
Anti-Tumor Effects of a Penetratin Peptide Targeting Transcription of E2F-1, 2 and 3a Is Enhanced When Used in Combination with Pemetrexed or Cisplatin

 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. D-Arg PEP in Combination with Cisplatin
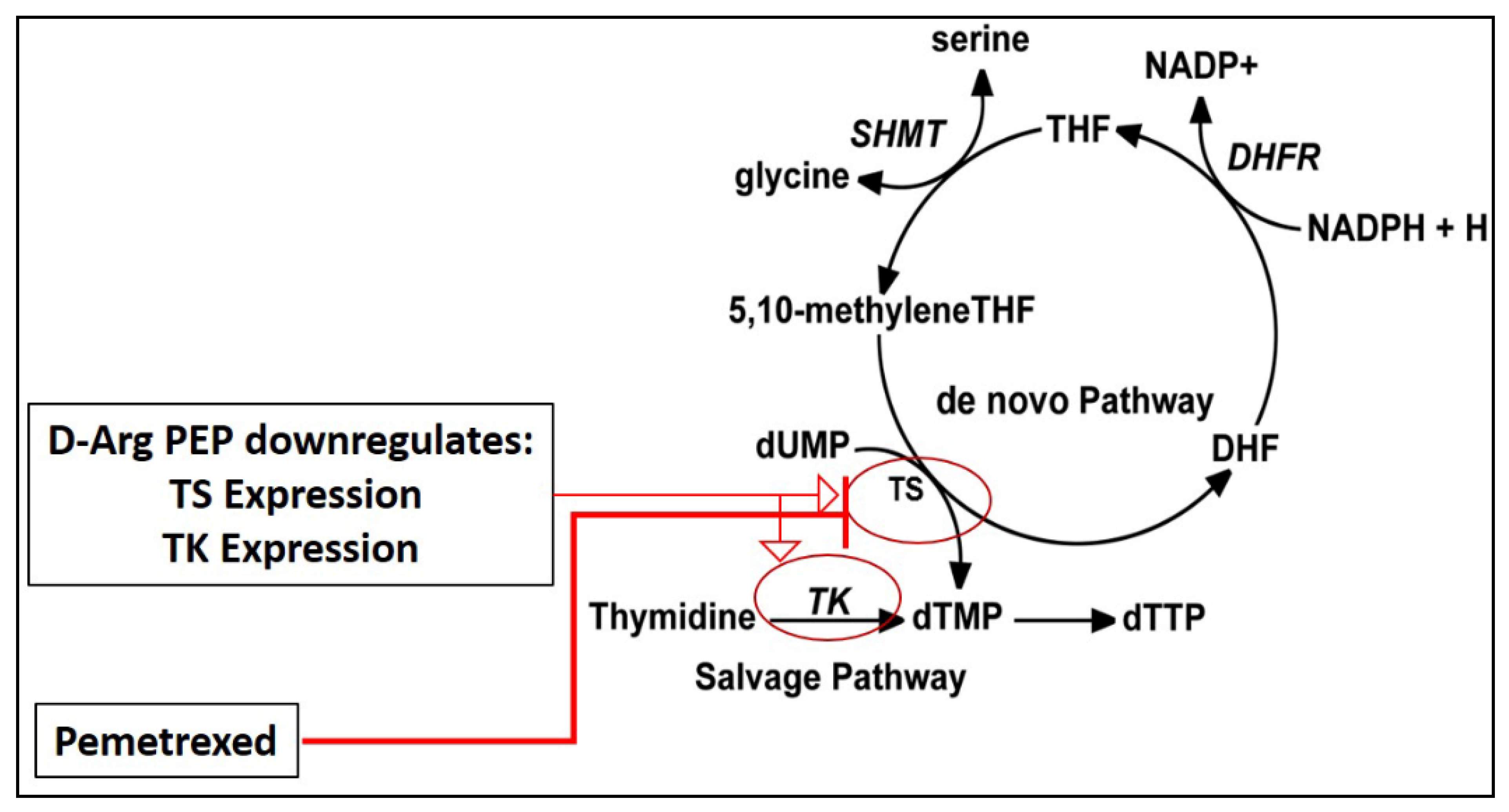
2.2. D-Arg PEP Showed a Marked Synergistic Anti-Cancer Effect with Pemetrexed against NSCLC Cell Lines
2.3. In-Vivo PL-PEP Combination Study with Pemetrexed
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cell Lines and Chemical Compounds
4.3. Cell Viability Assays
4.4. In-Vitro Combination Drug Treatments
4.5. Immunofluorescence Assay
4.6. Western Blotting
4.7. Preparation and Characterization of Liposomes
4.8. Loading Efficiency and Content of D-Arg PEP in the PEGylated Liposomes
4.9. D-Arg PEP Release Profile from PEGylated Liposomes
4.10. H2009 Xenograft Study
4.11. Histologic Preparation and Immunohistochemistry Staining
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ren, B.; Cam, H.; Takahashi, Y.; Volkert, T.; Terragni, J.; Young, R.A.; Dynlacht, B.D. E2F integrates cell cycle progression with DNA repair, replication, and G2/M checkpoints. Genes Dev. 2002, 16, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Z.; Tsai, S.Y.; Leone, G. Emerging roles of E2Fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammens, T.; Li, J.; Leone, G.; De Veylder, L. Atypical E2Fs: New players in the E2F transcription factor family. Trends Cell Biol. 2009, 19, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Hallstrom, T.C.; Mori, S.; Nevins, J.R. An E2F1-Dependent gene expression program that determines the balance between proliferation and cell death. Cancer Cell 2008, 13, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Engelmann, D.; Pützer, B.M. The dark side of E2F1: In transit beyond apoptosis. Cancer Res. 2012, 72, 571–575. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Harbour, J.W.; Dean, D.C. Rb function in cell-cycle regulation and apoptosis. Nat. Cell Biol. 2000, 2, E65–E67. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998, 12, 2245–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevins, J.R. The Rb/E2F pathway and cancer. Hum. Mol. Genet. 2001, 10, 699–703. [Google Scholar] [CrossRef]
- Gordon, G.M.; Du, W. Conserved RB functions in development and tumor suppression. Protein Cell 2011, 2, 864–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, M.; Banerjee, D.; Menon, L.G.; Jurkiewicz, A.; Rao, P.H.; Kemeny, N.E.; Fong, Y.; Jhanwar, S.C.; Gorlick, R.; Bertino, J.R. Overexpression of E2F-1 in lung and liver metastases of human colon cancer is associated with gene amplification. Cancer Biol. Ther. 2004, 3, 395–399. [Google Scholar] [CrossRef] [Green Version]
- Gorgoulis, V.G.; Zacharatos, P.; Mariatos, G.; Kotsinas, A.; Bouda, M.; Kletsas, D.; Asimacopoulos, P.J.; Agnantis, N.; Kittas, C.; Papavassiliou, A.G. Transcriptional factor E2F-1 acts as a growth-promoting factor and is associated with adverse prognosis in non-small cell lung carcinomas. J. Pathol. 2002, 198, 142–156. [Google Scholar] [CrossRef]
- Bertino, J.R.; Banerjee, D. E2F-1 as an anticancer drug target. Oncol. Rev. 2009, 3, 207–214. [Google Scholar] [CrossRef]
- Weinstein, I.B. Cancer. Addiction to Oncogenes--the Achilles Heal of Cancer. Science 2002, 297, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.M.; Alemany, R.; Fueyo, J.; Gomez-Manzano, C. E2F1 in gliomas: A paradigm of oncogene addiction. Cancer Lett. 2008, 263, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Knudsen, K.E. Tailoring to RB: Tumour suppressor status and therapeutic response. Nat. Rev. Cancer 2008, 8, 714–724. [Google Scholar] [CrossRef]
- Wu, L.; Timmers, C.; Maiti, B.; Saavedra, H.I.; Sang, L.; Chong, G.T.; Nuckolls, F.; Giangrande, P.; Wright, F.A.; Field, S.J.; et al. The E2F1-3 transcription factors are essential for cellular proliferation. Nature 2001, 414, 457–462. [Google Scholar] [CrossRef]
- Xie, X.; Kerrigan, J.E.; Minko, T.; Garbuzenko, O.; Lee, K.C.; Scarborough, A.; Abali, E.E.; Budak-Alpdogan, T.; Johnson-Farley, N.; Banerjee, D.; et al. Antitumor and modeling studies of a penetratin-peptide that targets E2F-1 in small cell lung cancer. Cancer Biol. Ther. 2013, 14, 742–751. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Bansal, N.; Shaik, T.; Kerrigan, J.E.; Minko, T.; Garbuzenko, O.; Abali, E.E.; Johnson-Farley, N.; Banerjee, D.; Scotto, K.W.; et al. A novel peptide that inhibits E2F transcription and regresses prostate tumor xenografts. Oncotarget 2014, 5, 901–907. [Google Scholar] [CrossRef] [Green Version]
- Shaik, T.; Rather, G.M.; Bansal, N.; Minko, T.; Garbuzenko, O.; Szekely, Z.; Abali, E.E.; Banerjee, D.; Kerrigan, J.E.; Scotto, K.W.; et al. Modeling and antitumor studies of a modified L–penetratin peptide targeting E2F in lung cancer and prostate cancer. Oncotarget 2018, 9, 33249–33257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, C.; La Thangue, N.B. The emerging role of E2F-1 in the DNA damage response and checkpoint control. DNA Repair 2004, 3, 1071–1079. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; MenckI, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics (Sao Paulo) 2018, 73 (Suppl. 1), e478s. [Google Scholar] [CrossRef]
- Taubert, S.; Gorrini, C.; Frank, S.R.; Parisi, T.; Fuchs, M.; Chan, H.M.; Livingston, D.M.; Amati, B. E2F-dependent histone acetylation and recruitment of the Tip60 acetyltransferase complex to chromatin in late G1. Mol. Cell Biol. 2004, 24, 4546–4556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putzer, B.M.; Engelmann, D. E2F1 apoptosis counterattacked: Evil strikes back. Trends Mol. Med. 2013, 19, 89–98. [Google Scholar] [CrossRef]
- Galvani, E.; Peters, G.J.; Giovannetti, E. Thymidylate synthase inhibitors for non-small cell lung cancer. Expert Opin. Investig. Drugs 2011, 20, 1343–1356. [Google Scholar] [CrossRef] [PubMed]
- Takezawa, K.; Okamoto, I.; Okamoto, W.; Takeda, M.; Sakai, K.; Tsukioka, S.; Kuwata, K.; Yamaguchi, H.; Nishio, K.; Nakagawaet, K. Thymidylate synthase as a determinant of pemetrexed sensitivity in non-small cell lung cancer. Br. J. Cancer 2011, 104, 1594–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellicano, F.; Park, L.; Hopcroft, L.E.M.; Shah, M.M.; Jackson, L.; Scott, M.T.; Clarke, C.J.; Sinclair, A.; Abraham, S.A.; Hair, A.; et al. hsa-mir183/EGR1 mediated regulation of E2F1 is required for CML stem/progenitor cell survival. Blood 2018, 131, 1532–1544. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.; Sheeley, S.; Sweedler, J.V. Analysis of endogenous D-Amino acid-containing peptides in Metazoa. Bioanal. Rev. 2009, 1, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Funke, S.A.; Willbold, D. Mirror image phage display—A method to generate D-peptide ligands for use in diagnostic or therapeutical applications. Mol. Biosyst. 2009, 5, 783–786. [Google Scholar] [CrossRef] [PubMed]
- Garbuzenko, O.B.; Saad, M.; Betigeri, S.; Zhang, M.; Vetcher, A.A.; Soldatenkov, V.A.; Reimer, D.C.; Pozharov, V.P.; Minko, T. Intratracheal versus intravenous liposomal delivery of siRNA, antisense oligonucleotides and anticancer drug. Pharm. Res. 2009, 26, 382–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbuzenko, O.B.; Mainelis, G.; Taratula, O.; Minko, T. Inhalation treatment of lung cancer: The influence of composition, size and shape of nanocarriers on their lung accumulation and retention. Cancer Biol. Med. 2014, 11, 44–55. [Google Scholar] [PubMed]
- Rather, G.M.; Anyanwu, M.; Kerrigan, J.E.; Scotto, K.W.; Garbuzenko, O.; Minko, T.; Zoltan, S.; Bertino, J.R. A modified L-penetratin peptide targeting e2f-1, 2 and 3 is effective in combination with cisplatin or pemetrexed against prostate and lung cancer cell lines. In Proceedings of the American Association for Cancer Research Annual Meeting 2019, Atlanta, GA, USA, 29 March–3 April 2019. [Google Scholar]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rather, G.M.; Anyanwu, M.; Minko, T.; Garbuzenko, O.; Szekely, Z.; Bertino, J.R. Anti-Tumor Effects of a Penetratin Peptide Targeting Transcription of E2F-1, 2 and 3a Is Enhanced When Used in Combination with Pemetrexed or Cisplatin. Cancers 2021, 13, 972. https://doi.org/10.3390/cancers13050972
Rather GM, Anyanwu M, Minko T, Garbuzenko O, Szekely Z, Bertino JR. Anti-Tumor Effects of a Penetratin Peptide Targeting Transcription of E2F-1, 2 and 3a Is Enhanced When Used in Combination with Pemetrexed or Cisplatin. Cancers. 2021; 13(5):972. https://doi.org/10.3390/cancers13050972
Chicago/Turabian StyleRather, Gulam Mohmad, Michael Anyanwu, Tamara Minko, Olga Garbuzenko, Zoltan Szekely, and Joseph R. Bertino. 2021. "Anti-Tumor Effects of a Penetratin Peptide Targeting Transcription of E2F-1, 2 and 3a Is Enhanced When Used in Combination with Pemetrexed or Cisplatin" Cancers 13, no. 5: 972. https://doi.org/10.3390/cancers13050972
APA StyleRather, G. M., Anyanwu, M., Minko, T., Garbuzenko, O., Szekely, Z., & Bertino, J. R. (2021). Anti-Tumor Effects of a Penetratin Peptide Targeting Transcription of E2F-1, 2 and 3a Is Enhanced When Used in Combination with Pemetrexed or Cisplatin. Cancers, 13(5), 972. https://doi.org/10.3390/cancers13050972