
A Broad Overview of Signaling in Ph-Negative Classic Myeloproliferative Neoplasms



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. JAK2-Related Canonical Signaling Pathways
2.1. JAK2/STAT Pathway
2.2. MAPK/ERK Pathway
2.3. PI3K/AKT Pathway
3. Non-Canonical Signaling Pathways
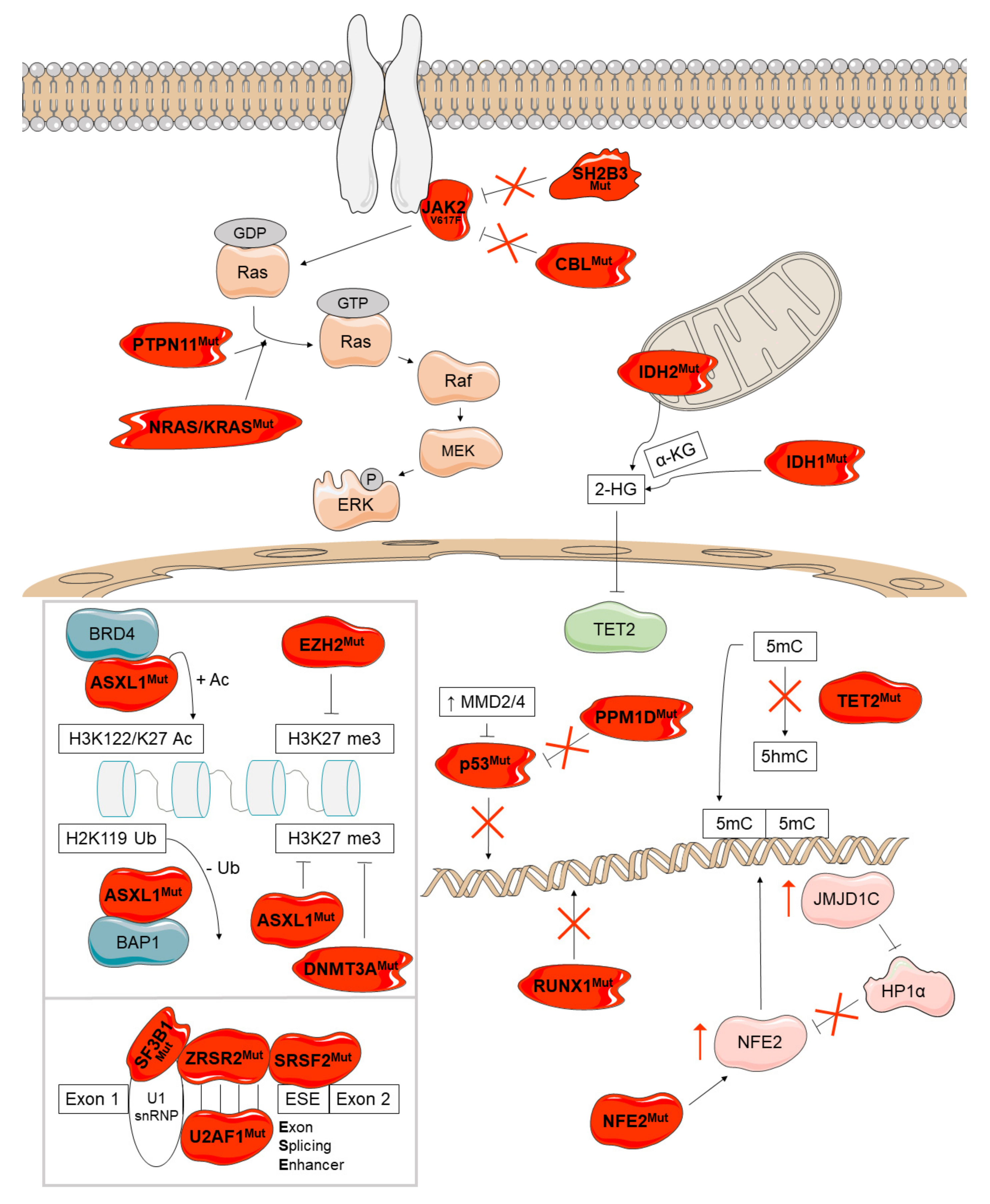
3.1. JAK2-Related Non-Canonical Signaling
3.2. CALR-Related Non-Canonical Signaling
3.3. Additional Non-Canonical Signaling
3.3.1. Inflammatory Signaling Pathways
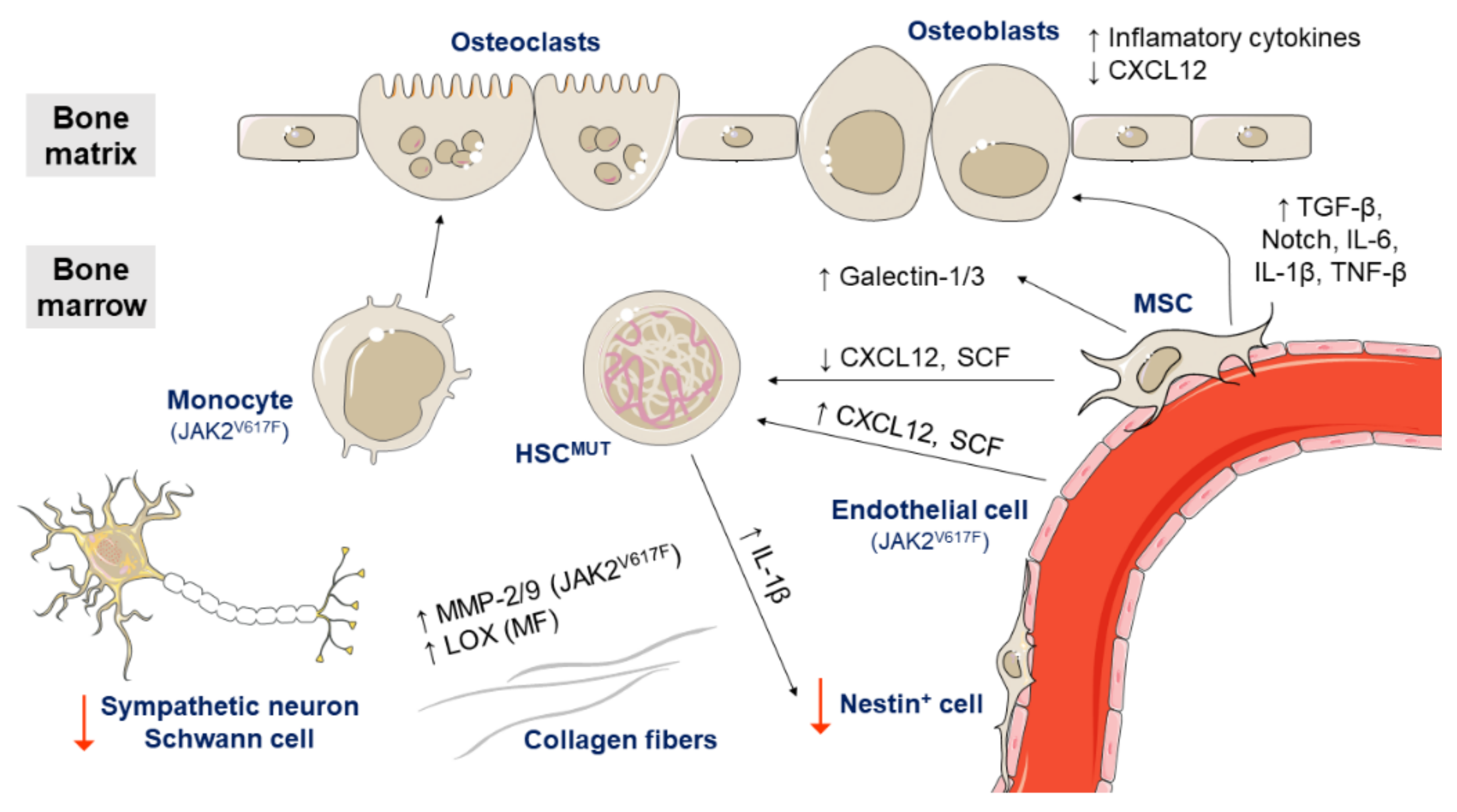
3.3.2. Bone Marrow Microenvironment
4. Disease Modifiers
4.1. Epigenetic Regulation
4.2. Tumor Suppression
4.3. Regulation of Transcription
4.4. Splicing
4.5. Additional Signaling Pathways
5. Additional Factors Involved in Disease
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tefferi, A.; Vardiman, J.W. Classification and diagnosis of myeloproliferative neoplasms: The 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia 2008, 22, 14–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Druker, B.J.; Sawyers, C.L.; Kantarjian, H.; Resta, D.J.; Reese, S.F.; Ford, J.M.; Capdeville, R.; Talpaz, M. Activity of a specific inhibitor of the bcr-abl tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N. Engl. J. Med. 2001, 344, 1038–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef] [Green Version]
- Druker, B.J. Inhibition of the Bcr-Abl tyrosine kinase as a therapeutic strategy for CML. Oncogene 2002, 21, 8541–8546. [Google Scholar] [CrossRef] [Green Version]
- Redner, R.L. Why doesn’t imatinib cure chronic myeloid leukemia? Oncologist 2010, 15, 182–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalmanti, L.; Saussele, S.; Lauseker, M.; Müller, M.C.; Dietz, C.; Heinrich, L.; Hanfstein, B.; Proetel, U.; Fabarius, A.; Krause, S.W.; et al. Safety and efficacy of imatinib in CML over a period of 10 years: Data from the randomized CML-study IV. Leukemia 2015, 29, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, A.; Larson, R.A.; Guilhot, F.; Radich, J.P.; Branford, S.; Hughes, T.P.; Baccarani, M.; Deininger, M.W.; Cervantes, F.; Fujihara, S.; et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017, 376, 917–927. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brkic, S.; Meyer, S.C. Challenges and perspectives for therapeutic targeting of myeloproliferative neoplasms. Hemasphere 2020, 5, e516. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Kiladjian, J.J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Mesa, R.; et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 2015, 372, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Deininger, M.; Radich, J.; Burn, T.C.; Huber, R.; Paranagama, D.; Verstovsek, S. The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood 2015, 126, 1551–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenfield, G.; McPherson, S.; Mills, K.; McMullin, M.F. The ruxolitinib effect: Understanding how molecular pathogenesis and epigenetic dysregulation impact therapeutic efficacy in myeloproliferative neoplasms. J. Transl. Med. 2018, 16, 1–16. [Google Scholar] [CrossRef]
- Martí-Carvajal, A.J.; Anand, V.; Solà, I. Janus kinase-1 and Janus kinase-2 inhibitors for treating myelofibrosis. Cochrane Database Syst. Rev. 2015, CD010298. [Google Scholar] [CrossRef]
- Cervantes, F.; Pereira, A. Does ruxolitinib prolong the survival of patients with myelofibrosis? Blood 2017, 129, 832–838. [Google Scholar] [CrossRef]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 2010, 363, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, J.B.; Stein, B.L.; McMahon, B.; Giles, F.J.; Platanias, L.C. Evolving therapeutic strategies for the classic philadelphia-negative myeloproliferative neoplasms. EBioMedicine 2016, 3, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, M.A.; Bannister, A.J.; Göttgens, B.; Foster, S.D.; Bartke, T.; Green, A.R.; Kouzarides, T. JAK2 phosphorylates histone H3Y41 and excludes HP1α from chromatin. Nature 2009, 461, 819–822. [Google Scholar] [CrossRef] [Green Version]
- De Koning, L.; Sauignoni, A.; Boumendil, C.; Rehman, H.; Asselain, B.; Sastre-Garau, X.; Almouzni, G. Heterochromatin protein lα: A hallmark of cell proliferation relevant to clinical oncology. EMBO Mol. Med. 2009, 1, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhao, X.; Perna, F.; Wang, L.; Koppikar, P.; Abdel-Wahab, O.; Harr, M.W.; Levine, R.L.; Xu, H.; Tefferi, A.; et al. JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell 2011, 19, 283–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Grandis, M.; Cambot, M.; Wautier, M.P.; Cassinat, B.; Chomienne, C.; Colin, Y.; Wautier, J.-L.; Le Van Kim, C.; El Nemer, W. JAK2V617F activates Lu/BCAM-mediated red cell adhesion in polycythemia vera through an EpoR-independent Rap1/Akt pathway. Blood 2013, 121, 658–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, H.Y.; Brooks, S.A.; Craver, B.M.; Morse, S.J.; Nguyen, T.K.; Haghighi, N.; Garbati, M.R.; Fleischman, A.G. Defective negative regulation of Toll-like receptor signaling leads to excessive TNF-α in myeloproliferative neoplasm. Blood Adv. 2019, 3, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; Vasudevan, K.B.; LaTocha, D.H.; Yang, F.; Press, R.D.; et al. TNFα facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood 2011, 118, 6392–6398. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT pathway activation in malignant and non-malignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015, 5, 316–331. [Google Scholar] [CrossRef] [Green Version]
- Stetka, J.; Vyhlidalova, P.; Lanikova, L.; Koralkova, P.; Gursky, J.; Hlusi, A.; Flodr, P.; Hubackova, S.; Bartek, J.; Hodny, Z.; et al. Addiction to DUSP1 protects JAK2V617F-driven polycythemia vera progenitors against inflammatory stress and DNA damage, allowing chronic proliferation. Oncogene 2019, 38, 5627–5642. [Google Scholar] [CrossRef]
- Salati, S.; Genovese, E.; Carretta, C.; Zini, R.; Bartalucci, N.; Prudente, Z.; Pennucci, V.; Ruberti, S.; Rossi, C.; Rontauroli, S.; et al. Calreticulin Ins5 and Del52 mutations impair unfolded protein and oxidative stress responses in K562 cells expressing CALR mutants. Sci. Rep. 2019, 9, 10558. [Google Scholar] [CrossRef] [Green Version]
- Pronier, E.; Cifani, P.; Merlinsky, T.R.; Berman, K.B.; Somasundara, A.V.H.; Rampal, R.K.; LaCava, J.; Wei, K.E.; Pastore, F.; Maag, J.L.; et al. Targeting the CALR interactome in myeloproliferative neoplasms. JCI Insight. 2018, 3, e122703. [Google Scholar] [CrossRef] [PubMed]
- Eder-Azanza, L.; Navarro, D.; Aranaz, P.; Novo, F.J.; Cross, N.C.P.; Vizmanos, J.L. Bioinformatic analyses of CALR mutations in myeloproliferative neoplasms support a role in signaling. Leukemia 2014, 28, 2106–2109. [Google Scholar] [CrossRef]
- Fucikova, J.; Spisek, R.; Kroemer, G.; Galluzzi, L. Calreticulin and cancer. Cell Res. 2021, 31, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Arshad, N.; Cresswell, P. Tumor-associated calreticulin variants functionally compromise the peptide loading complex and impair its recruitment of MHC-I. J. Biol. Chem. 2018, 293, 9555–9569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Zhao, L.; Loos, F.; Marty, C.; Xie, W.; Martins, I.; Lachkar, S.; Qu, B.; Waeckel-Énée, E.; Plo, I.; et al. Immunosuppression by mutated calreticulin released from malignant cells. Mol. Cell. 2020, 77, 748–760.e9. [Google Scholar] [CrossRef] [PubMed]
- Di Buduo, C.A.; Abbonante, V.; Marty, C.; Moccia, F.; Rumi, E.; Pietra, D.; Soprano, P.M.; Lim, D.; Cattaneo, D.; Iurlo, A.; et al. Defective interaction of mutant calreticulin and SOCE in megakaryocytes from patients with myeloproliferative neoplasms. Blood 2020, 135, 133–144. [Google Scholar] [CrossRef]
- Cabagnols, X.; Defour, J.P.; Ugo, V.; Ianotto, J.C.; Mossuz, P.; Mondet, J.; Girodon, F.; Alexandre, J.H.; Mansier, O.; Viallard, J.F.; et al. Differential association of calreticulin type 1 and type 2 mutations with myelofibrosis and essential thrombocytemia: Relevance for disease evolution. Leukemia 2015, 29, 249–252. [Google Scholar] [CrossRef]
- Pietra, D.; Rumi, E.; Ferretti, V.V.; Di Buduo, C.A.; Milanesi, C.; Cavalloni, C.; Sant’Antonio, E.; Abbonante, V.; Moccia, F.; Casetti, I.C.; et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 2016, 30, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Hoermann, G.; Greiner, G.; Valent, P. Cytokine regulation of microenvironmental cells in myeloproliferative neoplasms. Mediators Inflamm. 2015, 2015, 869242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, D.A.C.; Miner, C.A.; Engle, E.K.; Hu, H.; Collins, T.B.; Zhou, A.; Allen, M.J.; Malkova, O.; Oh, S.T. Cytokine production in myelofibrosis exhibits differential responsiveness to JAK-STAT, MAP Kinase, and NFκB signaling. Leukemia 2019, 33, 1978–1995. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Koche, R.; Zou, L.; van Galen, P.; Hill, C.E.; Dong, L.; De Groote, S.; Papalexi, E.; Hanasoge Somasundara, A.V.; Cordner, K.; et al. Dual targeting of oncogenic activation and inflammatory signaling increases therapeutic efficacy in myeloproliferative neoplasms. Cancer Cell 2018, 33, 785–787. [Google Scholar] [CrossRef]
- Wang, W.; Schwemmers, S.; Hexner, E.O.; Pahl, H.L. AML1 is overexpressed in patients with myeloproliferative neoplasms and mediates JAK2V617F-independent overexpression of NF-E2. Blood 2010, 116, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C.; Thomassen, M.; Riley, C.H.; Kjær, L.; Larsen, T.S.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Skov, V. Whole blood transcriptional profiling reveals deregulation of oxidative and antioxidative defence genes in myelofibrosis and related neoplasms. Potential implications of downregulation of Nrf2 for genomic instability and disease progression. PLoS ONE 2014, 9, e112786. [Google Scholar] [CrossRef] [PubMed]
- Wehrle, J.; Seeger, T.S.; Schwemmers, S.; Pfeifer, D.; Bulashevska, A.; Pahl, H.L. Transcription factor nuclear factor erythroid-2 mediates expression of the cytokine interleukin 8, a known predictor of inferior outcome in patients with myeloproliferative Neoplasms. Haematologica 2013, 98, 1073–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jutzi, J.S.; Bogeska, R.; Nikoloski, G.; Schmid, C.A.; Seeger, T.S.; Stegelmann, F.; Schwemmers, S.; Gründer, A.; Peeken, J.C.; Gothwal, M.; et al. MPN patients harbor recurrent truncating mutations in transcription factor NF-E2. J. Exp. Med. 2013, 210, 1003–1019. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Kimura, M.; Fujita, R.; Inoue, A.; Pan, X.; Takayama, M.; Katsuoka, F.; Aburatani, H.; Bresnick, E.H.; Yamamoto, M. NF-E2 domination over Nrf2 promotes ROS accumulation and megakaryocytic maturation. Blood 2010, 115, 677–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasselbalch, H.C. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk. Res. 2013, 37, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.; Lacout, C.; Marty, C.; Cuingnet, M.; Solary, E.; Vainchenker, W.; Villeval, J.-L. JAK2V617F expression in mice amplifies early hematopoietic cells and gives them a competitive advantage that is hampered by IFNα. Blood 2013, 22, 1464–1477. [Google Scholar] [CrossRef] [PubMed]
- Mullally, A.; Bruedigam, C.; Poveromo, L.; Heidel, F.H.; Purdon, A.; Vu, T.; Austin, R.; Heckl, D.; Breyfogle, L.J.; Kuhn, C.P.; et al. Depletion of Jak2V617F myeloproliferative neoplasm-propagating stem cells by interferon-α in a murine model of polycythemia vera. Blood 2013, 121, 3692–3702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmati, S.; Haque, T.; Gritsman, K. Inflammatory signaling pathways in preleukemic and leukemic stem cells. Front. Oncol. 2017, 7, 265. [Google Scholar] [CrossRef] [Green Version]
- Hérault, A.; Binnewies, M.; Leong, S.; Calero-Nieto, F.J.; Zhang, Y.; Kang, Y.; Wang, X.; Pietras, E.M.; Chu, S.H.; Barry-Holson, K.; et al. Myeloid progenitor cluster formation drives emergency and leukemic myelopoiesis. Nature 2017, 544, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Geyer, H.L.; Dueck, A.C.; Scherber, R.M.; Mesa, R.A. Impact of inflammation on myeloproliferative neoplasm symptom development. Mediators Inflamm. 2015, 2015, 284706. [Google Scholar] [CrossRef] [Green Version]
- Pourcelot, E.; Trocme, C.; Mondet, J.; Bailly, S.; Toussaint, B.; Mossuz, P. Cytokine profiles in polycythemia vera and essential thrombocythemia patients: Clinical implications. Exp. Hematol. 2014, 42, 360–368. [Google Scholar] [CrossRef]
- Hoermann, G.; Cerny-Reiterer, S.; Herrmann, H.; Blatt, K.; Bilban, M.; Gisslinger, H.; Müllauer, L.; Kralovics, R.; Mannhalter, C.; Valent, P.; et al. Identification of oncostatin M as a JAK2 V617F-dependent amplifier of cytokine production and bone marrow remodeling in myeloproliferative neoplasms. FASEB J. 2012, 26, 894–906. [Google Scholar] [CrossRef]
- Le Bousse-Kerdilès, M.C.; Chevillard, S.; Charpentier, A.; Romquin, N.; Clay, D.; Smadja-Joffe, F.; Praloran, V.; Dupriez, B.; Demory, J.L.; Jasmin, C.; et al. Differential expression of transforming growth factor-beta, basic fibroblast growth factor, and their receptors in CD34+ hematopoietic progenitor cells from patients with myelofibrosis and myeloid metaplasia. Blood 1996, 88, 4534–4546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martyré, M.-C.; Magdelenat, H.; Bryckaert, M.-C.; Laine-Bidron, C.; Calvo, F. Increased intraplatelet levels of platelet-derived growth factor and transforming growth factor-β in patients with myelofibrosis with myeloid metaplasia. Br. J. Haematol. 1991, 77, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Di Raimondo, F.; Azzaro, M.P.; Palumbo, G.A.; Bagnato, S.; Stagno, F.; Giustolisi, G.M.; Cacciola, E.; Sortino, G.; Guglielmo, P.; Giustolisi, R. Elevated vascular endothelial growth factor [VEGF] serum levels in idiopathic myelofibrosis. Leukemia 2001, 15, 976–980. [Google Scholar] [CrossRef] [Green Version]
- Bock, O.; Höftmann, J.; Theophile, K.; Hussein, K.; Wiese, B.; Schlué, J.; Kreipe, H. Bone morphogenetic proteins are overexpressed in the bone marrow of primary myelofibrosis and are apparently induced by fibrogenic cytokines. Am. J. Pathol. 2008, 172, 951–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.C.; Novetsky, A.; Chen, C.; Novetsky, A.D. Plasma matrix metalloproteinase and tissue inhibitor of metalloproteinase in patients with agnogenic myeloid metaplasia or idiopathic primary myelofibrosis. Br. J. Haematol. 2002, 119, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.K.; Holten-Andersen, M.N.; Riisbro, R.; De Nully Brown, P.; Larsen, M.B.; Kjeldsen, L.; Heickendorff, L.; Brünner, N.; Hasselbalch, H.C. Elevated plasma levels of TIMP-1 correlate with plasma suPAR/uPA in patients with chronic myeloproliferative disorders. Eur. J. Haematol. 2003, 71, 377–384. [Google Scholar] [CrossRef]
- Lu, M.; Xia, L.; Liu, Y.C.; Hochman, T.; Bizzari, L.; Aruch, D.; Lew, J.; Weinberg, R.; Goldberg, J.D.; Hoffman, R. Lipocalin produced by myelofibrosis cells affects the fate of both hematopoietic and marrow microenvironmental cells. Blood 2015, 126, 972–982. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, M.; Barrio, S.; Fernandez, M.; Paradela, A.; Arenas, A.; Toldos, O.; Ayala, R.; Albizua, E.; Jimenez, A.; Redondo, S.; et al. Proteomic analysis reveals heat shock protein 70 has a key role in polycythemia vera. Mol. Cancer 2013, 12, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asea, A.; Kraeft, S.K.; Kurt-Jones, E.A.; Stevenson, M.A.; Chen, L.B.; Finberg, R.W.; Koo, G.C.; Calderwood, S.K. HSP70 stimulates cytokine production through a CD 14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 2000, 6, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Sevin, M.; Girodon, F.; Garrido, C.; De Thonel, A. HSP90 and HSP70: Implication in inflammation processes and therapeutic approaches for myeloproliferative neoplasms. Mediators Inflamm. 2015, 2015, 970242. [Google Scholar] [CrossRef] [Green Version]
- Falanga, A.; Marchetti, M.; Evangelista, V.; Vignoli, A.; Licini, M.; Balicco, M.; Manarini, S.; Finazzi, G.; Cerletti, C.; Barbui, T. Polymorphonuclear leukocyte activation and hemostasis in patients with essential thrombocythemia and polycythemia vera. Blood 2000, 96, 4261–4266. [Google Scholar] [CrossRef]
- Villmow, T.; Kemkes-Matthes, B.; Matzdorff, A.C. Markers of platelet activation and platelet-leukocyte interaction in patients with myeloproliferative syndromes. Thromb. Res. 2002, 108, 139–145. [Google Scholar] [CrossRef]
- Falanga, A.; Marchetti, M.; Vignoli, A.; Balducci, D.; Barbui, T. Leukocyte-platelet interaction in patients with essential thrombocythemia and polycythemia vera. Exp. Hematol. 2005, 33, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Curto-Garcia, N.; Harrison, C.; McLornan, D.P. Bone marrow niche dysregulation in myeloproliferative neoplasms. Haematologica 2020, 105, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Zhan, H.; Lin, C.H.S.; Segal, Y.; Kaushansky, K. The JAK2V617F-bearing vascular niche promotes clonal expansion in myeloproliferative neoplasms. Leukemia 2018, 32, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.S.; Zhang, Y.; Kaushansky, K.; Zhan, H. JAK2V617F-bearing vascular niche enhances malignant hematopoietic regeneration following radiation injury. Haematologica 2018, 103, 1160–1168. [Google Scholar] [CrossRef] [Green Version]
- Ramos, T.L.; Sánchez-Abarca, L.I.; Rosón-Burgo, B.; Redondo, A.; Rico, A.; Preciado, S.; Ortega, R.; Rodríguez, C.; Muntión, S.; Hernández-Hernández, A.; et al. Mesenchymal stromal cells [MSC] from JAK2+ myeloproliferative neoplasms differ from normal MSC and contribute to the maintenance of neoplastic hematopoiesis. PLoS ONE 2017, 12, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Koopmans, S.M.; Bot, F.J.; Schouten, H.C.; Janssen, J.; van Marion, A.M. The involvement of Galectins in the modulation of the JAK/STAT pathway in myeloproliferative neoplasia. Am. J. Blood Res. 2012, 2, 119–127. [Google Scholar] [PubMed]
- Spanoudakis, E.; Papoutselis, M.; Bazdiara, I.; Lamprianidi, E.; Kordella, X.; Tilkeridis, C.; Tsatalas, C.; Kotsianidis, I. The JAK2V617F point mutation increases the osteoclast forming ability of monocytes in patients with chronic myeloproliferative neoplasms and makes their osteoclasts more susceptible to JAK2 inhibition. Mediterr. J. Hematol. Infect. Dis. 2018, 10, e2018058. [Google Scholar] [CrossRef]
- Korn, C.; Rak, J.; García-García, A.; Fielding, C.; Khorshed, R.; González-Antón, S.; Li, J.; Norfo, R.; Baxter, E.J.; McKerrell, T.; et al. Niche heterogeneity impacts evolution of myeloproliferative neoplasms driven by the same oncogenic pathway. Blood 2018, 132 (Suppl. 1), 98. [Google Scholar] [CrossRef]
- Arranz, L.; Sánchez-Aguilera, A.; Martín-Pérez, D.; Isern, J.; Langa, X.; Tzankov, A.; Lundberg, P.; Muntión, S.; Tzeng, Y.-S.; Lai, D.-M.; et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 2014, 512, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.M.; Zhang, L.J.; Fu, J.Z.; Liang, W.T.; Cheng, Z.Y.; Bai, P.; Bian, Y.S.; Wan, J.S. Regulation of Ruxolitinib on matrix metalloproteinase in JAK2V617F positive myeloroliferative neoplasms cells. Zhonghua Xue Ye Xue Za Zhi 2017, 38, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Tadmor, T.; Bejar, J.; Attias, D.; Mischenko, E.; Sabo, E.; Neufeld, G.; Vadasz, Z. The expression of lysyl-oxidase gene family members in myeloproliferative neoplasms. Am. J. Hematol. 2013, 88, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Elala, Y.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016, 1, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Score, J.; Mannarelli, C.; Pancrazzi, A.; Biamonte, F.; Pardanani, A.; Zoi, K.; Reiter, A.; et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: An international study of 797 patients. Leukemia 2014, 28, 1804–1810. [Google Scholar] [CrossRef]
- Grabek, J.; Straube, J.; Bywater, M.; Lane, S.W. MPN: The molecular drivers of disease initiation, progression and transformation and their effect on treatment. Cells 2020, 9, 1901. [Google Scholar] [CrossRef] [PubMed]
- Marneth, A.E.; Mullally, A. The molecular genetics of myeloproliferative neoplasms. Cold Spring Harb. Perspect. Med. 2020, 10, a034876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, J.; Mead, A.J. Heterogeneity in myeloproliferative neoplasms: Causes and consequences. Adv. Biol. Regul. 2019, 71, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couédic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Kralovics, R.; Teo, S.-S.; Li, S.; Theocharides, A.; Buser, A.S.; Tichelli, A.; Skoda, R.C. Acquisition of the V617F mutation of JAK2 is a late genetic event in a subset of patients with myeloproliferative disorders. Blood 2006, 108, 1377–1380. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.P.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Xing, S.; Li, Z.; Fu, X.; Li, Q.; Krantz, S.B.; Zhao, Z.J. Identification of an acquired JAK2 mutation in polycythemia vera. J. Biol. Chem. 2005, 280, 22788–22792. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2019 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2019, 94, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Passamonti, F.; Elena, C.; Schnittger, S.; Skoda, R.C.; Green, A.R.; Girodon, F.; Kiladjian, J.-J.; McMullin, M.F.; Ruggeri, M.; Besses, C.; et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood 2011, 117, 2813–2816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. MPL515 mutations in myeloproliferative and other myeloid disorders: A study of 1182 patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef] [Green Version]
- Defour, J.P.; Itaya, M.; Gryshkova, V.; Brett, I.C.; Pecquet, C.; Sato, T.; Smith, S.O.; Constantinescu, S.N. Tryptophan at the transmembrane-cytosolic junction modulates thrombopoietin receptor dimerization and activation. Proc. Natl. Acad. Sci. USA 2013, 110, 2540–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nangalia, J.; Grinfeld, J.; Green, A.R. Pathogenesis of myeloproliferative disorders. Annu. Rev. Pathol. 2016, 11, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef] [Green Version]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalak, M.; Corbett, E.F.; Mesaeli, N.; Nakamura, K.; Opas, M. Calreticulin: One protein, one gene, many functions. Biochem. J. 1999, 344, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.A.; Groenendyk, J.; Michalak, M. Calreticulin signaling in health and disease. Int. J. Biochem. Cell Biol. 2012, 44, 842–846. [Google Scholar] [CrossRef] [PubMed]
- Shivarov, V.; Ivanova, M.; Tiu, R.V. Mutated calreticulin retains structurally disordered C terminus that cannot bind Ca[2+]: Some mechanistic and therapeutic implications. Blood Cancer J. 2014, 4, e185. [Google Scholar] [CrossRef] [Green Version]
- Araki, M.; Yang, Y.; Masubuchi, N.; Hironaka, Y.; Takei, H.; Morishita, S.; Mizukami, Y.; Kan, S.; Shirane, S.; Edahiro, Y.; et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood 2016, 127, 1307–1316. [Google Scholar] [CrossRef]
- Chachoua, I.; Pecquet, C.; El-Khoury, M.; Nivarthi, H.; Albu, R.I.; Marty, C.; Gryshkova, V.; Defour, J.-P.; Vertenoeil, G.; Ngo, A.; et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 2016, 127, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Elf, S.; Abdelfattah, N.S.; Chen, E.; Perales-Patón, J.; Rosen, E.A.; Ko, A.; Peisker, F.; Florescu, N.; Giannini, S.; Wolach, O.; et al. Mutant calreticulin requires both its mutant C-terminus and the thrombopoietin receptor for oncogenic transformation. Cancer Discov. 2016, 6, 368–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [Green Version]
- Elf, S.; Abdelfattah, N.S.; Baral, A.J.; Beeson, D.; Rivera, J.F.; Ko, A.; Florescu, N.; Birrane, G.; Chen, E.; Mullally, A. Defining the requirements for the pathogenic interaction between mutant calreticulin and MPL in MPN. Blood 2018, 131, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Araki, M.; Komatsu, N. The role of calreticulin mutations in myeloproliferative neoplasms. Int. J. Hematol. 2020, 111, 200–205. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Schubert, C.; Köhler, J.; Schemionek, M.; Isfort, S.; Brümmendorf, T.H.; Koschmieder, S.; Chatain, N. Calreticulin-mutant proteins induce megakaryocytic signaling to transform hematopoietic cells and undergo accelerated degradation and Golgi-mediated secretion. J. Hematol. Oncol. 2016, 9, 45. [Google Scholar] [CrossRef] [Green Version]
- Pecquet, C.; Balligand, T.; Chachoua, I.; Roy, A.; Vertenoeil, G.; Colau, D.; Fertig, E.; Marty, C.; Nivarthi, H.; Defour, J.-P.; et al. Secreted mutant calreticulins as rogue cytokines trigger thrombopoietin receptor activation specifically in CALR mutated cells: Perspectives for MPN therapy. Blood 2018, 132 (Suppl. 1), 4. [Google Scholar] [CrossRef]
- Rampal, R.; Al-Shahrour, F.; Abdel-Wahab, O.; Patel, J.; Brunel, J.-P.; Mermel, C.H.; Bass, A.J.; Pretz, J.; Ahn, J.; Hricik, T.; et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 2014, 123, e123–e133. [Google Scholar] [CrossRef] [PubMed]
- Reuther, G.W. Myeloproliferative neoplasms: Molecular drivers and therapeutics. Prog. Mol. Biol. Transl. Sci. 2016, 144, 437–484. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: A comprehensive cytokine profiling study. J. Clin. Oncol. 2011, 29, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Kiladjian, J.J.; Giraudier, S.; Cassinat, B. Interferon-alpha for the therapy of myeloproliferative neoplasms: Targeting the malignant clone. Leukemia 2016, 30, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Kagoya, Y.; Yoshimi, A.; Tsuruta-Kishino, T.; Arai, S.; Satoh, T.; Akira, S.; Kurokawa, M. JAK2V617F+ myeloproliferative neoplasm clones evoke paracrine DNA damage to adjacent normal cells through secretion of lipocalin-2. Blood 2014, 124, 2996–3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decker, M.; Martinez-Morentin, L.; Wang, G.; Lee, Y.; Liu, Q.; Leslie, J.; Ding, L. Leptin-receptor-expressing bone marrow stromal cells are myofibroblasts in primary myelofibrosis. Nat. Cell Biol. 2017, 19, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.K.; Mullally, A.; Dugourd, A.; Peisker, F.; Hoogenboezem, R.; Van Strien, P.M.H.; Bindels, E.M.; Heckl, D.; Büsche, G.; Fleck, D.; et al. Gli1+ mesenchymal stromal cells are a key driver of bone marrow fibrosis and an important cellular therapeutic target. Cell. Stem Cell. 2017, 20, 785–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef] [Green Version]
- Asada, S.; Goyama, S.; Inoue, D.; Shikata, S.; Takeda, R.; Fukushima, T.; Yonezawa, T.; Fujino, T.; Hayashi, Y.; Kawabata, K.C.; et al. Mutant ASXL1 cooperates with BAP1 to promote myeloid leukaemogenesis. Nat. Commun. 2018, 9, 2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramani, A.; Larjo, A.; Bassein, J.A.; Chang, X.; Hastie, R.B.; Togher, S.M.; Lähdesmäki, H.; Rao, A. Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex. Nat. Commun. 2015, 6, 7307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Kurtenbach, S.; Guo, Y.; Lohse, I.; Durante, M.A.; Li, J.; Li, Z.; Al-Ali, H.; Li, L.; Chen, Z.; et al. Gain of function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies. Blood 2018, 131, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Saika, M.; Inoue, D.; Nagase, R.; Sato, N.; Tsuchiya, A.; Yabushita, T.; Kitamura, T.; Goyama, S. ASXL1 and SETBP1 mutations promote leukaemogenesis by repressing TGFβ pathway genes through histone deacetylation. Sci. Rep. 2018, 8, 15873. [Google Scholar] [CrossRef]
- Cho, Y.-S.; Kim, E.-J.; Park, U.-H.; Sin, H.-S.; Um, S.-J. Additional sex comb-like 1 [ASXL1], in cooperation with SRC-1, acts as a ligand-dependent coactivator for retinoic acid receptor. J. Biol. Chem. 2006, 281, 17588–17598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, U.-H.; Yoon, S.-K.; Park, T.; Kim, E.-J.; Um, S.-J. Additional sex comb-like [ASXL] proteins 1 and 2 play opposite roles in adipogenesis via reciprocal regulation of peroxisome proliferator-activated receptor γ. J. Biol. Chem. 2011, 286, 1354–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.N.; Jankowska, A.M.; Mahfouz, R.; Dunbar, A.J.; Sugimoto, Y.; Hosono, N.; Hu, Z.; Cheriyath, V.; Vatolin, S.; Przychodzen, B.; et al. Multiple mechanisms deregulate EZH2 and histone H3 lysine 27 epigenetic changes in myeloid malignancies. Leukemia 2013, 27, 1301–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinke, J.; Chase, A.; Cross, N.C.P.; Hochhaus, A.; Ernst, T. EZH2 in myeloid malignancies. Cells 2020, 9, 1639. [Google Scholar] [CrossRef]
- Anteneh, H.; Fang, J.; Song, J. Structural basis for impairment of DNA methylation by the DNMT3A R882H mutation. Nat. Commun. 2020, 11, 2294. [Google Scholar] [CrossRef] [PubMed]
- Jacquelin, S.; Straube, J.; Cooper, L.; Vu, T.; Song, A.; Bywater, M.; Baxter, E.; Heidecker, M.; Wackrow, B.; Porter, A.; et al. Jak2V617F and Dnmt3a loss cooperate to induce myelofibrosis through activated enhancer-driven inflammation. Blood 2018, 132, 2707–2721. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, O.; Mullally, A.; Hedvat, C.; Garcia-Manero, G.; Patel, J.; Wadleigh, M.; Malinge, S.; Yao, J.; Kilpivaara, O.; Bhat, R.; et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009, 114, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Ostrander, E.L.; Kramer, A.C.; Mallaney, C.; Celik, H.; Koh, W.K.; Fairchild, J.; Haussler, E.; Zhang, C.R.C.; Challen, G.A. Divergent effects of Dnmt3a and Tet2 mutations on hematopoietic progenitor cell fitness. Stem Cell Reports 2020, 14, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Lasho, T.L.; Abdel-Wahab, O.; Guglielmelli, P.; Patel, J.; Caramazza, D.; Pieri, L.; Finke, C.M.; Kilpivaara, O.; Wadleigh, M.; et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia 2010, 24, 1302–1309. [Google Scholar] [CrossRef]
- Pardanani, A.; Lasho, T.L.; Finke, C.M.; Mai, M.; McClure, R.F.; Tefferi, A. IDH1 and IDH2 mutation analysis in chronic-and blast-phase myeloproliferative neoplasms. Leukemia 2010, 24, 1146–1151. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Yoshimi, A.; Lin, K.T.; Wiseman, D.H.; Rahman, M.A.; Pastore, A.; Wang, B.; Lee, S.C.; Micol, J.B.; Zhang, X.J.; de Botton, S.; et al. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature 2019, 574, 273–277. [Google Scholar] [CrossRef]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Elf, S.E.; Miyata, Y.; Sashida, G.; Liu, Y.; Huang, G.; Di Giandomenico, S.; Lee, J.M.; Deblasio, A.; Menendez, S.; et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009, 4, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcellino, B.K.; Hoffman, R.; Tripodi, J.; Lu, M.; Kosiorek, H.; Mascarenhas, J.; Rampal, R.K.; Dueck, A.; Najfeld, V. Advanced forms of MPNs are accompanied by chromosomal abnormalities that lead to dysregulation of TP53. Blood Adv. 2018, 2, 3581–3589. [Google Scholar] [CrossRef] [Green Version]
- Fiscella, M.; Zhang, H.; Fan, S.; Sakaguchi, K.; Shen, S.; Mercer, W.E.; Vande Woude, G.F.; O’Connor, P.M.; Appella, E. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc. Natl. Acad. Sci. USA 1997, 94, 6048–6053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, J.D.; Miller, P.G.; Silver, A.J.; Sellar, R.S.; Bhatt, S.; Gibson, C.; McConkey, M.; Adams, D.; Mar, B.; Mertins, P.; et al. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 2018, 132, 1095–1105. [Google Scholar] [CrossRef]
- Sood, R.; Kamikubo, Y.; Liu, P. Role of RUNX1 in hematological malignancies. Blood 2017, 129, 2070–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeken, J.C.; Jutzi, J.S.; Wehrle, J.; Koellerer, C.; Staehle, H.F.; Becker, H.; Schoenwandt, E.; Seeger, T.S.; Schanne, D.H.; Gothwal, M.; et al. Epigenetic regulation of NFE2 overexpression in myeloproliferative neoplasms. Blood 2018, 131, 2065–2073. [Google Scholar] [CrossRef] [Green Version]
- Will, C.L.; Lührmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.B.; Murphy, M.E.; et al. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.C.; North, K.; Kim, E.; Jang, E.; Obeng, E.; Lu, S.X.; Liu, B.; Inoue, D.; Yoshimi, A.; Ki, M.; et al. Synthetic lethal and convergent biological effects of cancer-associated spliceosomal gene mutations. Cancer Cell 2018, 34, 225–241.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Chen, J.Y.; Huang, Y.J.; Gu, Y.; Qiu, J.; Qian, H.; Shao, C.; Zhang, X.; Hu, J.; Li, H.; et al. The augmented R-loop is a unifying mechanism for myelodysplastic syndromes induced by high-risk splicing factor mutations. Mol. Cell 2018, 69, 412–425.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.D.; Leong, W.Y.; Li, W.; Reddy, P.N.G.; Sullivan, J.D.; Walter, M.J.; Zou, L.; Graubert, T.A. Spliceosome mutations induce R loop-associated sensitivity to ATR inhibition in myelodysplastic syndromes. Cancer Res. 2018, 78, 5363–5374. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, H.; Harada, Y.; Ogata, Y.; Kagiyama, Y.; Shingai, N.; Doki, N.; Ohashi, K.; Kitamura, T.; Komatsu, N.; Harada, H. Overexpression of RUNX1 short isoform has an important role in the development of myelodysplastic/myeloproliferative neoplasms. Blood Adv. 2017, 1, 1382–1386. [Google Scholar] [CrossRef] [Green Version]
- Xiao, R.; Sun, Y.; Ding, J.H.; Lin, S.; Rose, D.W.; Rosenfeld, M.G.; Fu, X.D.; Li, X. Splicing regulator SC35 is essential for genomic stability and cell proliferation during mammalian organogenesis. Mol. Cell Biol. 2007, 27, 5393–5402. [Google Scholar] [CrossRef] [Green Version]
- Soares, L.M.; Zanier, K.; Mackereth, C.; Sattler, M.; Valcárcel, J. Intron removal requires proofreading of U2AF/3′ splice site recognition by DEK. Science 2006, 312, 1961–1965. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. U2AF1 mutation types in primary myelofibrosis: Phenotypic and prognostic distinctions. Leukemia 2018, 32, 2274–2278. [Google Scholar] [CrossRef] [PubMed]
- Graubert, T.A.; Shen, D.; Ding, L.; Okeyo-Owuor, T.; Lunn, C.L.; Shao, J.; Krysiak, K.; Harris, C.C.; Koboldt, D.C.; Larson, D.E.; et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 2011, 44, 53–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, C.J.; Wise, J.A. The splicing factor U2AF small subunit is functionally conserved between fission yeast and humans. Mol. Cell Biol. 2004, 24, 4229–4240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palangat, M.; Anastasakis, D.G.; Fei, D.L.; Lindblad, K.E.; Bradley, R.; Hourigan, C.S.; Hafner, M.; Larson, D.R. The splicing factor U2AF1 contributes to cancer progression through a noncanonical role in translation regulation. Genes Dev. 2019, 33, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Kanojia, D.; Li, J.; Okamoto, R.; Sato-Otsubo, A.; Kohlmann, A.; Sanada, M.; Grossmann, V.; Sundaresan, J.; Shiraishi, Y.; et al. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat. Commun. 2015, 6, 6042. [Google Scholar] [CrossRef]
- Cazzola, M.; Rossi, M.; Malcovati, L. Biologic and clinical significance of somatic mutations of SF3B1 in myeloid and lymphoid neoplasms. Blood 2013, 121, 260–269. [Google Scholar] [CrossRef] [Green Version]
- Obeng, E.A.; Chappell, R.J.; Seiler, M.; Chen, M.C.; Campagna, D.R.; Schmidt, P.J.; Schneider, R.K.; Lord, A.M.; Wang, L.; Gambe, R.G.; et al. Physiologic expression of Sf3b1[K700E] causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Cancer Cell 2016, 30, 404–417. [Google Scholar] [CrossRef] [Green Version]
- Tong, W.; Zhang, J.; Lodish, H.F. Lnk inhibits erythropoiesis and Epo-dependent JAK2 activation and downstream signaling pathways. Blood 2005, 105, 4604–4612. [Google Scholar] [CrossRef] [PubMed]
- Bersenev, A.; Wu, C.; Balcerek, J.; Tong, W. Lnk controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. J. Clin. Investig. 2008, 118, 2832–2844. [Google Scholar] [CrossRef] [Green Version]
- Simon, C.; Dondi, E.; Chaix, A.; de Sepulveda, P.; Kubiseski, T.J.; Varin-Blank, N.; Velazquez, L. Lnk adaptor protein down-regulates specific Kit-induced signaling pathways in primary mast cells. Blood 2008, 112, 4039–4047. [Google Scholar] [CrossRef] [PubMed]
- Takaki, S.; Sauer, K.; Iritani, B.M.; Chien, S.; Ebihara, Y.; Tsuji, K.; Takatsu, K.; Perlmutter, R.M. Control of B cell production by the adaptor protein Lnk: Definition of a conserved family of signal-modulating proteins. Immunity 2000, 13, 599–609. [Google Scholar] [CrossRef] [Green Version]
- Takaki, S.; Morita, H.; Tezuka, Y.; Takatsu, K. Enhanced hematopoiesis by hematopoietic progenitor cells lacking intracellular adaptor protein, Lnk. J. Exp. Med. 2002, 195, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velazquez, L.; Cheng, A.M.; Fleming, H.E.; Furlonger, C.; Vesely, S.; Bernstein, A.; Paige, C.J.; Pawson, T. Cytokine signaling and hematopoietic homeostasis are disrupted in Lnk-deficient mice. J. Exp. Med. 2002, 195, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Thien, C.B.; Langdon, W.Y. Negative regulation of PTK signalling by Cbl proteins. Growth Factors. 2005, 23, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, B.; Ahmad, G.; Nadeau, S.; Zutshi, N.; An, W.; Scheffe, S.; Dong, L.; Feng, D.; Goetz, B.; Arya, P.; et al. Protein tyrosine kinase regulation by ubiquitination: Critical roles of Cbl-family ubiquitin ligases. Biochim. Biophys. Acta. 2013, 1833, 122–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, K.; Jiang, J.; Donaghy, R.; Riling, C.R.; Cheng, Y.; Chandra, V.; Rozenova, K.; An, W.; Mohapatra, B.C.; Goetz, B.T.; et al. CBL family E3 ubiquitin ligases control JAK2 ubiquitination and stability in hematopoietic stem cells and myeloid malignancies. Genes Dev. 2017, 31, 1007–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanada, M.; Suzuki, T.; Shih, L.Y.; Otsu, M.; Kato, M.; Yamazaki, S.; Tamura, A.; Honda, H.; Sakata-Yanagimoto, M.; Kumano, K.; et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature 2009, 460, 904–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer. 2007, 7, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Bunda, S.; Burrell, K.; Heir, P.; Zeng, L.; Alamsahebpour, A.; Kano, Y.; Raught, B.; Zhang, Z.Y.; Zadeh, G.; Ohh, M. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat. Commun. 2015, 6, 8859. [Google Scholar] [CrossRef] [Green Version]
- Niihori, T.; Aoki, Y.; Ohashi, H.; Kurosawa, K.; Kondoh, T.; Ishikiriyama, S.; Kawame, H.; Kamasaki, H.; Yamanaka, T.; Takada, F.; et al. Functional analysis of PTPN11/SHP-2 mutants identified in Noonan syndrome and childhood leukemia. J. Hum. Genet. 2005, 50, 192–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Z.Q.; Yu, D.H.; Park, M.; Marshall, M.; Feng, G.S. Molecular mechanism for the Shp-2 tyrosine phosphatase function in promoting growth factor stimulation of Erk activity. Mol. Cell Biol. 2000, 20, 1526–1536. [Google Scholar] [CrossRef] [Green Version]
- Eaves, C.J. Hematopoietic stem cells: Concepts, definitions, and the new reality. Blood 2015, 125, 2605–2613. [Google Scholar] [CrossRef] [Green Version]
- Mead, A.J.; Mullally, A. Myeloproliferative neoplasm stem cells. Blood 2017, 129, 1607–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, A.V.; Chase, A.; Silver, R.T.; Oscier, D.; Zoi, K.; Wang, Y.L.; Cario, H.; Pahl, H.L.; Collins, A.; Reiter, A.; et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat. Genet. 2009, 41, 446–449. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.V.; Campbell, P.J.; Beer, P.A.; Schnittger, S.; Vannucchi, A.M.; Zoi, K.; Percy, M.J.; McMullin, M.F.; Scott, L.M.; Tapper, W.; et al. The JAK2 46/1 haplotype predisposes to MPL-mutated myeloproliferative neoplasms. Blood 2010, 115, 4517–4523. [Google Scholar] [CrossRef]
- Tapper, W.; Jones, A.V.; Kralovics, R.; Harutyunyan, A.S.; Zoi, K.; Leung, W.; Godfrey, A.L.; Guglielmelli, P.; Callaway, A.; Ward, D.; et al. Genetic variation at MECOM, TERT, JAK2 and HBS1L-MYB predisposes to myeloproliferative neoplasms. Nat. Commun. 2015, 6, 6691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifa, A.P.; Bănescu, C.; Bojan, A.S.; Voina, C.M.; Popa, Ș.; Vișan, S.; Ciubean, A.D.; Tripon, F.; Dima, D.; Popov, V.M.; et al. MECOM, HBS1L-MYB, THRB-RARB, JAK2, and TERT polymorphisms defining the genetic predisposition to myeloproliferative neoplasms: A study on 939 patients. Am. J. Hematol. 2018, 93, 100–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinds, D.A.; Barnholt, K.E.; Mesa, R.A.; Kiefer, A.K.; Do, C.B.; Eriksson, N.; Mountain, J.L.; Francke, U.; Tung, J.Y.; Nguyen, H.M.; et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood 2016, 128, 1121–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godfrey, A.L.; Chen, E.; Pagano, F.; Silber, Y.; Campbell, P.J.; Green, A.R. Clonal analyses reveal associations of JAK2V617F homozygosity with hematologic features, age and gender in polycythemia vera and essential thrombocythemia. Haematologica 2013, 98, 718–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geyer, H.L.; Kosiorek, H.; Dueck, A.C.; Scherber, R.; Slot, S.; Zweegman, S.; Te Boekhorst, P.A.; Senyak, Z.; Schouten, H.C.; Sackmann, F.; et al. Associations between gender, disease features and symptom burden in patients with myeloproliferative neoplasms: An analysis by the MPN QOL International Working Group. Haematologica 2017, 102, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Larrán, A.; Cervantes, F.; Bellosillo, B.; Giralt, M.; Juliá, A.; Hernández-Boluda, J.C.; Bosch, A.; Hernández-Nieto, L.; Clapés, V.; Burgaleta, C.; et al. Essential thrombocythemia in young individuals: Frequency and risk factors for vascular events and evolution to myelofibrosis in 126 patients. Leukemia 2007, 21, 1218–2123. [Google Scholar] [CrossRef] [PubMed]
- Srour, S.A.; Devesa, S.S.; Morton, L.M.; Check, D.P.; Curtis, R.E.; Linet, M.S.; Dores, G.M. Incidence and patient survival of myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms in the United States, 2001–2012. Br. J. Haematol. 2016, 174, 382–396. [Google Scholar] [CrossRef] [Green Version]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and personalized prognosis in myeloproliferative neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.A.; Duncombe, A.S.; Hughes, M.; Mills, M.E.; Wilson, J.C.; McMullin, M.F. Environmental, lifestyle, and familial/ethnic factors associated with myeloproliferative neoplasms. Am. J. Hematol. 2012, 87, 175–182. [Google Scholar] [CrossRef]
- Jayasuriya, N.A.; Ellervik, C.; Hasselbalch, H.C.; Sørensen, A. Cigarette smoking, complete blood count, and myeloproliferative neoplasms—A meta-analysis. Blood 2017, 130, 4199. [Google Scholar] [CrossRef]
- Jayasuriya, N.A.; Kjaergaard, A.D.; Pedersen, K.M.; Sørensen, A.L.; Bak, M.; Larsen, M.K.; Nordestgaard, B.G.; Bojesen, S.E.; Çolak, Y.; Skov, V.; et al. Smoking, blood cells and myeloproliferative neoplasms: Meta-analysis and Mendelian randomization of 2·3 million people. Br. J. Haematol. 2020, 189, 323–334. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guijarro-Hernández, A.; Vizmanos, J.L. A Broad Overview of Signaling in Ph-Negative Classic Myeloproliferative Neoplasms. Cancers 2021, 13, 984. https://doi.org/10.3390/cancers13050984
Guijarro-Hernández A, Vizmanos JL. A Broad Overview of Signaling in Ph-Negative Classic Myeloproliferative Neoplasms. Cancers. 2021; 13(5):984. https://doi.org/10.3390/cancers13050984
Chicago/Turabian StyleGuijarro-Hernández, Ana, and José Luis Vizmanos. 2021. "A Broad Overview of Signaling in Ph-Negative Classic Myeloproliferative Neoplasms" Cancers 13, no. 5: 984. https://doi.org/10.3390/cancers13050984
APA StyleGuijarro-Hernández, A., & Vizmanos, J. L. (2021). A Broad Overview of Signaling in Ph-Negative Classic Myeloproliferative Neoplasms. Cancers, 13(5), 984. https://doi.org/10.3390/cancers13050984