
Oxidative Stress in the Tumor Microenvironment and Its Relevance to Cancer Immunotherapy

 ,
,  and
and {kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. The Impact of Oxidative Stress on Cancer Cells
2.1. ROS in Tumor Initiation, Progression, and Survival
2.2. ROS in Tumor Angiogenesis, Metastasis, and Chemoresistance
3. The Impact of Oxidative Stress on Immune Cells in the TME
3.1. The Impact of ROS on T Cells and NK Cells
3.2. Oxidative Stress and Antigen Presentation
3.3. Oxidative Stress and Immunosuppressor Cells
4. Cancer Therapies Targeting Tumor Redox
4.1. The Use of Antioxidants for Cancer Prevention
4.2. The Use of Pro-Oxidants in Cancer Therapy
5. The Impact of Cancer Immunotherapies on Oxidative Stress in the TME
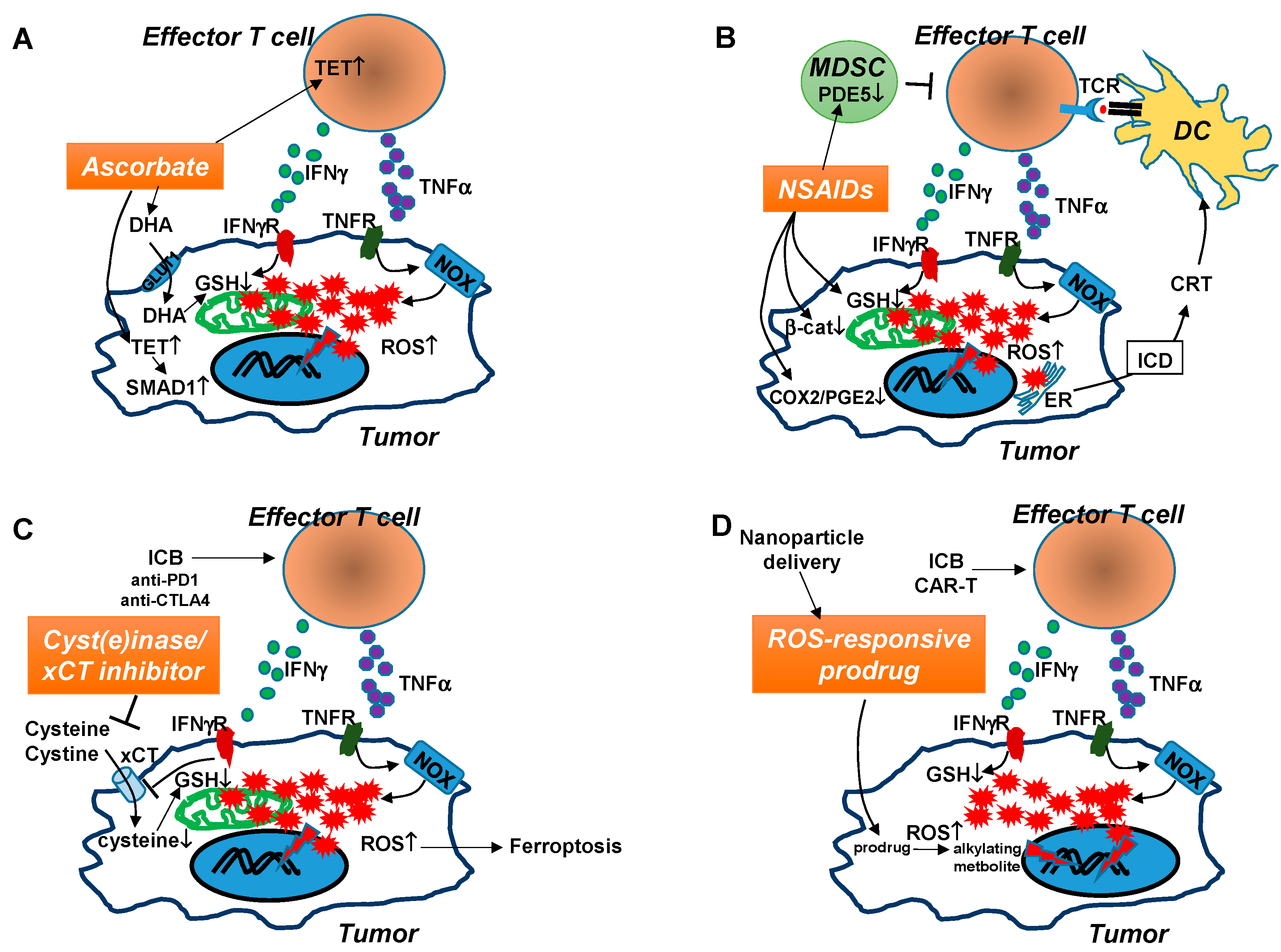
6. Emerging ROS-Modulating Agents with the Potential to Enhance the Efficacy of Cancer Immunotherapy
6.1. High Dose Ascorbate (Vitamin C)
6.2. Non-Steroid Anti-Inflammatory Drugs (NSAIDs)
6.3. xCT Inhibitors and Cyst(e)inase
6.4. ROS-Responsive Prodrugs
7. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Chio, I.I.C.; Tuveson, D.A. ROS in Cancer: The Burning Question. Trends Mol. Med. 2017, 23, 411–429. [Google Scholar] [CrossRef] [Green Version]
- Reczek, C.R.; Chandel, N.S. The Two Faces of Reactive Oxygen Species in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 79–98. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Yoo, Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Nogueira, V.; Hay, N. Molecular pathways: Reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrend, L.; Henderson, G.; Zwacka, R.M. Reactive oxygen species in oncogenic transformation. Biochem. Soc. Trans. 2003, 31, 1441–1444. [Google Scholar] [CrossRef]
- Hu, Y.; Rosen, D.G.; Zhou, Y.; Feng, L.; Yang, G.; Liu, J.; Huang, P. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: Role in cell proliferation and response to oxidative stress. J. Biol. Chem. 2005, 280, 39485–39492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pervaiz, S.; Clement, M.V. Tumor intracellular redox status and drug resistance--serendipity or a causal relationship? Curr. Pharm. Des. 2004, 10, 1969–1977. [Google Scholar] [CrossRef]
- Clerkin, J.S.; Naughton, R.; Quiney, C.; Cotter, T.G. Mechanisms of ROS modulated cell survival during carcinogenesis. Cancer Lett. 2008, 266, 30–36. [Google Scholar] [CrossRef]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, B.; Radeke, H.H.; Selle, S.; Younes, M.; Sies, H.; Resch, K.; Habermehl, G.G. Human fibroblasts release reactive oxygen species in response to interleukin-1 or tumour necrosis factor-alpha. Biochem. J. 1989, 263, 539–545. [Google Scholar] [CrossRef]
- Ohba, M.; Shibanuma, M.; Kuroki, T.; Nose, K. Production of hydrogen peroxide by transforming growth factor-beta 1 and its involvement in induction of egr-1 in mouse osteoblastic cells. J. Cell Biol. 1994, 126, 1079–1088. [Google Scholar] [CrossRef]
- Lo, Y.Y.; Cruz, T.F. Involvement of reactive oxygen species in cytokine and growth factor induction of c-fos expression in chondrocytes. J. Biol. Chem. 1995, 270, 11727–11730. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef] [Green Version]
- Roy, D.; Sarkar, S.; Felty, Q. Levels of IL-1 beta control stimulatory/inhibitory growth of cancer cells. Front. Biosci. 2006, 11, 889–898. [Google Scholar] [CrossRef] [Green Version]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; GoldschmidtClermont, P.J. Mitogenic signaling mediated by oxidants in ras-transformed fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar] [CrossRef]
- Mitsushita, J.; Lambeth, J.D.; Kamata, T. The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer Res. 2004, 64, 3580–3585. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogrunc, M.; Di Micco, R.; Liontos, M.; Bombardelli, L.; Mione, M.; Fumagalli, M.; Gorgoulis, V.G.; d’Adda di Fagagna, F. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 2014, 21, 998–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, Y.S.; Kang, S.W.; Seo, M.S.; Baines, I.C.; Tekle, E.; Chock, P.B.; Rhee, S.G. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide—Role in EGF receptor-mediated tyrosine phosphorylation. J. Biol. Chem. 1997, 272, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Sirokmány, G.; Pató, A.; Zana, M.; Donkó, Á.; Bíró, A.; Nagy, P.; Geiszt, M. Epidermal growth factor-induced hydrogen peroxide production is mediated by dual oxidase 1. Free Radic. Biol. Med. 2016, 97, 204–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmeen, A.; Andersen, J.N.; Myers, M.P.; Meng, T.C.; Hinks, J.A.; Tonks, N.K.; Barford, D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 2003, 423, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Phosphoinositide 3-Kinase/Akt Signaling and Redox Metabolism in Cancer. Front. Oncol. 2018, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a Putative Protein Tyrosine Phosphatase Gene Mutated in Human Brain, Breast, and Prostate Cancer. Science 1997, 275, 1943. [Google Scholar] [CrossRef]
- Wu, H.; Goel, V.; Haluska, F.G. PTEN signaling pathways in melanoma. Oncogene 2003, 22, 3113–3122. [Google Scholar] [CrossRef] [Green Version]
- Verrastro, I.; Tveen-Jensen, K.; Woscholski, R.; Spickett, C.M.; Pitt, A.R. Reversible oxidation of phosphatase and tensin homolog (PTEN) alters its interactions with signaling and regulatory proteins. Free Radic. Biol. Med. 2016, 90, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Seth, D.; Rudolph, J. Redox Regulation of MAP Kinase Phosphatase 3. Biochemistry 2006, 45, 8476–8487. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.; Cheong, Y.-K.; Kim, N.-H.; Chung, H.-T.; Kang, D.; Pae, H.-O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal. Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef]
- Zhang, J.; Johnston, G.; Stebler, B.; Keller, E.T. Hydrogen Peroxide Activates NFκB and the Interleukin-6 Promoter Through NFκB-Inducing Kinase. Antioxid. Redox Signal. 2001, 3, 493–504. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.-g. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lingappan, K. NF-κB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Zhang, J.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Kumar, A.P.; Ahn, K.S.; Sethi, G. NF-kappaB in cancer therapy. Arch. Toxicol. 2015, 89, 711–731. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Nakamura, Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008, 266, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.S. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006, 25, 695–705. [Google Scholar] [CrossRef]
- Nishikawa, M. Reactive oxygen species in tumor metastasis. Cancer Lett. 2008, 266, 53–59. [Google Scholar] [CrossRef]
- Xia, C.; Meng, Q.; Liu, L.-Z.; Rojanasakul, Y.; Wang, X.-R.; Jiang, B.-H. Reactive Oxygen Species Regulate Angiogenesis and Tumor Growth through Vascular Endothelial Growth Factor. Cancer Res. 2007, 67, 10823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, D.; Kato, M.; Nakayama, J.; Miyagawa, S.; Kamata, T. NADPH oxidase 1 plays a critical mediating role in oncogenic Ras-induced vascular endothelial growth factor expression. Oncogene 2008, 27, 4724–4732. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef] [Green Version]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Schumacker, P.T.; Chandel, N.S. Mitochondrial reactive oxygen species trigger hypoxia-inducible factor-dependent extension of the replicative life span during hypoxia. Mol. Cell Biol. 2007, 27, 5737–5745. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia-inducible factors: Coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. EMBO J. 2017, 36, 252–259. [Google Scholar] [CrossRef]
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-Modified Cancer Cell Metabolism. Front. Cell Dev. Biol. 2019, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binker, M.G.; Binker-Cosen, A.A.; Richards, D.; Oliver, B.; Cosen-Binker, L.I. EGF promotes invasion by PANC-1 cells through Rac1/ROS-dependent secretion and activation of MMP-2. Biochem. Biophys. Res. Commun. 2009, 379, 445–450. [Google Scholar] [CrossRef]
- Kar, S.; Subbaram, S.; Carrico, P.M.; Melendez, J.A. Redox-control of matrix metalloproteinase-1: A critical link between free radicals, matrix remodeling and degenerative disease. Respir. Physiol. Neurobiol. 2010, 174, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Ryoo, I.-g.; Lee, S.-h.; Kwak, M.-K. Redox Modulating NRF2: A Potential Mediator of Cancer Stem Cell Resistance. Oxidative Med. Cell. Longev. 2016, 2016, 2428153. [Google Scholar] [CrossRef] [Green Version]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Zimta, A.-A.; Cenariu, D.; Irimie, A.; Magdo, L.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. The Role of Nrf2 Activity in Cancer Development and Progression. Cancers 2019, 11, 1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, N.M.; Kleeberger, S.R.; Yamamoto, M.; Kensler, T.W.; Scollick, C.; Biswal, S.; Reddy, S.P. Genetic dissection of the Nrf2-dependent redox signaling-regulated transcriptional programs of cell proliferation and cytoprotection. Physiol. Genom. 2007, 32, 74–81. [Google Scholar] [CrossRef] [Green Version]
- Nioi, P.; Hayes, J.D. Contribution of NAD(P)H:quinone oxidoreductase 1 to protection against carcinogenesis, and regulation of its gene by the Nrf2 basic-region leucine zipper and the arylhydrocarbon receptor basic helix-loop-helix transcription factors. Mutat. Res. 2004, 555, 149–171. [Google Scholar] [CrossRef]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. A catalogue of somatic NRF2 gain-of-function mutations in cancer. Sci. Rep. 2018, 8, 12846. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Hayes, J.D.; McMahon, M. The double-edged sword of Nrf2: Subversion of redox homeostasis during the evolution of cancer. Mol. Cell 2006, 21, 732–734. [Google Scholar] [CrossRef]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef] [PubMed]
- Fiocchetti, M.; Fernandez, V.S.; Montalesi, E.; Marino, M. Neuroglobin: A Novel Player in the Oxidative Stress Response of Cancer Cells. Oxid Med. Cell Longev 2019, 2019, 6315034. [Google Scholar] [CrossRef] [Green Version]
- Solar Fernandez, V.; Cipolletti, M.; Ascenzi, P.; Marino, M.; Fiocchetti, M. Neuroglobin As Key Mediator in the 17beta-Estradiol-Induced Antioxidant Cell Response to Oxidative Stress. Antioxid Redox Signal. 2020, 32, 217–227. [Google Scholar] [CrossRef]
- Fiocchetti, M.; Solar Fernandez, V.; Segatto, M.; Leone, S.; Cercola, P.; Massari, A.; Cavaliere, F.; Marino, M. Extracellular Neuroglobin as a Stress-Induced Factor Activating Pre-Adaptation Mechanisms against Oxidative Stress and Chemotherapy-Induced Cell Death in Breast Cancer. Cancers 2020, 12, 2451. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Valacchi, G.; Virgili, F.; Cervellati, C.; Pecorelli, A. OxInflammation: From Subclinical Condition to Pathological Biomarker. Front. Physiol. 2018, 9, 858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, C.; Cunningham-Bussel, A. Beyond oxidative stress: An immunologist’s guide to reactive oxygen species. Nat. Rev. Immunol. 2013, 13, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Song, M.; Zhang, B.; Zhang, Y. Reactive Oxygen Species Regulate T Cell Immune Response in the Tumor Microenvironment. Oxid Med. Cell Longev 2016, 2016, 1580967. [Google Scholar] [CrossRef] [Green Version]
- Franchina, D.G.; Dostert, C.; Brenner, D. Reactive Oxygen Species: Involvement in T Cell Signaling and Metabolism. Trends Immunol. 2018, 39, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Chandel, N.S. Regulation of redox balance in cancer and T cells. J. Biol. Chem. 2018, 293, 7499–7507. [Google Scholar] [CrossRef]
- Kotsafti, A.; Scarpa, M.; Castagliuolo, I.; Scarpa, M. Reactive Oxygen Species and Antitumor Immunity-From Surveillance to Evasion. Cancers 2020, 12, 1748. [Google Scholar] [CrossRef]
- Mullen, L.; Mengozzi, M.; Hanschmann, E.M.; Alberts, B.; Ghezzi, P. How the redox state regulates immunity. Free Radic. Biol. Med. 2020, 157, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Muri, J.; Kopf, M. Redox regulation of immunometabolism. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Van Loenhout, J.; Peeters, M.; Bogaerts, A.; Smits, E.; Deben, C. Oxidative Stress-Inducing Anticancer Therapies: Taking a Closer Look at Their Immunomodulating Effects. Antioxidants 2020, 9, 1188. [Google Scholar] [CrossRef]
- Reth, M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat. Immunol. 2002, 3, 1129–1134. [Google Scholar] [CrossRef]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Hildeman, D.A.; Mitchell, T.; Teague, T.K.; Henson, P.; Day, B.J.; Kappler, J.; Marrack, P.C. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity 1999, 10, 735–744. [Google Scholar] [CrossRef] [Green Version]
- Kono, K.; Salazar-Onfray, F.; Petersson, M.; Hansson, J.; Masucci, G.; Wasserman, K.; Nakazawa, T.; Anderson, P.; Kiessling, R. Hydrogen peroxide secreted by tumor-derived macrophages down-modulates signal-transducing zeta molecules and inhibits tumor-specific T cell-and natural killer cell-mediated cytotoxicity. Eur. J. Immunol. 1996, 26, 1308–1313. [Google Scholar] [CrossRef]
- Nakamura, K.; Matsunaga, K. Susceptibility of natural killer (NK) cells to reactive oxygen species (ROS) and their restoration by the mimics of superoxide dismutase (SOD). Cancer Biother. Radiopharm. 1998, 13, 275–290. [Google Scholar] [CrossRef]
- Malmberg, K.J.; Arulampalam, V.; Ichihara, F.; Petersson, M.; Seki, K.; Andersson, T.; Lenkei, R.; Masucci, G.; Pettersson, S.; Kiessling, R. Inhibition of activated/memory (CD45RO(+)) T cells by oxidative stress associated with block of NF-kappaB activation. J. Immunol. 2001, 167, 2595–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, T.W.; Grusdat, M.; Duncan, G.S.; Dostert, C.; Nonnenmacher, Y.; Cox, M.; Binsfeld, C.; Hao, Z.; Brustle, A.; Itsumi, M.; et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunity 2017, 46, 1089–1090. [Google Scholar] [CrossRef]
- Kesarwani, P.; Al-Khami, A.A.; Scurti, G.; Thyagarajan, K.; Kaur, N.; Husain, S.; Fang, Q.; Naga, O.S.; Simms, P.; Beeson, G.; et al. Promoting thiol expression increases the durability of antitumor T-cell functions. Cancer Res. 2014, 74, 6036–6047. [Google Scholar] [CrossRef] [Green Version]
- Scheffel, M.J.; Scurti, G.; Simms, P.; Garrett-Mayer, E.; Mehrotra, S.; Nishimura, M.I.; Voelkel-Johnson, C. Efficacy of Adoptive T-cell Therapy Is Improved by Treatment with the Antioxidant N-Acetyl Cysteine, Which Limits Activation-Induced T-cell Death. Cancer Res. 2016, 76, 6006–6016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilipow, K.; Scamardella, E.; Puccio, S.; Gautam, S.; De Paoli, F.; Mazza, E.M.; De Simone, G.; Polletti, S.; Buccilli, M.; Zanon, V.; et al. Antioxidant metabolism regulates CD8+ T memory stem cell formation and antitumor immunity. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Scheffel, M.J.; Scurti, G.; Wyatt, M.M.; Garrett-Mayer, E.; Paulos, C.M.; Nishimura, M.I.; Voelkel-Johnson, C. N-acetyl cysteine protects anti-melanoma cytotoxic T cells from exhaustion induced by rapid expansion via the downmodulation of Foxo1 in an Akt-dependent manner. Cancer Immunol. Immunother. 2018, 67, 691–702. [Google Scholar] [CrossRef]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed Catalase Protects Chimeric Antigen Receptor-Redirected T Cells as well as Bystander Cells from Oxidative Stress-Induced Loss of Antitumor Activity. J. Immunol. 2016, 196, 759–766. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Neo, S.Y.; Chen, Z.; Cui, W.; Chen, Y.; Guo, M.; Wang, Y.; Xu, H.; Kurzay, A.; Alici, E.; et al. Thioredoxin activity confers resistance against oxidative stress in tumor-infiltrating NK cells. J. Clin. Invest. 2020, 130, 5508–5522. [Google Scholar] [CrossRef] [PubMed]
- Mantegazza, A.R.; Savina, A.; Vermeulen, M.; Perez, L.; Geffner, J.; Hermine, O.; Rosenzweig, S.D.; Faure, F.; Amigorena, S. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood 2008, 112, 4712–4722. [Google Scholar] [CrossRef] [Green Version]
- Weiskopf, D.; Schwanninger, A.; Weinberger, B.; Almanzar, G.; Parson, W.; Buus, S.; Lindner, H.; Grubeck-Loebenstein, B. Oxidative stress can alter the antigenicity of immunodominant peptides. J. Leukoc. Biol. 2010, 87, 165–172. [Google Scholar] [CrossRef]
- Trujillo, J.A.; Croft, N.P.; Dudek, N.L.; Channappanavar, R.; Theodossis, A.; Webb, A.I.; Dunstone, M.A.; Illing, P.T.; Butler, N.S.; Fett, C.; et al. The cellular redox environment alters antigen presentation. J. Biol. Chem. 2014, 289, 27979–27991. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kepp, O.; Menger, L.; Vacchelli, E.; Locher, C.; Adjemian, S.; Yamazaki, T.; Martins, I.; Sukkurwala, A.Q.; Michaud, M.; Senovilla, L.; et al. Crosstalk between ER stress and immunogenic cell death. Cytokine Growth Factor Rev. 2013, 24, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Panaretakis, T.; Kepp, O.; Brockmeier, U.; Tesniere, A.; Bjorklund, A.C.; Chapman, D.C.; Durchschlag, M.; Joza, N.; Pierron, G.; van Endert, P.; et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009, 28, 578–590. [Google Scholar] [CrossRef]
- Deng, H.; Zhou, Z.; Yang, W.; Lin, L.S.; Wang, S.; Niu, G.; Song, J.; Chen, X. Endoplasmic Reticulum Targeting to Amplify Immunogenic Cell Death for Cancer Immunotherapy. Nano Lett. 2020, 20, 1928–1933. [Google Scholar] [CrossRef]
- Kazama, H.; Ricci, J.E.; Herndon, J.M.; Hoppe, G.; Green, D.R.; Ferguson, T.A. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 2008, 29, 21–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, H.; Yang, W.; Zhou, Z.; Tian, R.; Lin, L.; Ma, Y.; Song, J.; Chen, X. Targeted scavenging of extracellular ROS relieves suppressive immunogenic cell death. Nat. Commun. 2020, 11, 4951. [Google Scholar] [CrossRef] [PubMed]
- Kraaij, M.D.; Savage, N.D.; van der Kooij, S.W.; Koekkoek, K.; Wang, J.; van den Berg, J.M.; Ottenhoff, T.H.; Kuijpers, T.W.; Holmdahl, R.; van Kooten, C.; et al. Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 17686–17691. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Garg, S.K.; Banerjee, R. Regulatory T cells interfere with glutathione metabolism in dendritic cells and T cells. J. Biol. Chem. 2010, 285, 41525–41532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efimova, O.; Szankasi, P.; Kelley, T.W. Ncf1 (p47phox) is essential for direct regulatory T cell mediated suppression of CD4+ effector T cells. PLoS ONE 2011, 6, e16013. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.; Shimojima, Y.; Shirai, T.; Li, Y.; Ju, J.; Yang, Z.; Tian, L.; Goronzy, J.J.; Weyand, C.M. NADPH oxidase deficiency underlies dysfunction of aged CD8+ Tregs. J. Clin. Invest. 2016, 126, 1953–1967. [Google Scholar] [CrossRef] [PubMed]
- Mougiakakos, D.; Johansson, C.C.; Kiessling, R. Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood 2009, 113, 3542–3545. [Google Scholar] [CrossRef] [Green Version]
- Mougiakakos, D.; Johansson, C.C.; Jitschin, R.; Bottcher, M.; Kiessling, R. Increased thioredoxin-1 production in human naturally occurring regulatory T cells confers enhanced tolerance to oxidative stress. Blood 2011, 117, 857–861. [Google Scholar] [CrossRef] [Green Version]
- Kurniawan, H.; Franchina, D.G.; Guerra, L.; Bonetti, L.; Baguet, L.S.; Grusdat, M.; Schlicker, L.; Hunewald, O.; Dostert, C.; Merz, M.P.; et al. Glutathione Restricts Serine Metabolism to Preserve Regulatory T Cell Function. Cell Metab 2020, 31, 920–936. [Google Scholar] [CrossRef]
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wang, W.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W.; et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 2017, 18, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef]
- Schmielau, J.; Finn, O.J. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. 2001, 61, 4756–4760. [Google Scholar] [PubMed]
- Aarts, C.E.M.; Hiemstra, I.H.; Beguin, E.P.; Hoogendijk, A.J.; Bouchmal, S.; van Houdt, M.; Tool, A.T.J.; Mul, E.; Jansen, M.H.; Janssen, H.; et al. Activated neutrophils exert myeloid-derived suppressor cell activity damaging T cells beyond repair. Blood Adv. 2019, 3, 3562–3574. [Google Scholar] [CrossRef] [Green Version]
- Otsuji, M.; Kimura, Y.; Aoe, T.; Okamoto, Y.; Saito, T. Oxidative stress by tumor-derived macrophages suppresses the expression of CD3 zeta chain of T-cell receptor complex and antigen-specific T-cell responses. Proc. Natl. Acad. Sci. USA 1996, 93, 13119–13124. [Google Scholar] [CrossRef] [Green Version]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8(+) T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Wei, J.; Guo, G.; Zhou, J. Norepinephrine-induced myeloid-derived suppressor cells block T-cell responses via generation of reactive oxygen species. Immunopharmacol. Immunotoxicol. 2015, 37, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Corzo, C.A.; Cotter, M.J.; Cheng, P.Y.; Cheng, F.D.; Kusmartsev, S.; Sotomayor, E.; Padhya, T.; McCaffrey, T.V.; McCaffrey, J.C.; Gabrilovich, D.I. Mechanism Regulating Reactive Oxygen Species in Tumor-Induced Myeloid-Derived Suppressor Cells. J. Immunol. 2009, 182, 5693–5701. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Gabrilovich, D.I. Inhibition of myeloid cell differentiation in cancer: The role of reactive oxygen species. J. Leukoc Biol. 2003, 74, 186–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beury, D.W.; Carter, K.A.; Nelson, C.; Sinha, P.; Hanson, E.; Nyandjo, M.; Fitzgerald, P.J.; Majeed, A.; Wali, N.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cell Survival and Function Are Regulated by the Transcription Factor Nrf2. J. Immunol. 2016, 196, 3470–3478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusmartsev, S.; Nefedova, Y.; Yoder, D.; Gabrilovich, D.I. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J. Immunol. 2004, 172, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Gabrilovich, D.I. Molecular pathways: Tumor-infiltrating myeloid cells and reactive oxygen species in regulation of tumor microenvironment. Clin. Cancer Res. 2012, 18, 4877–4882. [Google Scholar] [CrossRef] [Green Version]
- Ohl, K.; Tenbrock, K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front. Immunol. 2018, 9, 2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrand-Rosenberg, S.; Beury, D.W.; Parker, K.H.; Horn, L.A. Survival of the fittest: How myeloid-derived suppressor cells survive in the inhospitable tumor microenvironment. Cancer Immunol. Immunother. 2020, 69, 215–221. [Google Scholar] [CrossRef]
- Wang, J.; Yi, J. Cancer cell killing via ROS: To increase or decrease, that is the question. Cancer Biol. Ther. 2008, 7, 1875–1884. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Raza, M.H.; Siraj, S.; Arshad, A.; Waheed, U.; Aldakheel, F.; Alduraywish, S.; Arshad, M. ROS-modulated therapeutic approaches in cancer treatment. J. Cancer Res. Clin. Oncol. 2017, 143, 1789–1809. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxid Med. Cell Longev 2019, 2019, 5381692. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Khan, N.; Afaq, F.; Mukhtar, H. Cancer chemoprevention through dietary antioxidants: Progress and promise. Antioxid Redox Signal. 2008, 10, 475–510. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Zhang, H.; Dinavahi, R.; Li, F.; Xiang, Y.; Raman, V.; Bhujwalla, Z.M.; Felsher, D.W.; Cheng, L.; Pevsner, J.; et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell 2007, 12, 230–238. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, M.; Hyoudou, K.; Kobayashi, Y.; Umeyama, Y.; Takakura, Y.; Hashida, M. Inhibition of metastatic tumor growth by targeted delivery of antioxidant enzymes. J. Control. Release 2005, 109, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, S.; Jiang, X.; Weydert, C.; Zhang, Y.; Zhang, H.J.; Goswami, P.C.; Ritchie, J.M.; Oberley, L.W.; Buettner, G.R. Manganese superoxide dismutase overexpression inhibits the growth of androgen-independent prostate cancer cells. Oncogene 2005, 24, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Du, J.; Zhang, Y.; Sun, W.; Smith, B.J.; Oberley, L.W.; Cullen, J.J. Suppression of the malignant phenotype in pancreatic cancer by overexpression of phospholipid hydroperoxide glutathione peroxidase. Hum. Gene Ther. 2006, 17, 105–116. [Google Scholar] [CrossRef]
- Nelson, S.K.; Bose, S.K.; Grunwald, G.K.; Myhill, P.; McCord, J.M. The induction of human superoxide dismutase and catalase in vivo: A fundamentally new approach to antioxidant therapy. Free Radic. Biol. Med. 2006, 40, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Teoh-Fitzgerald, M.L.; Fitzgerald, M.P.; Zhong, W.; Askeland, R.W.; Domann, F.E. Epigenetic reprogramming governs EcSOD expression during human mammary epithelial cell differentiation, tumorigenesis and metastasis. Oncogene 2014, 33, 358–368. [Google Scholar] [CrossRef] [Green Version]
- The Effect of Vitamin E and Beta Carotene on the Incidence of Lung Cancer and Other Cancers in Male Smokers. N. Engl. J. Med. 1994, 330, 1029–1035. [CrossRef]
- Omenn, G.S.; Goodman, G.E.; Thornquist, M.D.; Balmes, J.; Cullen, M.R.; Glass, A.; Keogh, J.P.; Meyskens, F.L.; Valanis, B.; Williams, J.H.; et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N. Engl. J. Med. 1996, 334, 1150–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Andrea, G.M. Use of antioxidants during chemotherapy and radiotherapy should be avoided. CA Cancer J. Clin. 2005, 55, 319–321. [Google Scholar] [CrossRef]
- Seifried, H.E.; Anderson, D.E.; Fisher, E.I.; Milner, J.A. A review of the interaction among dietary antioxidants and reactive oxygen species. J. Nutr. Biochem. 2007, 18, 567–579. [Google Scholar] [CrossRef]
- Klein, E.A.; Thompson, I.M., Jr.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef]
- Chandel, N.S.; Tuveson, D.A. The promise and perils of antioxidants for cancer patients. N. Engl. J. Med. 2014, 371, 177–178. [Google Scholar] [CrossRef]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyurek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re308. [Google Scholar] [CrossRef] [PubMed]
- Peiris-Pages, M.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Metastasis and Oxidative Stress: Are Antioxidants a Metabolic Driver of Progression? Cell Metab. 2015, 22, 956–958. [Google Scholar] [CrossRef] [Green Version]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T.; et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 2019, 178, 330–345. [Google Scholar] [CrossRef]
- Firczuk, M.; Bajor, M.; Graczyk-Jarzynka, A.; Fidyt, K.; Goral, A.; Zagozdzon, R. Harnessing altered oxidative metabolism in cancer by augmented prooxidant therapy. Cancer Lett. 2020, 471, 1–11. [Google Scholar] [CrossRef]
- Berndtsson, M.; Hagg, M.; Panaretakis, T.; Havelka, A.M.; Shoshan, M.C.; Linder, S. Acute apoptosis by cisplatin requires induction of reactive oxygen species but is not associated with damage to nuclear DNA. Int. J. Cancer 2007, 120, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Hwang, P.M.; Bunz, F.; Yu, J.; Rago, C.; Chan, T.A.; Murphy, M.P.; Kelso, G.F.; Smith, R.A.; Kinzler, K.W.; Vogelstein, B. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat. Med. 2001, 7, 1111–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, I.; Niethammer, D.; Bruchelt, G. Anthracycline-derived chemotherapeutics in apoptosis and free radical cytotoxicity (Review). Int. J. Mol. Med. 1998, 1, 491–494. [Google Scholar] [CrossRef]
- De Boo, S.; Kopecka, J.; Brusa, D.; Gazzano, E.; Matera, L.; Ghigo, D.; Bosia, A.; Riganti, C. iNOS activity is necessary for the cytotoxic and immunogenic effects of doxorubicin in human colon cancer cells. Mol. Cancer 2009, 8, 108. [Google Scholar] [CrossRef] [Green Version]
- Varbiro, G.; Veres, B.; Gallyas, F., Jr.; Sumegi, B. Direct effect of Taxol on free radical formation and mitochondrial permeability transition. Free Radic. Biol. Med. 2001, 31, 548–558. [Google Scholar] [CrossRef]
- Ramanathan, B.; Jan, K.Y.; Chen, C.H.; Hour, T.C.; Yu, H.J.; Pu, Y.S. Resistance to paclitaxel is proportional to cellular total antioxidant capacity. Cancer Res. 2005, 65, 8455–8460. [Google Scholar] [CrossRef] [Green Version]
- Chiu, W.H.; Luo, S.J.; Chen, C.L.; Cheng, J.H.; Hsieh, C.Y.; Wang, C.Y.; Huang, W.C.; Su, W.C.; Lin, C.F. Vinca alkaloids cause aberrant ROS-mediated JNK activation, Mcl-1 downregulation, DNA damage, mitochondrial dysfunction, and apoptosis in lung adenocarcinoma cells. Biochem. Pharmacol. 2012, 83, 1159–1171. [Google Scholar] [CrossRef]
- Chou, W.C.; Jie, C.; Kenedy, A.A.; Jones, R.J.; Trush, M.A.; Dang, C.V. Role of NADPH oxidase in arsenic-induced reactive oxygen species formation and cytotoxicity in myeloid leukemia cells. Proc. Natl. Acad. Sci. USA 2004, 101, 4578–4583. [Google Scholar] [CrossRef] [Green Version]
- Griffith, O.W.; Meister, A. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine). J. Biol. Chem. 1979, 254, 7558–7560. [Google Scholar] [CrossRef]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and Thioredoxin Antioxidant Pathways Synergize to Drive Cancer Initiation and Progression (vol 27, pg 211, 2015). Cancer Cell 2015, 27, 314. [Google Scholar] [CrossRef]
- Teppo, H.R.; Soini, Y.; Karihtala, P. Reactive Oxygen Species-Mediated Mechanisms of Action of Targeted Cancer Therapy. Oxid Med. Cell Longev. 2017, 2017, 1485283. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Hernandez, M.A.; de la Cruz-Ojeda, P.; Lopez-Grueso, M.J.; Navarro-Villaran, E.; Requejo-Aguilar, R.; Castejon-Vega, B.; Negrete, M.; Gallego, P.; Vega-Ochoa, A.; Victor, V.M.; et al. Integrated molecular signaling involving mitochondrial dysfunction and alteration of cell metabolism induced by tyrosine kinase inhibitors in cancer. Redox. Biol. 2020, 36, 101510. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Zhou, Y.; Zhang, H.; Demizu, Y.; Chen, Z.; Pelicano, H.; Chiao, P.J.; Achanta, G.; Arlinghaus, R.B.; Liu, J.S.; et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 2006, 10, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.X.; Yun, B.S.; Ma, M.; Basu, H.S.; Church, D.R.; Ingenhorst, G.; Huang, Y.; Yang, D.; Lohman, J.R.; Tang, G.L.; et al. Leinamycin E1 acting as an anticancer prodrug activated by reactive oxygen species. Proc. Natl Acad Sci USA 2015, 112, 8278–8283. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Winslow, M.M.; Magendantz, M.; Ouyang, C.; Dowdle, J.; Subramanian, A.; Lewis, T.A.; Maglathin, R.L.; Tolliday, N.; Jacks, T. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc. Natl. Acad. Sci. USA 2011, 108, 8773–8778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogura, A.; Oowada, S.; Kon, Y.; Hirayama, A.; Yasui, H.; Meike, S.; Kobayashi, S.; Kuwabara, M.; Inanami, O. Redox regulation in radiation-induced cytochrome c release from mitochondria of human lung carcinoma A549 cells. Cancer Lett. 2009, 277, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization. Int. J. Mol. Sci. 2016, 17, 102. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.E.; Mackall, C.L. CAR T cell therapy: Inroads to response and resistance. Nat. Rev. Immunol 2019, 19, 73–74. [Google Scholar] [CrossRef] [PubMed]
- Habtetsion, T.; Ding, Z.C.; Pi, W.; Li, T.; Lu, C.; Chen, T.; Xi, C.; Spartz, H.; Liu, K.; Hao, Z.; et al. Alteration of Tumor Metabolism by CD4+ T Cells Leads to TNF-alpha-Dependent Intensification of Oxidative Stress and Tumor Cell Death. Cell Metab. 2018, 28, 228–242. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Green, M.; Choi, J.E.; Gijon, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kryczek, I.; Dostal, L.; Lin, H.; Tan, L.; Zhao, L.; Lu, F.; Wei, S.; Maj, T.; Peng, D.; et al. Effector T Cells Abrogate Stroma-Mediated Chemoresistance in Ovarian Cancer. Cell 2016, 165, 1092–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cramer, S.L.; Saha, A.; Liu, J.; Tadi, S.; Tiziani, S.; Yan, W.; Triplett, K.; Lamb, C.; Alters, S.E.; Rowlinson, S.; et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med. 2017, 23, 120–127. [Google Scholar] [CrossRef]
- Tang, J.; Ramis-Cabrer, D.; Wang, X.; Barreiro, E. Immunotherapy with Monoclonal Antibodies in Lung Cancer of Mice: Oxidative Stress and Other Biological Events. Cancers 2019, 11, 1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 2012, 1826, 443–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Chapman, J.; Levine, M.; Polireddy, K.; Drisko, J.; Chen, Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci. Transl. Med. 2014, 6, 222ra218. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Xu, Y.; Xu, F.F.; Chaiswing, L.; Schnell, D.; Noel, T.; Wang, C.; Chen, J.; St Clair, D.K.; St Clair, W.H. RelB Expression Determines the Differential Effects of Ascorbic Acid in Normal and Cancer Cells. Cancer Res. 2017, 77, 1345–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, N.; Creagan, E.; Witzig, T.; Levine, M. Ascorbic Acid in Cancer Treatment: Let the Phoenix Fly. Cancer Cell 2018, 34, 700–706. [Google Scholar] [CrossRef] [Green Version]
- Vissers, M.C.M.; Das, A.B. Potential Mechanisms of Action for Vitamin C in Cancer: Reviewing the Evidence. Front. Physiol 2018, 9, 809. [Google Scholar] [CrossRef] [Green Version]
- Creagan, E.T.; Moertel, C.G.; O’Fallon, J.R.; Schutt, A.J.; O’Connell, M.J.; Rubin, J.; Frytak, S. Failure of high-dose vitamin C (ascorbic acid) therapy to benefit patients with advanced cancer. A controlled trial. N. Engl. J. Med. 1979, 301, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Moertel, C.G.; Fleming, T.R.; Creagan, E.T.; Rubin, J.; O’Connell, M.J.; Ames, M.M. High-dose vitamin C versus placebo in the treatment of patients with advanced cancer who have had no prior chemotherapy. A randomized double-blind comparison. N. Engl. J. Med. 1985, 312, 137–141. [Google Scholar] [CrossRef]
- Padayatty, S.J.; Sun, H.; Wang, Y.; Riordan, H.D.; Hewitt, S.M.; Katz, A.; Wesley, R.A.; Levine, M. Vitamin C pharmacokinetics: Implications for oral and intravenous use. Ann. Internt. Med. 2004, 140, 533–537. [Google Scholar] [CrossRef]
- Verrax, J.; Calderon, P.B. Pharmacologic concentrations of ascorbate are achieved by parenteral administration and exhibit antitumoral effects. Free Radic. Biol. Med. 2009, 47, 32–40. [Google Scholar] [CrossRef]
- Nauman, G.; Gray, J.C.; Parkinson, R.; Levine, M.; Paller, C.J. Systematic Review of Intravenous Ascorbate in Cancer Clinical Trials. Antioxidants 2018, 7, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenfeld, J.D.; Sibenaller, Z.A.; Mapuskar, K.A.; Wagner, B.A.; Cramer-Morales, K.L.; Furqan, M.; Sandhu, S.; Carlisle, T.L.; Smith, M.C.; Abu Hejleh, T.; et al. O2(-) and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell 2017, 32, 268. [Google Scholar] [CrossRef] [PubMed]
- Spitz, D.R.; Sim, J.E.; Ridnour, L.A.; Galoforo, S.S.; Lee, Y.J. Glucose deprivation-induced oxidative stress in human tumor cells. A fundamental defect in metabolism? Ann. N Y Acad. Sci. 2000, 899, 349–362. [Google Scholar] [CrossRef]
- Aykin-Burns, N.; Ahmad, I.M.; Zhu, Y.; Oberley, L.W.; Spitz, D.R. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem. J. 2009, 418, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Caltagirone, A.; Weiss, G.; Pantopoulos, K. Modulation of cellular iron metabolism by hydrogen peroxide. Effects of H2O2 on the expression and function of iron-responsive element-containing mRNAs in B6 fibroblasts. J. Biol. Chem. 2001, 276, 19738–19745. [Google Scholar] [CrossRef] [Green Version]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef]
- Shenoy, N.; Bhagat, T.; Nieves, E.; Stenson, M.; Lawson, J.; Choudhary, G.S.; Habermann, T.; Nowakowski, G.; Singh, R.; Wu, X.; et al. Upregulation of TET activity with ascorbic acid induces epigenetic modulation of lymphoma cells. Blood Cancer J. 2017, 7, e587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, N.; Bhagat, T.D.; Cheville, J.; Lohse, C.; Bhattacharyya, S.; Tischer, A.; Machha, V.; Gordon-Mitchell, S.; Choudhary, G.; Wong, L.F.; et al. Ascorbic acid-induced TET activation mitigates adverse hydroxymethylcytosine loss in renal cell carcinoma. J. Clin. Invest. 2019, 129, 1612–1625. [Google Scholar] [CrossRef] [Green Version]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luchtel, R.A.; Bhagat, T.; Pradhan, K.; Jacobs, W.R., Jr.; Levine, M.; Verma, A.; Shenoy, N. High-dose ascorbic acid synergizes with anti-PD1 in a lymphoma mouse model. Proc. Natl. Acad. Sci. USA 2020, 117, 1666–1677. [Google Scholar] [CrossRef] [Green Version]
- Magri, A.; Germano, G.; Lorenzato, A.; Lamba, S.; Chila, R.; Montone, M.; Amodio, V.; Ceruti, T.; Sassi, F.; Arena, S.; et al. High-dose vitamin C enhances cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaay8707. [Google Scholar] [CrossRef]
- Brune, K.; Patrignani, P. New insights into the use of currently available non-steroidal anti-inflammatory drugs. J. Pain Res. 2015, 8, 105–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janne, P.A.; Mayer, R.J. Chemoprevention of colorectal cancer. N. Engl. J. Med. 2000, 342, 1960–1968. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.J.; Giardiello, F.M. Chemoprevention strategies using NSAIDs and COX-2 inhibitors. Cancer Biol Ther 2003, 2, S140–S149. [Google Scholar] [CrossRef]
- Rao, C.V.; Reddy, B.S. NSAIDs and chemoprevention. Curr. Cancer Drug Targets 2004, 4, 29–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Dubois, R.N. Prostaglandins and cancer. Gut 2006, 55, 115–122. [Google Scholar] [CrossRef]
- Finetti, F.; Travelli, C.; Ercoli, J.; Colombo, G.; Buoso, E.; Trabalzini, L. Prostaglandin E2 and Cancer: Insight into Tumor Progression and Immunity. Biology 2020, 9, 434. [Google Scholar] [CrossRef]
- Tinsley, H.N.; Gary, B.D.; Keeton, A.B.; Lu, W.; Li, Y.; Piazza, G.A. Inhibition of PDE5 by sulindac sulfide selectively induces apoptosis and attenuates oncogenic Wnt/beta-catenin-mediated transcription in human breast tumor cells. Cancer Prev. Res. 2011, 4, 1275–1284. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Xi, Y.; Tinsley, H.N.; Gurpinar, E.; Gary, B.D.; Zhu, B.; Li, Y.; Chen, X.; Keeton, A.B.; Abadi, A.H.; et al. Sulindac selectively inhibits colon tumor cell growth by activating the cGMP/PKG pathway to suppress Wnt/beta-catenin signaling. Mol. Cancer Ther. 2013, 12, 1848–1859. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Chen, X.; Zhu, B.; Ramirez-Alcantara, V.; Canzoneri, J.C.; Lee, K.; Sigler, S.; Gary, B.; Li, Y.; Zhang, W.; et al. Suppression of beta-catenin/TCF transcriptional activity and colon tumor cell growth by dual inhibition of PDE5 and 10. Oncotarget 2015, 6, 27403–27415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Lee, K.; Xi, Y.; Zhu, B.; Gary, B.D.; Ramirez-Alcantara, V.; Gurpinar, E.; Canzoneri, J.C.; Fajardo, A.; Sigler, S.; et al. Phosphodiesterase 10A: A novel target for selective inhibition of colon tumor cell growth and beta-catenin-dependent TCF transcriptional activity. Oncogene 2015, 34, 1499–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsutsumi, S.; Gotoh, T.; Tomisato, W.; Mima, S.; Hoshino, T.; Hwang, H.J.; Takenaka, H.; Tsuchiya, T.; Mori, M.; Mizushima, T. Endoplasmic reticulum stress response is involved in nonsteroidal anti-inflammatory drug-induced apoptosis. Cell Death Differ. 2004, 11, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Adachi, M.; Sakamoto, H.; Kawamura, R.; Wang, W.; Imai, K.; Shinomura, Y. Nonsteroidal anti-inflammatory drugs and oxidative stress in cancer cells. Histol. Histopathol. 2007, 22, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.C.; Yang, C.R.; Cheng, C.L.; Raung, S.L.; Hung, Y.Y.; Chen, C.J. Indomethacin induces apoptosis in 786-O renal cell carcinoma cells by activating mitogen-activated protein kinases and AKT. Eur. J. Pharmacol. 2007, 563, 49–60. [Google Scholar] [CrossRef]
- Du, H.; Li, W.; Wang, Y.; Chen, S.; Zhang, Y. Celecoxib induces cell apoptosis coupled with up-regulation of the expression of VEGF by a mechanism involving ER stress in human colorectal cancer cells. Oncol. Rep. 2011, 26, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Lee, S.H.; Min, K.W.; McEntee, M.F.; Jeong, J.B.; Li, Q.; Baek, S.J. The involvement of endoplasmic reticulum stress in the suppression of colorectal tumorigenesis by tolfenamic acid. Cancer Prev. Res. 2013, 6, 1337–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, W.; Park, S.W.; Kwon, T.K.; Hah, J.H.; Sung, M.W. Endoplasmic reticulum stress response as a possible mechanism of cyclooxygenase-2-independent anticancer effect of celecoxib. Anticancer Res. 2014, 34, 1731–1735. [Google Scholar] [PubMed]
- Ralph, S.J.; Pritchard, R.; Rodriguez-Enriquez, S.; Moreno-Sanchez, R.; Ralph, R.K. Hitting the Bull’s-Eye in Metastatic Cancers-NSAIDs Elevate ROS in Mitochondria, Inducing Malignant Cell Death. Pharmaceuticals 2015, 8, 62–106. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, M.; Resnick, L.; Gamliel, E.; Kesaraju, S.; Weissbach, H.; Binninger, D. Sulindac enhances the killing of cancer cells exposed to oxidative stress. PLoS ONE 2009, 4, e5804. [Google Scholar] [CrossRef] [Green Version]
- Raza, H.; John, A.; Benedict, S. Acetylsalicylic acid-induced oxidative stress, cell cycle arrest, apoptosis and mitochondrial dysfunction in human hepatoma HepG2 cells. Eur. J. Pharmacol. 2011, 668, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Tse, A.K.; Cao, H.H.; Cheng, C.Y.; Kwan, H.Y.; Yu, H.; Fong, W.F.; Yu, Z.L. Indomethacin sensitizes TRAIL-resistant melanoma cells to TRAIL-induced apoptosis through ROS-mediated upregulation of death receptor 5 and downregulation of survivin. J. Invest. Dermatol. 2014, 134, 1397–1407. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, R.; Rodriguez-Enriquez, S.; Pacheco-Velazquez, S.C.; Bortnik, V.; Moreno-Sanchez, R.; Ralph, S. Celecoxib inhibits mitochondrial O2 consumption, promoting ROS dependent death of murine and human metastatic cancer cells via the apoptotic signalling pathway. Biochem. Pharmacol. 2018, 154, 318–334. [Google Scholar] [CrossRef] [Green Version]
- Ralph, S.J.; Nozuhur, S.; Moreno-Sánchez, R.; Rodríguez-Enríquez, S.; Pritchard, R. NSAID celecoxib: A potent mitochondrial pro-oxidant cytotoxic agent sensitizing metastatic cancers and cancer stem cells to chemotherapy. J. Cancer Metastasis Treat. 2018, 4, 49. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Rigas, B. The thioredoxin system mediates redox-induced cell death in human colon cancer cells: Implications for the mechanism of action of anticancer agents. Cancer Res. 2008, 68, 8269–8277. [Google Scholar] [CrossRef] [Green Version]
- Rousar, T.; Parik, P.; Kucera, O.; Bartos, M.; Cervinkova, Z. Glutathione reductase is inhibited by acetaminophen-glutathione conjugate in vitro. Physiol. Res. 2010, 59, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Raza, H.; John, A. Implications of altered glutathione metabolism in aspirin-induced oxidative stress and mitochondrial dysfunction in HepG2 cells. PLoS ONE 2012, 7, e36325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, M.; Javeed, A.; Ashraf, M.; Al-Zaubai, N.; Stewart, A.; Mukhtar, M.M. Non-steroidal anti-inflammatory drugs, tumour immunity and immunotherapy. Pharmacol. Res. 2012, 66, 7–18. [Google Scholar] [CrossRef]
- Serafini, P.; Meckel, K.; Kelso, M.; Noonan, K.; Califano, J.; Koch, W.; Dolcetti, L.; Bronte, V.; Borrello, I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J. Exp. Med. 2006, 203, 2691–2702. [Google Scholar] [CrossRef]
- Noonan, K.A.; Ghosh, N.; Rudraraju, L.; Bui, M.; Borrello, I. Targeting immune suppression with PDE5 inhibition in end-stage multiple myeloma. Cancer Immunol. Res. 2014, 2, 725–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, R.; Tong, J.; Risnik, D.; Leibowitz, B.J.; Wang, Y.-J.; Concha-Benavente, F.; DeLiberty, J.M.; Stolz, D.B.; Pai, R.K.; Ferris, R.L.; et al. Non-steroidal anti-inflammatory drugs induce immunogenic cell death in suppressing colorectal tumorigenesis. Oncogene 2021. [Google Scholar] [CrossRef] [PubMed]
- Majchrzak, K.; Nelson, M.H.; Bowers, J.S.; Bailey, S.R.; Wyatt, M.M.; Wrangle, J.M.; Rubinstein, M.P.; Varela, J.C.; Li, Z.; Himes, R.A.; et al. beta-catenin and PI3Kdelta inhibition expands precursor Th17 cells with heightened stemness and antitumor activity. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelenay, S.; van der Veen, A.G.; Bottcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Xia, X.; Huang, P. xCT: A Critical Molecule That Links Cancer Metabolism to Redox Signaling. Mol. Ther. 2020, 28, 2358–2366. [Google Scholar] [CrossRef]
- Huang, Y.; Dai, Z.; Barbacioru, C.; Sadee, W. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. 2005, 65, 7446–7454. [Google Scholar] [CrossRef] [Green Version]
- Lo, M.; Wang, Y.Z.; Gout, P.W. The x(c)- cystine/glutamate antiporter: A potential target for therapy of cancer and other diseases. J. Cell Physiol. 2008, 215, 593–602. [Google Scholar] [CrossRef]
- Daher, B.; Vucetic, M.; Pouyssegur, J. Cysteine Depletion, a Key Action to Challenge Cancer Cells to Ferroptotic Cell Death. Front. Oncol. 2020, 10, 723. [Google Scholar] [CrossRef]
- Wang, W.; Zou, W. Amino Acids and Their Transporters in T Cell Immunity and Cancer Therapy. Mol. Cell 2020, 80, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2020. [Google Scholar] [CrossRef]
- Gout, P.W.; Buckley, A.R.; Simms, C.R.; Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: A new action for an old drug. Leukemia 2001, 15, 1633–1640. [Google Scholar] [CrossRef] [Green Version]
- Nagane, M.; Kanai, E.; Shibata, Y.; Shimizu, T.; Yoshioka, C.; Maruo, T.; Yamashita, T. Sulfasalazine, an inhibitor of the cystine-glutamate antiporter, reduces DNA damage repair and enhances radiosensitivity in murine B16F10 melanoma. PLoS ONE 2018, 13, e0195151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iida, Y.; Okamoto-Katsuyama, M.; Maruoka, S.; Mizumura, K.; Shimizu, T.; Shikano, S.; Hikichi, M.; Takahashi, M.; Tsuya, K.; Okamoto, S.; et al. Effective ferroptotic small-cell lung cancer cell death from SLC7A11 inhibition by sulforaphane. Oncol. Lett. 2021, 21, 71. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC(-)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, M.; Kusumi, R.; Hamashima, S.; Kobayashi, S.; Sasaki, S.; Komiyama, Y.; Izumikawa, T.; Conrad, M.; Bannai, S.; Sato, H. The ferroptosis inducer erastin irreversibly inhibits system xc- and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Sci. Rep. 2018, 8, 968. [Google Scholar] [CrossRef] [Green Version]
- Cobler, L.; Zhang, H.; Suri, P.; Park, C.; Timmerman, L.A. xCT inhibition sensitizes tumors to gamma-radiation via glutathione reduction. Oncotarget 2018, 9, 32280–32297. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, Y.; Zhang, R.; Wang, F.; Wang, T.; Jiao, Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. Oncol. Targets Ther. 2020, 13, 5429–5441. [Google Scholar] [CrossRef]
- Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.M.; et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020, 368, 85–89. [Google Scholar] [CrossRef]
- Arensman, M.D.; Yang, X.S.; Leahy, D.M.; Toral-Barza, L.; Mileski, M.; Rosfjord, E.C.; Wang, F.; Deng, S.; Myers, J.S.; Abraham, R.T.; et al. Cystine-glutamate antiporter xCT deficiency suppresses tumor growth while preserving antitumor immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 9533–9542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, X.; Green, M.D.; Wang, W.; Yu, J.; Choi, J.E.; Jiang, L.; Liao, P.; Zhou, J.; Zhang, Q.; Dow, A.; et al. Radiotherapy and Immunotherapy Promote Tumoral Lipid Oxidation and Ferroptosis via Synergistic Repression of SLC7A11. Cancer Discov. 2019, 9, 1673–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Saw, P.E.; Tao, W.; Li, Y.; Ji, X.; Bhasin, S.; Liu, Y.; Ayyash, D.; Rasmussen, J.; Huo, M.; et al. ROS-Responsive Polyprodrug Nanoparticles for Triggered Drug Delivery and Effective Cancer Therapy. Adv. Mater. 2017, 29, 1700141. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; He, Z. ROS-responsive drug delivery systems for biomedical applications. Asian J. Pharm. Sci. 2018, 13, 101–112. [Google Scholar] [CrossRef]
- Peiro Cadahia, J.; Previtali, V.; Troelsen, N.S.; Clausen, M.H. Prodrug strategies for targeted therapy triggered by reactive oxygen species. Medchemcomm 2019, 10, 1531–1549. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Ru, Z.; Song, W.; Chen, L.; Ma, H.; Sun, L. A ROS-responsive polymeric prodrug nanosystem with self-amplified drug release for PSMA (-) prostate cancer specific therapy. J. Nanobiotechnol. 2019, 17, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Gao, J.; Pei, Q.; Xu, H.; Yu, H. Engineering Prodrug Nanomedicine for Cancer Immunotherapy. Adv. Sci. (Weinh) 2020, 7, 2002365. [Google Scholar] [CrossRef]
- Gong, Y.; Chen, M.; Tan, Y.; Shen, J.; Jin, Q.; Deng, W.; Sun, J.; Wang, C.; Liu, Z.; Chen, Q. Injectable Reactive Oxygen Species-Responsive SN38 Prodrug Scaffold with Checkpoint Inhibitors for Combined Chemoimmunotherapy. ACS Appl. Mater. Interfaces 2020, 12, 50248–50259. [Google Scholar] [CrossRef]
- Kuang, Y.; Balakrishnan, K.; Gandhi, V.; Peng, X. Hydrogen peroxide inducible DNA cross-linking agents: Targeted anticancer prodrugs. J. Am. Chem. Soc. 2011, 133, 19278–19281. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Gandhi, V. ROS-activated anticancer prodrugs: A new strategy for tumor-specific damage. Ther. Deliv. 2012, 3, 823–833. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Fan, H.; Balakrishnan, K.; Wang, Y.; Sun, H.; Fan, Y.; Gandhi, V.; Arnold, L.A.; Peng, X. Discovery and Optimization of Novel Hydrogen Peroxide Activated Aromatic Nitrogen Mustard Derivatives as Highly Potent Anticancer Agents. J. Med. Chem. 2018, 61, 9132–9145. [Google Scholar] [CrossRef]
- Chen, W.; Balakrishnan, K.; Kuang, Y.; Han, Y.; Fu, M.; Gandhi, V.; Peng, X. Reactive oxygen species (ROS) inducible DNA cross-linking agents and their effect on cancer cells and normal lymphocytes. J. Med. Chem. 2014, 57, 4498–4510. [Google Scholar] [CrossRef] [PubMed]
- Hagen, H.; Marzenell, P.; Jentzsch, E.; Wenz, F.; Veldwijk, M.R.; Mokhir, A. Aminoferrocene-based prodrugs activated by reactive oxygen species. J. Med. Chem. 2012, 55, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Schikora, M.; Reznikov, A.; Chaykovskaya, L.; Sachinska, O.; Polyakova, L.; Mokhir, A. Activity of aminoferrocene-based prodrugs against prostate cancer. Bioorg. Med. Chem. Lett. 2015, 25, 3447–3450. [Google Scholar] [CrossRef]
- Daum, S.; Reshetnikov, M.S.V.; Sisa, M.; Dumych, T.; Lootsik, M.D.; Bilyy, R.; Bila, E.; Janko, C.; Alexiou, C.; Herrmann, M.; et al. Lysosome-Targeting Amplifiers of Reactive Oxygen Species as Anticancer Prodrugs. Angew. Chem. Int. Ed. Engl. 2017, 56, 15545–15549. [Google Scholar] [CrossRef] [PubMed]
- Reshetnikov, V.; Daum, S.; Janko, C.; Karawacka, W.; Tietze, R.; Alexiou, C.; Paryzhak, S.; Dumych, T.; Bilyy, R.; Tripal, P.; et al. ROS-Responsive N-Alkylaminoferrocenes for Cancer-Cell-Specific Targeting of Mitochondria. Angew. Chem. Int. Ed. Engl. 2018, 57, 11943–11946. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.J.; Liu, Y.; Wang, L.; Schubert, M.L.; Hoffmann, J.M.; Wang, S.; Neuber, B.; Huckelhoven-Krauss, A.; Gern, U.; Schmitt, A.; et al. Tumor-Specific Reactive Oxygen Species Accelerators Improve Chimeric Antigen Receptor T Cell Therapy in B Cell Malignancies. Int. J. Mol. Sci. 2019, 20, 2469. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aboelella, N.S.; Brandle, C.; Kim, T.; Ding, Z.-C.; Zhou, G. Oxidative Stress in the Tumor Microenvironment and Its Relevance to Cancer Immunotherapy. Cancers 2021, 13, 986. https://doi.org/10.3390/cancers13050986
Aboelella NS, Brandle C, Kim T, Ding Z-C, Zhou G. Oxidative Stress in the Tumor Microenvironment and Its Relevance to Cancer Immunotherapy. Cancers. 2021; 13(5):986. https://doi.org/10.3390/cancers13050986
Chicago/Turabian StyleAboelella, Nada S., Caitlin Brandle, Timothy Kim, Zhi-Chun Ding, and Gang Zhou. 2021. "Oxidative Stress in the Tumor Microenvironment and Its Relevance to Cancer Immunotherapy" Cancers 13, no. 5: 986. https://doi.org/10.3390/cancers13050986
APA StyleAboelella, N. S., Brandle, C., Kim, T., Ding, Z.-C., & Zhou, G. (2021). Oxidative Stress in the Tumor Microenvironment and Its Relevance to Cancer Immunotherapy. Cancers, 13(5), 986. https://doi.org/10.3390/cancers13050986