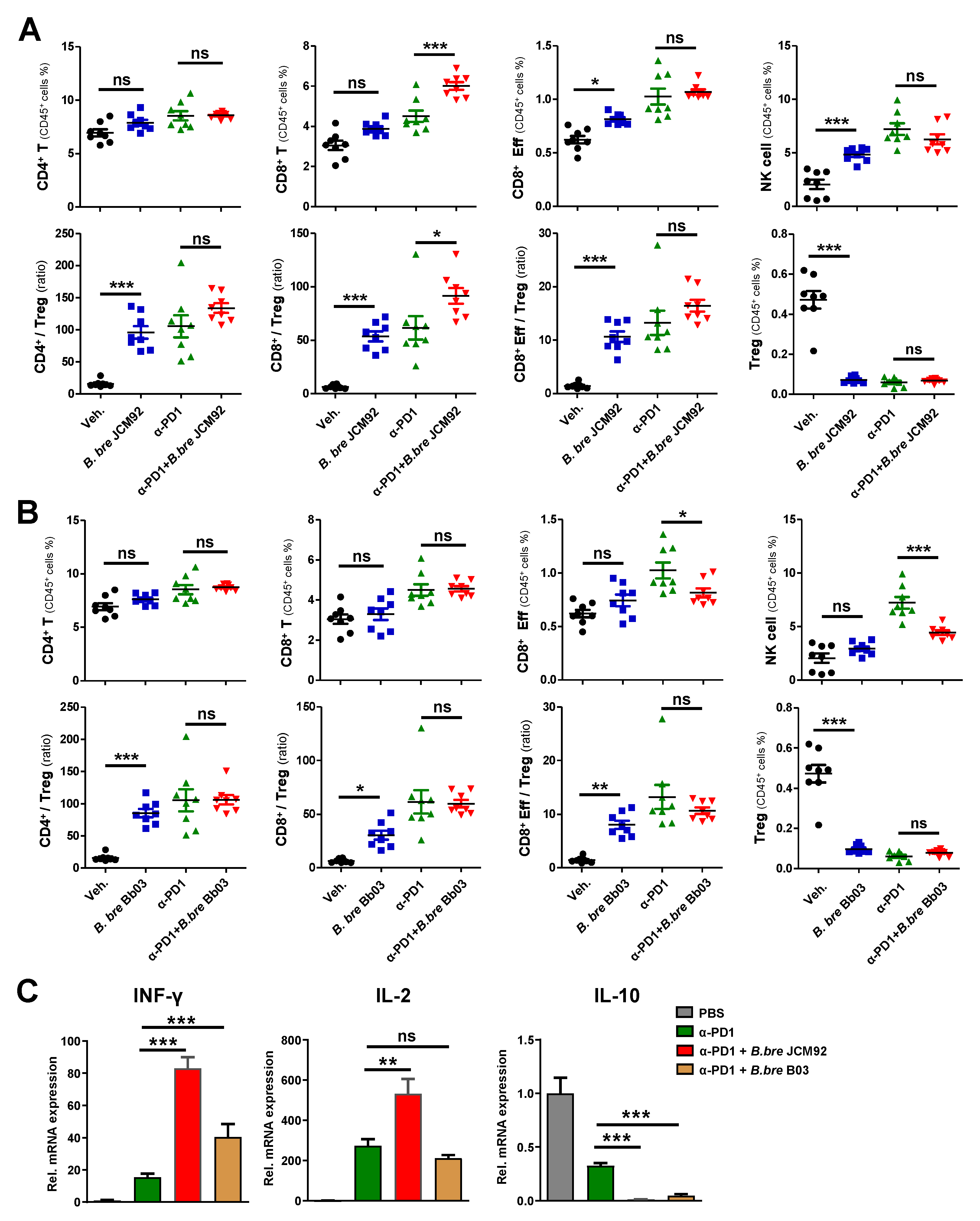
3.3. B. bre JCM92 Boosts the Efficacy of PD-1 Blockade by Enhancing Anti-Tumor Immunity
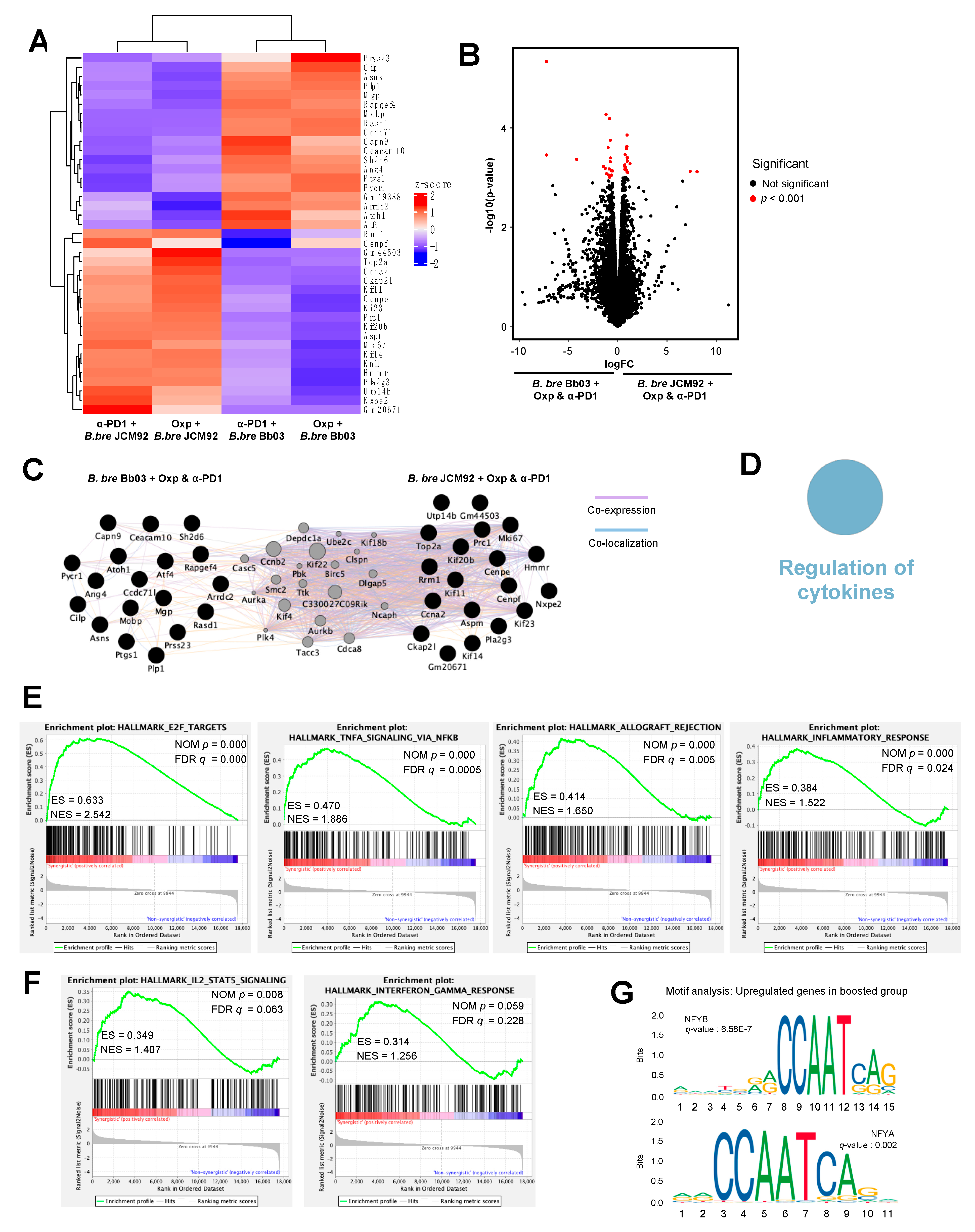
3.4. Transcriptome Analysis of Intestinal Tissue Reveals That B. bre JCM92 Has the Ability to Boost the Effects of Cancer Therapeutics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Keefe, S.J. Nutrition and colonic health: The critical role of the microbiota. Curr. Opin. Gastroenterol. 2008, 24, 51–58. [Google Scholar] [CrossRef]
- Kundu, P.; Blacher, E.; Elinav, E.; Pettersson, S. Our gut microbiome: The evolving inner self. Cell 2017, 171, 1481–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejea, C.M.; Wick, E.C.; Hechenbleikner, E.M.; White, J.R.; Welch, J.L.M.; Rossetti, B.J.; Peterson, S.N.; Snesrud, E.C.; Borisy, G.G.; Lazarev, M. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 18321–18326. [Google Scholar] [CrossRef] [Green Version]
- Mima, K.; Sukawa, Y.; Nishihara, R.; Qian, Z.R.; Yamauchi, M.; Inamura, K.; Kim, S.A.; Masuda, A.; Nowak, J.A.; Nosho, K. Fusobacterium nucleatum and T cells in colorectal carcinoma. Jama Oncol. 2015, 1, 653–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA A Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejea, C.M.; Fathi, P.; Craig, J.M.; Boleij, A.; Taddese, R.; Geis, A.L.; Wu, X.; Shields, C.E.D.; Hechenbleikner, E.M.; Huso, D.L. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 2018, 359, 592–597. [Google Scholar] [CrossRef] [Green Version]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Adolph, T.E.; Gerner, R.R.; Moschen, A.R. The intestinal microbiota in colorectal cancer. Cancer Cell 2018, 33, 954–964. [Google Scholar] [CrossRef] [Green Version]
- Daillère, R.; Vétizou, M.; Waldschmitt, N.; Yamazaki, T.; Isnard, C.; Poirier-Colame, V.; Duong, C.P.; Flament, C.; Lepage, P.; Roberti, M.P. Enterococcus hirae and Barnesiella intestinihominis facilitate cyclophosphamide-induced therapeutic immunomodulatory effects. Immunity 2016, 45, 931–943. [Google Scholar] [CrossRef] [Green Version]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti–PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrieta, M.-C.; Stiemsma, L.T.; Amenyogbe, N.; Brown, E.M.; Finlay, B. The intestinal microbiome in early life: Health and disease. Front. Immunol. 2014, 5, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuki, T.; Watanabe, K.; Tanaka, R.; Fukuda, M.; Oyaizu, H. Distribution of bifidobacterial species in human intestinal microflora examined with 16S rRNA-gene-targeted species-specific primers. Appl. Environ. Microbiol. 1999, 65, 4506–4512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, F.; Ouwehand, A.C.; Isolauri, E.; Hashimoto, H.; Benno, Y.; Salminen, S. Comparison of mucosal adhesion and species identification of bifidobacteria isolated from healthy and allergic infants. Fems Immunol. Med Microbiol. 2001, 30, 43–47. [Google Scholar] [CrossRef]
- Patole, S.K.; Rao, S.C.; Keil, A.D.; Nathan, E.A.; Doherty, D.A.; Simmer, K.N. Benefits of Bifidobacterium breve M-16V supplementation in preterm neonates-a retrospective cohort study. PLoS ONE 2016, 11, e0150775. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, S.E.; Tobin, J.M.; Opie, G.F.; Donath, S.; Tabrizi, S.N.; Pirotta, M.; Morley, C.J.; Garland, S.M. Probiotic effects on late-onset sepsis in very preterm infants: A randomized controlled trial. Pediatrics 2013, 132, 1055–1062. [Google Scholar] [CrossRef] [Green Version]
- Ling, X.; Linglong, P.; Weixia, D.; Hong, W. Protective effects of bifidobacterium on intestinal barrier function in LPS-induced enterocyte barrier injury of Caco-2 monolayers and in a rat NEC model. PLoS ONE 2016, 11, e0161635. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- André, T.; Boni, C.; Mounedji-Boudiaf, L.; Navarro, M.; Tabernero, J.; Hickish, T.; Topham, C.; Zaninelli, M.; Clingan, P.; Bridgewater, J. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N. Engl. J. Med. 2004, 350, 2343–2351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Gramont, A.d.; Figer, A.; Seymour, M.; Homerin, M.; Hmissi, A.; Cassidy, J.; Boni, C.; Cortes-Funes, H.; Cervantes, A.; Freyer, G. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J. Clin. Oncol. 2000, 18, 2938–2947. [Google Scholar] [CrossRef] [PubMed]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darnell, J.E. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740–749. [Google Scholar] [CrossRef]
- Lee, T.I.; Young, R.A. Transcriptional regulation and its misregulation in disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [Green Version]
- Habib, N.; Kaplan, T.; Margalit, H.; Friedman, N. A novel Bayesian DNA motif comparison method for clustering and retrieval. PLoS Comput. Biol. 2008, 4, e1000010. [Google Scholar] [CrossRef] [Green Version]
- Li-Weber, M.; Davydov, I.V.; Krafft, H.; Krammer, P.H. The role of NF-Y and IRF-2 in the regulation of human IL-4 gene expression. J. Immunol. 1994, 153, 4122–4133. [Google Scholar]
- Szabo, S.J.; Gold, J.S.; Murphy, T.L.; Murphy, K.M. Identification of cis-acting regulatory elements controlling interleukin-4 gene expression in T cells: Roles for NF-Y and NF-ATc. Mol. Cell. Biol. 1993, 13, 4793–4805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wills-Karp, M.; Finkelman, F.D. Untangling the complex web of IL-4–and IL-13–mediated signaling pathways. Sci. Signal. 2008, 1, pe55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Chen, L.; Qin, Z. Paradoxical roles of IL-4 in tumor immunity. Cell. Mol. Immunol. 2009, 6, 415–422. [Google Scholar] [CrossRef]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Xuan, C.; Shamonki, J.M.; Chung, A.; DiNome, M.L.; Chung, M.; Sieling, P.A.; Lee, D.J. Microbial dysbiosis is associated with human breast cancer. PLoS ONE 2014, 9, e83744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavasani, S.; Dzhambazov, B.; Nouri, M.; Fåk, F.; Buske, S.; Molin, G.; Thorlacius, H.; Alenfall, J.; Jeppsson, B.; Weström, B. A novel probiotic mixture exerts a therapeutic effect on experimental autoimmune encephalomyelitis mediated by IL-10 producing regulatory T cells. PLoS ONE 2010, 5, e9009. [Google Scholar] [CrossRef]
- De Oliveira, G.L.V.; Leite, A.Z.; Higuchi, B.S.; Gonzaga, M.I.; Mariano, V.S. Intestinal dysbiosis and probiotic applications in autoimmune diseases. Immunology 2017, 152, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Toh, H.; Hase, K.; Oshima, K.; Nakanishi, Y.; Yoshimura, K.; Tobe, T.; Clarke, J.M.; Topping, D.L.; Suzuki, T. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 2011, 469, 543–547. [Google Scholar] [CrossRef]
- Duranti, S.; Gaiani, F.; Mancabelli, L.; Milani, C.; Grandi, A.; Bolchi, A.; Santoni, A.; Lugli, G.A.; Ferrario, C.; Mangifesta, M. Elucidating the gut microbiome of ulcerative colitis: Bifidobacteria as novel microbial biomarkers. Fems Microbiol. Ecol. 2016, 92, fiw191. [Google Scholar] [CrossRef] [Green Version]
- Hadrup, S.; Donia, M.; Thor Straten, P. Effector CD4 and CD8 T cells and their role in the tumor microenvironment. Cancer Microenviron. 2013, 6, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Bronte, V.; Pittet, M.J. The spleen in local and systemic regulation of immunity. Immunity 2013, 39, 806–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.N.; Walker, A. Bacterial colonization and the development of intestinal defences. Can. J. Gastroenterol. 2004, 18, 493–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, W.A. The importance of appropriate initial bacterial colonization of the intestine in newborn, child, and adult health. Pediatric Res. 2017, 82, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishnan, V.; Spencer, C.; Nezi, L.; Reuben, A.; Andrews, M.; Karpinets, T.; Prieto, P.; Vicente, D.; Hoffman, K.; Wei, S. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Kent, L.N.; Leone, G. The broken cycle: E2F dysfunction in cancer. Nat. Rev. Cancer 2019, 19, 326–338. [Google Scholar] [CrossRef]
- Chen, H.-Z.; Tsai, S.-Y.; Leone, G. Emerging roles of E2Fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Dyson, N.J. RB1: A prototype tumor suppressor and an enigma. Genes Dev. 2016, 30, 1492–1502. [Google Scholar] [CrossRef] [Green Version]
- Müller, H.; Helin, K. The E2F transcription factors: Key regulators of cell proliferation. Biochim. Et Biophys. Acta (Bba)-Rev. Cancer 2000, 1470, M1–M12. [Google Scholar]
- Kent, L.N.; Rakijas, J.B.; Pandit, S.K.; Westendorp, B.; Chen, H.-Z.; Huntington, J.T.; Tang, X.; Bae, S.; Srivastava, A.; Senapati, S. E2f8 mediates tumor suppression in postnatal liver development. J. Clin. Investig. 2016, 126, 2955–2969. [Google Scholar] [CrossRef]
- Van Horssen, R.; Ten Hagen, T.L.; Eggermont, A.M. TNF-α in cancer treatment: Molecular insights, antitumor effects, and clinical utility. Oncologist 2006, 11, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef] [Green Version]
- Roberts, N.J.; Zhou, S.; Diaz Jr, L.A.; Holdhoff, M. Systemic use of tumor necrosis factor alpha as an anticancer agent. Oncotarget 2011, 2, 739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, R.B.; Isaacs, J.T.; Coffey, D.S. Tumor necrosis factor enhances the in vitro and in vivo efficacy of chemotherapeutic drugs targeted at DNA topoisomerase II in the treatment of murine bladder cancer. J. Urol. 1987, 138, 427–429. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Eessalu, T.E.; Hass, P.E. Characterization of receptors for human tumour necrosis factor and their regulation by γ-interferon. Nature 1985, 318, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.E.; Kitagawa, M.; Su, W.-C.S.; You, Z.-H.; Iwamoto, Y.; Fu, X.-Y. Cell growth arrest and induction of cyclin-dependent kinase inhibitor p21WAF1/CIP1 mediated by STAT1. Science 1996, 272, 719–722. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Horvath, C.M.; Wen, Z.; Schreiber, R.D.; Darnell, J.E. Transcriptionally active Stat1 is required for the antiproliferative effects of both interferon alpha and interferon gamma. Proc. Natl. Acad. Sci. USA 1996, 93, 7673–7678. [Google Scholar] [CrossRef] [Green Version]
- Chin, Y.E.; Kitagawa, M.; Kuida, K.; Flavell, R.A.; Fu, X.-Y. Activation of the STAT signaling pathway can cause expression of caspase 1 and apoptosis. Mol. Cell. Biol. 1997, 17, 5328–5337. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Fu, X.-Y.; Plate, J.; Chong, A.S. IFN-γ induces cell growth inhibition by Fas-mediated apoptosis: Requirement of STAT1 protein for up-regulation of Fas and FasL expression. Cancer Res. 1998, 58, 2832–2837. [Google Scholar]
- Groom, J.R.; Luster, A.D. CXCR3 ligands: Redundant, collaborative and antagonistic functions. Immunol. Cell Biol. 2011, 89, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Melero, I.; Rouzaut, A.; Motz, G.T.; Coukos, G. T-cell and NK-cell infiltration into solid tumors: A key limiting factor for efficacious cancer immunotherapy. Cancer Discov. 2014, 4, 522–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celada, A.; Gray, P.W.; Rinderknecht, E.; Schreiber, R. Evidence for a gamma-interferon receptor that regulates macrophage tumoricidal activity. J. Exp. Med. 1984, 160, 55–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kursunel, M.A.; Esendagli, G. The untold story of IFN-γ in cancer biology. Cytokine Growth Factor Rev. 2016, 31, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Lu, J. Interferon gamma in cancer immunotherapy. Cancer Med. 2018, 7, 4509–4516. [Google Scholar] [CrossRef]
- Campanella, G.S.; Colvin, R.A.; Luster, A.D. CXCL10 can inhibit endothelial cell proliferation independently of CXCR3. PLoS ONE 2010, 5, e12700. [Google Scholar] [CrossRef]
- Feldman, E.D.; Weinreich, D.M.; Carroll, N.M.; Burness, M.L.; Feldman, A.L.; Turner, E.; Xu, H.; Alexander, H.R. Interferon γ–inducible protein 10 selectively inhibits proliferation and induces apoptosis in endothelial cells. Ann. Surg. Oncol. 2006, 13, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 2016, 167, 397–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N. Identification of essential genes for cancer immunotherapy. Nature 2017, 548, 537. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, Y.; Kim, G.; Jeon, B.-N.; Fang, S.; Park, H. Bifidobacterium Strain-Specific Enhances the Efficacy of Cancer Therapeutics in Tumor-Bearing Mice. Cancers 2021, 13, 957. https://doi.org/10.3390/cancers13050957
Yoon Y, Kim G, Jeon B-N, Fang S, Park H. Bifidobacterium Strain-Specific Enhances the Efficacy of Cancer Therapeutics in Tumor-Bearing Mice. Cancers. 2021; 13(5):957. https://doi.org/10.3390/cancers13050957
Chicago/Turabian StyleYoon, Youngmin, Gihyeon Kim, Bu-Nam Jeon, Sungsoon Fang, and Hansoo Park. 2021. "Bifidobacterium Strain-Specific Enhances the Efficacy of Cancer Therapeutics in Tumor-Bearing Mice" Cancers 13, no. 5: 957. https://doi.org/10.3390/cancers13050957
APA StyleYoon, Y., Kim, G., Jeon, B.-N., Fang, S., & Park, H. (2021). Bifidobacterium Strain-Specific Enhances the Efficacy of Cancer Therapeutics in Tumor-Bearing Mice. Cancers, 13(5), 957. https://doi.org/10.3390/cancers13050957