Simple Summary
Although effective in the majority of patients, the progression-free survival (PFS) to imatinib treatment can vary widely in effectiveness. Based on the known predictive role of tyrosine kinase (KIT) and platelet-derived growth factor receptor α (PDGRA) tumor genotypes, the differential clinical response to first-line imatinib treatment might be related to the different types and gene locations of the mutations. In our study, metastatic patients with gastrointestinal stromal tumors (GIST)-carrying KIT exon 11 deletion or a deletion/insertion involving codons 557/558 showed significantly shorter PFS to imatinib compared with those with deletion in codons other than 557/558 and patients with exon 11 duplication, insertion or single nucleotide variants (SNVs). Conversely, the latter subgroup showed the longest PFS first-line to imatinib. These results highlight the predictive role of pathogenic variant (PV) type and codon location in GIST, and can support stratification via mutational status in future clinical trials.
Abstract
In previous studies on localized GISTs, KIT exon 11 deletions and mutations involving codons 557/558 showed an adverse prognostic influence on recurrence-free survival. In the metastatic setting, there are limited data on how mutation type and codon location might contribute to progression-free survival (PFS) variability to first-line imatinib treatment. We analyzed the type and gene location of KIT and PDGFRA mutations for 206 patients from a GIST System database prospectively collected at an Italian reference center between January 2005 and September 2020. By describing the mutational landscape, we focused on clinicopathological characteristics according to the critical mutations and investigated the predictive role of type and gene location of the KIT exon 11 mutations in metastatic patients treated with first-line imatinib. Our data showed a predictive impact of KIT exon 11 pathogenic variant on PFS to imatinib treatment: patients with deletion or insertion/deletion (delins) in 557/558 codons had a shorter PFS (median PFS: 24 months) compared to the patients with a deletion in other codons, or duplication/insertion/SNV (median PFS: 43 and 49 months, respectively) (p < 0.001). These results reached an independent value in the multivariate model, which showed that the absence of exon 11 deletions or delins 557/558, the female gender, primitive tumor diameter (≤5 cm) and polymorphonuclear leucocytosis (>7.5 109/L) were significant prognostic factors for longer PFS. Analysis of the predictive role of PDGFRA PVs showed no significant results. Our results also confirm the aggressive biology of 557/558 deletions/delins in the metastatic setting and allow for prediction at the baseline which GIST patients would develop resistance to first-line imatinib treatment earlier.
1. Introduction
The discovery of constitutive activating mutations in the proto-oncogene receptor tyrosine kinase (KIT) or platelet-derived growth factor receptor α (PDGFRA) oncogenes as molecular drivers of most gastrointestinal stromal tumors (GISTs) [1,2] has radically changed our knowledge on their tumor biology. Activating mutations in KIT (70–80% of GISTs) or PDGFRA (5–10% of GISTs) disrupt the normal autoinhibitory state of receptor tyrosine kinase (RTK), leading to the constitutive, ligand-independent activation of Ras/Raf/MAPK, JAK/STAT3 and PI3K/Akt/mTOR downstream pathways, ultimately resulting in increased cell proliferation [3,4]. The resulting aberrant receptor tyrosine kinases, known to be clinically excellent therapeutic targets, have transformed GISTs into a model for successful molecular targeted therapy [5,6]. Indeed, KIT and PDGRFA represent not only key diagnostic markers but, more importantly, they also present prognostic and predictive significance, providing a powerful tool for the therapy selection process [7,8,9,10,11]. Tumor mutational status is particularly important because GIST patients show great variability in response to medical treatment, depending on the mutation’s location [12]. GISTs with KIT exon 11 mutations are the most responsive to standard first-line imatinib 400 treatment, with a response rate of 80% [13], whereas KIT exon 9 mutations are associated with a lower response rate of 45%. These patients benefit from the higher dose of 800 mg/die [13,14]. Although most GISTs with KIT exon 11 mutations are highly sensitive to front-line imatinib, progression-free survival (PFS) can vary widely in this subset of patients [15,16,17]. In previous studies on localized GISTs treated with surgery and adjuvant imatinib, different types of KIT exon 11 mutations (mainly deletions or mutations that involve codons 557 and/or 558) showed an adverse prognostic influence on recurrence-free survival (RFS) [18,19,20]. Indeed, there are limited data on how mutation type and codon location might contribute to clinical behavior and the variability of response to therapy in metastatic GISTs treated with first-line imatinib. A previous study investigated the predictive role of KIT exon 11 mutations in a cohort of advanced patients, with a primary interest in pathogenic variant (PV) location. This showed that patients with alterations affecting codons 557/558 developed secondary resistance more rapidly compared to patients carrying the most distal exon 11 mutations (codon 559 or downstream) [21]. However, the potential predictive impact of specific PV type needs further elucidation in the metastatic setting.
Based on this rationale, we focused on the clinicopathological characteristics according to critical mutations by describing the mutational landscape in a broad cohort of GIST patients and investigated the predictive role of the type and codon location of the KIT exon 11 mutations in metastatic GIST patients treated with first-line imatinib.
2. Results
2.1. Pathogenic Variant Classification of GIST Patients
PV classification was assessed for 206 GIST patients; in Figure 1, we describe the genetic landscape of GIST patients according to exact PV type and codon location.
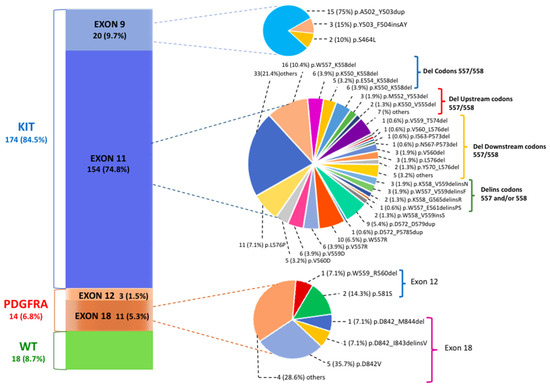
Figure 1.
The mutational landscape of gastrointestinal stromal tumor (GIST) patients according to pathogenic variant (PV) gene location. The most common tyrosine kinase (KIT) exon 11 PVs are classified into deletions involving codons 557/558, deletions upstream of codons 557/558, deletions downstream of codons 557/558 and deletions/insertions affecting codons 557 and/or 558.
Of 206 patients, 188 showed PV mutually exclusive of KIT (174 patients, 84.5%) or PDGFRA (14 patients, 6.8%) genes. In the group of 154 patients with KIT exon 11 mutations (74.8%), 95 deletions (del) (61.7%), 38 single nucleotide variants (SNVs) (24.7%), 10 duplications (6.5%), 9 insertions/deletions (delins) (5.8%) and 2 insertions (1.3%) were identified. The most common site of KIT exon 11 mutations included codons 550–560. Sixty out of 154 (39%) patients carried deletions that spanned the critical codons 557 and/or 558. These deletions were lost either as specific isolated 557/558 deletions, such as the most common W557_K558del (n.16 patients; 7.8% of all GIST patients), or as part of larger deletions that included mutation positions upstream of codon 557 (p.K550_K558del; p.K550_E554del) and downstream of codon 558. Among the latter are included several deletions involving codons 568–570, which are located in the distal part of the exon and which seem to have an important and specific effect on KIT signaling pathways [22]. These PVs, however, are characterized by a low frequency (with less than three cases for each PV). Among the other types of KIT exon 11 PVs, SNVs mainly affecting codons 557–560 were particularly frequent. The p.W557R, which leads to a missense substitution in position 557 of the KIT protein with the substitution of tryptophan (W) with arginine (R), was found in 10 GIST patients (4.9%). This variant, also known as c.1669T>C, results in a single nucleotide change with the substitution of a thymine fora cytosine in position 1669 of the coding sequence of the gene.
In KIT exon 9, which encodes for the extracellular domain, the most common PV identified was the A502_Y503 duplication (n. 15 patients; 75% of exon 9 PVs; 7.3% of all GIST patients), which mimics the conformational change that the extracellular KIT receptor undergoes upon ligand bounding.
Concerning the PDGFRA gene, most mutations were identified in exon 18 (n.11 patients; 78.6% of PDGFRA PVs; 5.3% of the total GIST patients). The most prevalent PV was the known imatinib-resistant p.D842V (n.5 patients, 1.9% of all patients; 45.4% of all exon 18 PDGFRA mutated GISTs), a SNV involving the second kinase domain and consisting of a single substitution at position 842 in the A-loop of an aspartic acid (D) by a valine (V). Rarely, in-frame deletions (p.D842_M844del) or deletions/insertions (p.D842_I843delinsV) of different lengths or other SNVs (p.N848K) were detected. Finally, the PDGFRA exon 12 PVs 3 (1.5%) were equally rare, with one deletion (p.W559_R560del) and two SNVs (p.P581S) observed. The detailed number of patients for each mutation type and the description of the corresponding pathogenic variants are included in Table 1.

Table 1.
Number of GIST patients with known KIT and PDGFRA mutational status. The description is based on affected gene and exon, mutation type (deletion, deletion/insertion, duplication, insertion and single nucleotide variant) and exact KIT or PDGFRA pathogenic variant.
2.2. Metastatic GIST Patients
2.2.1. Clinicopathological Characteristics According to Critical Mutations
In the group of 80 metastatic patients, 60 (75%) harbored a KIT exon 11 PV and 10 (12.5%) a KIT exon 9 PV, while PDGRFA mutations were found in 7 patients (8.7%) and KIT/PDGFRA wild-type genotypes were found in 3 patients (3.8%). Among the KIT exon 11 mutated patients, 25 (31.3%) showed exon 11 deletions or delins involving 557 and/or 558 codons and 35 patients (43.7%) had a deletion in codons other than 557/558 or other PV types (duplication, insertion or SNV).
The patients with GIST who carried deletions or delins in codons 557/558 were predominantly male (64%), with gastric GISTs (60%), a primitive tumor diameter of >5 cm (92%) and a mitotic index of >5/50 HPF (76%), without predominant hepatic (40%) or peritoneal (44%) metastatic involvement. Patients with GIST who carried KIT exon 11 PVs not including 557/558 deletions or delins showed tumors that most frequently originated in the small bowel (site of origin, small bowel 51.5% vs. stomach 22.8%), most frequently had large primitive tumors (diameter > 5 cm 77.2% vs. diameter ≤ 5 cm 22.8% of patients) and most frequently had a high median mitotic rate (mitosis >5/50 HPF 62.8% vs. mitosis ≤ 5/50 HPF 37.2% of patients).
The most common KIT exon 9 PV was the A502_Y503dup exon (8 out of 10 patients, 10% of all GISTs). These patients were mainly women (62.5%), with tumors frequently of small bowel origin (five out of eight tumors; 62.5%), a median mitotic rate of >5/50 HPF and a primitive tumor diameter of >5 cm in 75% of patients. The remainder of patients had PDGFRA-mutated GISTs, including two patients with D842V PV, and only three (3.8%) of these were KIT/PDGFRA WT GISTs.
Baseline clinical and pathological characteristics according to critical mutations are summarized in Table 2.
Table 2.
The clinical and pathological characteristics of metastatic GIST patients, according to the type and location of the critical mutations. The patients were classified based on genotyping as follows: (i) KIT exon 11 deletion or deletion/insertion involving 557 and/or 558 codons versus other KIT exon 11 PVs; (ii) KIT exon 9 A502-Y503 codon duplication versus other KIT exon 9 PVs; and (iii) the PDGFRA D842V single nucleotide variant versus other PDGFRA PVs.
2.2.2. Outcome Analysis and Objective Response in KIT Exon 11 Metastatic Patients
We focused on the KIT exon 11 genotypic landscape because these are the most common molecular alterations in GISTs and, at the same time, they show wide variability in the duration of their response to imatinib. We classified the patients treated with first-line imatinib and KIT exon 11 mutation in two groups: (i) KIT Exon 11 deletion or insertion/deletion in codons 557 and/or 558 (named “D-557/8”); (ii) mutations other than those in D-557/8 (named “No-D-557/8”). Twenty-five (25) patients (41.7%) were in the D-557/8 Group, and 35 patients (58.3%) were in in the No-D-557/8 Group. The exact PV types and codon locations of the exon 11 study population are shown in Figure 2.
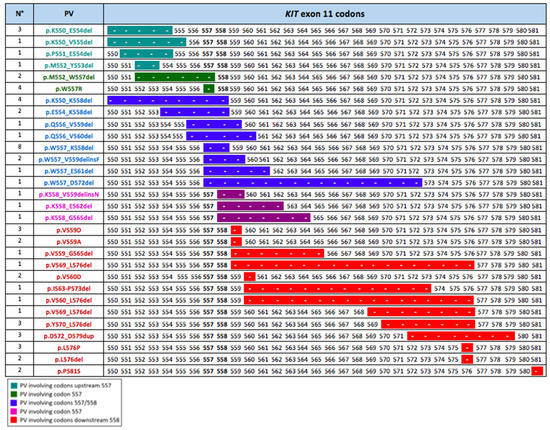
Figure 2.
Codon location of the PVs in the population of metastatic KIT exon 11 patients treated with first-line imatinib. The different colors indicate the PVs positions as related to 557/558 critical codons: PVs involving codons upstream of codon 557; PVs involving codon 557; PVs involving codons 557 and 558; PVs involving codon 558; and PVs involving codons downstream of codon 558. The length of the line indicates the number of codons involved in each KIT exon 11 mutation.
A total of 41 events (progression or death) were observed (68.3%): 18 events in the D-557/8 group of 25 patients (72%) and 23 events in the No-D-557/8 group of 35 patients (65.7%). The overall median PFS was 37 months (95% confidence interval (CI): 26.8–47.1). The median PFS was 24 months (95% CI: 21.3, 26.7) for the D-557/8 Group and 49 months (95% CI: 39.4, 58.6) for the No-D-557/8 Group (p < 0.001) (Figure 3A).
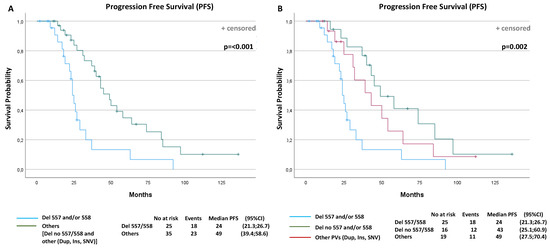
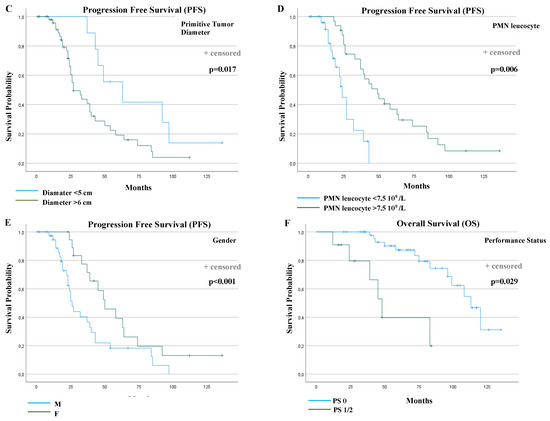
Figure 3.
Progression-free survival (PFS) and overall survival (OS) curves according to prognostic factors that resulted statistically significantly from multivariable analyses. PFS according to: (A) KIT Exon 11 deletion and delins in codons 557/558 or other PV types (p < 0.001); (B) KIT Exon 11 deletion and delins in codons 557/558, deletion in other codons or other PV types (duplication, insertion and single nucleotide variant (SNV)) (p = 0.002); (C) primitive tumor diameter (p = 0.017); (D) polymorphonuclear (PMN) leucocyte (p = 0.006); (E) gender (p < 0.001); (F) OS according to performance status (p = 0.029).
Subsequently, in the next analysis, we have divided the group of other PVs than 557/558 deletions (NoD-557/8) into two further subgroups, to investigate the potential different impact on PFS: (i) patients with KIT Exon 11 deletion or delins in codons other than 557/558; (ii) patients with duplication, insertion or SNV of all codons. The median PFS was 43 months (95% CI: 25.1, 60.9) for the patients with deletion or delins in other codons than 557/558, and 49 months (95% CI: 27.5, 70.4) for patients with duplication, insertion or SNV (p = 0.002) (Figure 3B).
After a median follow-up of 107 months, 18 events (deaths) were observed (30%): 10 in 25 of D-557/8 patients (40%) and 8 in 35 (23%) of No-D-557/8 patients. The overall median OS was 102 months (95% CI: 89.8, 114.5). The median OS was 99 months (95% CI: 65.8, 132.2) for the exon 11 557/558 deletions or delins patients, and not reached (NR) for the group of patients with other mutations (p = 0.14).
Regarding the associations of KIT Exon 11 mutation type and best overall response (BOR) in metastatic GIST patients treated with first-line imatinib, patients with deletion or delins regardless of codon regions had a significantly better CR rate (557/558: 40%; other codons: 37.5%) compared to the patients with other PVs (Dup, Ins or SNV) (5.3%) (p = 0.006).
2.2.3. Univariate and Multivariate Analysis
Table 3 summarizes the results of the univariable and multivariable prognostic factor analysis for PFS and overall survival (OS). Gender, performance status, the diameter of the primitive tumor, lymphopenia, polymorphonuclear (PMN) leucocytosis and KIT exon 11 PV type were found to be statistically significantly associated with PFS in univariable analyses. In the final multivariable Cox regression model, the following factors were significant: the female gender (median PFS: 26 versus 50 months, p < 0.001, HR: 0.23), a primitive tumor diameter ≤ 5 cm (median PFS: 27 versus 63 months, p = 0.017, HR: 3.39), a PMN leucocyte > 7.5 × 109 (median PFS: 24 versus 49 months, p = 0.006, HR: 0.27) and the absence of KIT Exon 11 Del or Delins 557/558 (median PFS: 24 versus 45 months, p < 0.001, HR: 0.12).
Table 3.
Univariate and multivariate analysis of prognostic factors of PFS and OS in GIST exon 11 mutated patients treated with first-line imatinib. Gender, performance status, primitive tumor diameter, number of mitoses, liver and peritoneal metastatic involvement, lymphopenia, polymorphonuclear leucocytosis and KIT exon 11 codon 557 and/or 558 deletion or deletion/insertion were evaluated in the Cox regression model.
Regarding OS, performance status, lymphopenia and PMN leucocytosis were statistically significantly associated in univariable analyses. In the final multivariable model, only performance status remained statistically significant (median OS: 113 vs. 48 months, p = 0.029, HR: 3.97).
Therefore, these results showed that, in the population of metastatic KIT exon 11 mutated patients treated with first-line imatinib, the absence of KIT exon 11 deletions or delins 557/558, the female gender, a primitive tumor diameter ≤ 5 cm and polymorphonuclear leucocytosis > 7.5 × 109/L were significant independent prognostic factors for longer PFS, while the performance status was the only significant prognostic factor for OS.
The PFS and OS curves were plotted according to each independent prognostic factor (Figure 3C–F).
3. Discussion
GIST oncogenic dependence on KIT and PDGFRA receptor is a paradigmatic model of oncogene addiction [22,23,24]. Imatinib is clinically efficacious in the treatment of advanced GIST patients, resulting in a median PFS of 20 months and a median overall survival (OS) of 57 months [14,25,26]. Although effective in the vast majority of patients, PFS to imatinib treatment may vary widely [27,28,29]. We know that in metastatic GIST patients, the most important predictive factors for response to imatinib are the KIT and PDGFRA genotypes [16,22]. We have learned that GIST patients harboring mutations in KIT exon 11 are highly sensitive to imatinib and show a deep and prolonged response. GISTs with KIT exon 9 mutation also benefit from imatinib, but are less sensitive to the standard dose of 400 mg/die and benefit from the increased dose of imatinib 800 mg/die [13,30]. The exon 11 mutations are the most common KIT mutations, and patients carrying these PVs represent a heterogeneous subgroup in terms of biological and clinical behavior, with 10% of patients remaining sensitive to imatinib despite the drug-selective pressure and keepingprogression-free after 10 years of treatment [16,31]. The lack of relevant predictive or prognostic clinical factors, according to the available data, makes the presence of this subset of long-term survivors of high biological and clinical interest.
Based on the known predictive role of genotype, the variable clinical outcome in KIT exon 11 mutated patients could be related to intrinsic molecular features, such as a different PV type and the gene location of exon 11 KIT mutations. Previous findings on localized GIST provide the rationale to better investigate this hypothesis [18,19,20]. Indeed, several studies showed the prognostic significance of different types of KIT exon 11 mutations for recurrence-free survival in localized GIST patients treated with surgery [18]. KIT exon 11 deletions and deletions affecting KIT exon 11 codons 557 and/or 558 are associated with a poor prognosis and have been described as independent adverse prognostic factors for relapse [18,23]; conversely, other exon 11 KIT mutations, such as insertion and duplication mutations, are generally associated with a favorable outcome [18].
In terms of metastatic disease, previous studies have addressed the predictive role of KIT exon 11 PVs on response and survival to imatinib. However, to our knowledge, very few studies have further subdivided the KIT exon 11 mutated population-based on PVs.
A first study compared the outcomes of patients with the most frequent deletion of KIT exon 11, the PV delWK557-558 and the deletion of Tyr568-570, which was selected because these tyrosines represent the first residues to be phosphorylated during activation and, thus, may be associated with specific effects on KIT signaling pathways. Metastatic GISTs, carrying one of two types of PVs (n.22 vs. n.14 patients, respectively) and treated with imatinib, showed no significant difference in terms of response rates, PFS and OS [23]. However, no other PVs were included in the analysis conducted for this study.
A subsequent study investigated the predictive role of KIT exon 11 mutations in a broader cohort of advanced patients, but with its main focus on PVs’ mutational locations. The researchers compared the molecular subgroups of advanced GIST treated in the BFR14 study of the French Sarcoma Group divided in terms of PV position: 557/558 codons, upstream or downstream exon 11 codons. Long-term outcome analysis showed that patients with alterations involving 557/558 codons developed secondary resistance more rapidly compared to the patients carrying the most distal exon 11 mutations (codon 559 or more) [21]. However, the role of a PV’s type, beyond its location, as a predictive biomarker for imatinib response has not been fully elucidated in metastatic patients.
Our study provides real-world evidence of the impact of type and location of KIT exon 11 PVs on clinical outcomes and response to therapy in advanced GIST patients treated with first-line imatinib 400. We show in a real-world population that the group of 557/558 mutations is not a homogeneous group in terms of response to imatinib: not all of the codons 557/558 PVs develop gain secondary resistance to imatinib more rapidly than other KIT PVs, as a markedly poorer PFS curve is shown for deletions involving codons 557/558. This result reaches an independent value in the multivariate model. Patients with deletion or insertion/deletion in codons 557/558 had a median PFS of 24 months; this PFS is in line with literature data in the metastatic setting [14], but is shorter compared to patients with a deletion in other codons (median PFS: 43 months) or patients with duplication, insertion or SNV (median PFS: 49 months) (p = 0.002). Multivariable prognostic factor analysis performed to assess the impact of the different baseline covariates on PFS and OS, showed that the absence of KIT Exon 11 del or delins 557/558, the female gender, a primitive tumor diameter ≤5 cm and PMN leucocytosis (>7.5 × 109/L) were significant prognostic factors for longer PFS to first-line imatinib; only performance status remains statistically significant in the multivariate model for OS.
These data, combined with previous findings in patients with localized diseases, confirm an aggressive biology of 557/558 exon 11 deletions or delins even in the metastatic setting, particularly for specific PVs such as the KIT p.W557_K558del, which explains their high prevalence in advanced GISTs [32]. The shorter PFS associated with 557/558 deletions could be biologically explained by their specifically designated role in the process of tyrosine kinase activation of the different segments of the KIT receptor. It is known from previous studies that the oncogenic KIT signaling mechanisms varied depending on the exact type and location of the PVs [33,34,35]. The 557/558 codon region contains residues that exert a critical autoinhibitory role on kinase activity, repressing the ligand-independent activation. The exon 11 codons 557/558 deletions may perturb KIT kinase autoinhibition by controlling the a-helical conformation, essentially leading to increased spontaneous receptor phosphorylation, the activation of Ras/Raf/MAPK, JAK/STAT3 and PI3K/Akt/mTOR downstream signaling pathways, ultimately leading tumor cell proliferation and the inhibition of apoptosis [36].
To our knowledge and based on the best data available today, this is the study that has provided the most representative picture of the predictive role of KIT exon 11 pathogenic variants in metastatic patients treated with first-line imatinib. It remains to be elucidated whether other convincing factors (such as microenvironment immunological characteristics or clinical factors not yet investigated) may have a further relevant impact on tumor response and progression-free survival.
4. Patients and Methods
4.1. Study Population
In the current study, clinicopathological variables and mutational status data were analyzed from a GIST System database prospectively collected in an Italian referral center for diagnosis and treatment of soft tissue sarcoma and GIST: the “Sicilian Regional Center for the Prevention, Diagnosis, and Treatment of Rare and Heredo–Familial Tumors” of the Section of Medical Oncology of University Hospital Policlinico “P. Giaccone” of Palermo. In the study the patients afferent to the center between January 2005 and September 2020 were included, based on retrievable tumor specimens in order to centrally repeat mutational analysis. The metastatic GIST patients treated with first-line imatinib 400 therapy were included in the analyses of the predictive role of mutation type and codon location. The information collected from these patients included gender, age, site of the origin of primary tumors, primitive tumor diameter and mitosis, KIT and PDGFRA pathogenic variant (PV) classification, data on active disease sites, best overall response (BOR) to imatinib (progression disease (PD), stable disease (SD), partial response (PR), complete response (CR)) assessed according to response evaluation criteria in solid tumors (RECIST version 1.1.), progression-free survival (PFS) to imatinib treatment and overall survival (OS). The association between KIT Exon 11 mutation type and BOR, PFS and OS was evaluated. The clinical, pathological and genetic information were anonymously recorded for all patients who previously provided written informed consent. This study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines.
4.2. Mutation Analysis
The diagnosis of GIST was made based on histopathologic assessment and immunohistochemical staining for CD117 antigen expression from local pathology testing of diagnostic core biopsies or tumor resections for clinical use. The pathologists, with special interest in sarcoma pathology, also reported tumor mitoses from 50 HPFs and diameter lesions. Four (4)-μm thick FFPE tissue sections were deparaffinized with a Deparaffinization Solution (Qiagen, Hilden, Germany) and genomic DNA was extracted using a QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Extracted DNA was quantified using a Qubit® 3.0 fluorometer (Thermofisher Scientific, Waltham, MA, USA) and its quality was assessed by using a SeqStudio Genetic Analyzer (Applied Biosystem). All GISTs were centrally examined for somatic mutations in KIT (exons 9, 11, 13 and 17) and PDGFRA (exons 12, 14 and 18) by polymerase chain reaction (PCR) amplification and direct Sanger sequencing. PCR reactions were run on Veriti Thermal Cycler (Applide Biosystem) and Sanger sequencing wasperformed using a BigDye Terminator 3.1 Cycle Sequencing Kit (Life Technologies, Carlsbad, CA, USA) on the SeqStudio Genetic Analyzer (Applied Biosystem, Foster City, CA, USA), according to manufacturer’s protocols. PVs were confirmed using Sanger sequencing with a BigDye Terminator 3.1 Cycle Sequencing Kit (Life Technologies) on the SeqStudio Genetic Analyzer (Applied Biosystem). Samples that scored negative were further profiled using a targeted next generation sequencing (NGS) panel for the presence of hot spot mutations in H/K/N RAS, BRAF and NTRK using an Ion Torrent S5 (Thermofisher Scientific) instrument. For all detected PVs found, a gene name, a nucleotide change (c.notation) and an amino acid change (p.notation) were codified. The classification of the variant was performed after consulting the databases “Catalogue Of Somatic Mutations In Cancer” (COSMIC) and ClinVar. According to the aim of the study, only the KIT and PDGFRA PVs were included in the following analyses.
4.3. Statistical Analysis
PFS was calculated from beginning of the imatinib treatment to death by any cause, disease progression or last follow-up (censored patients). OS was calculated from the beginning of the imatinib treatment to death by any cause or last follow-up (censored patients). The analyses of PFS and OS between groups were compared using the Kaplan–Meier method and a log-rank test. To identify independent prognostic factors for PFS and OS, univariate and multivariate Cox proportional hazard regression models were built. All tests were performed with a significance level of p = 0.05. Statistical analyses were conducted using IBM SPSS Statistics for Windows Version 25.0 (IBM Corporation, Armonk, NY, USA).
5. Conclusions
From previous studies on GIST, we have learned that, in metastatic patients, the main predictive factor for the duration of response to imatinib is the genotype [13]. In the KIT exon 11 mutated patients, who are the most common molecular-based subgroup of advanced GIST patients, our results reveal the important impact of the PVs’ type and location on PFS. We detected a significantly shorter PFS for those patients carrying deletions involving 557/558 codons compared to the patients with deletions in other codons or patients with duplications, insertions or SNVs. These data, combined with the existing evidence on the negative role of 557/558 deletion for RFS in localized patients treated with surgery [18], confirm the aggressive biology of 557/558 exon 11 deletions or delins in the metastatic setting as well. These findings have potential clinical utility because they only allow an understanding of why a subgroup of patients responds to imatinib better than other groups, but also predict at baseline which GIST patients would experience resistance to treatment earlier. This study also supports the use of stratification by mutational type and codon location in future clinical trials.
Author Contributions
Conceptualization, L.I., D.F., A.R., B.V. and G.B.; investigation, I.D.L., T.V.B., R.C., D.C., G.B. and G.P.; data curation and analysis, L.I., D.F. and B.V.; writing L.I. and G.B.; supervision, A.R., V.B. and G.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and Good Clinical Practice guidelines, and approved by the Ethics Committee of the University Hospital A.O.U.P. “P. Giaccone” of Palermo (protocol code: G-Land 2017; and date of approval: 01-03-2017).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Corless, C.L.; Barnett, C.M.; Heinrich, M.C. Gastrointestinal stromal tumours: Origin and molecular oncology. Nat. Rev. Cancer 2011, 11, 865–878. [Google Scholar] [CrossRef]
- Pantuso, G.; Macaione, I.; Taverna, A.; Guercio, G.; Incorvaia, L.; Di Piazza, M.; Di Grado, F.; Cilluffo, G.; Badalamenti, G.; Cipolla, C. Surgical treatment of primary gastrointestinal stromal tumors (GISTs): Management and prognostic role of R1 resections. Am. J. Surg. 2020, 220, 359–364. [Google Scholar] [CrossRef]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Corless, C.L.; Duensing, A.; McGreevey, L.; Chen, C.J.; Joseph, N.; Singer, S.; Griffith, D.J.; Haley, A.; Town, A.; et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003, 299, 708–710. [Google Scholar] [CrossRef] [PubMed]
- Corless, C.L.; Schroeder, A.; Griffith, D.; Town, A.; McGreevey, L.; Harrell, P.; Shiraga, S.; Bainbridge, T.; Morich, J.; Heinrich, M.C. PDGFRA Mutations in Gastrointestinal Stromal Tumors: Frequency, Spectrum and In Vitro Sensitivity to Imatinib. J. Clin. Oncol. 2005, 23, 5357–5364. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-E.; Tzen, C.-Y.; Wang, S.-Y.; Yeh, C.-N. Clinical Diagnosis of Gastrointestinal Stromal Tumor (GIST): From the Molecular Genetic Point of View. Cancers 2019, 11, 679. [Google Scholar] [CrossRef]
- Boikos, S.A.; Pappo, A.S.; Killian, J.K.; LaQuaglia, M.P.; Weldon, C.B.; George, S.; Trent, J.C.; Von Mehren, M.; Wright, J.A.; Schiffman, J.D.; et al. Molecular Subtypes of KIT/PDGFRA Wild-Type Gastrointestinal Stromal Tumors: A Report From the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol. 2016, 2, 922–928. [Google Scholar] [CrossRef]
- Vanden Bempt, I.; Vander Borght, S.; Sciot, R.; Spans, L.; Claerhout, S.; Brems, H.; Lehnert, S.; Dehaspe, L.; Fransis, S.; Neuville, B.; et al. Comprehensive targeted NGS approach in the molecular diagnosis of GIST. Genes Chromosomes Cancer 2020, 60, 239–249. [Google Scholar] [CrossRef]
- Miettinen, M.; Killian, J.K.; Wang, Z.-F.; Lasota, J.; Lau, C.; Jones, L.; Walker, R.; Pineda, M.; Zhu, Y.J.; Kim, S.Y.; et al. Immunohistochemical Loss of Succinate Dehydrogenase Subunit A (SDHA) in Gastrointestinal Stromal Tumors (GISTs) Signals SDHA Germline Mutation. Am. J. Surg. Pathol. 2013, 37, 234–240. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Li, F.; Huynh, H.; Li, X.; Ruddy, D.A.; Wang, Y.; Ong, R.; Chow, P.; Qiu, S.; Tam, A.; Rakiec, D.P.; et al. FGFR-Mediated Reactivation of MAPK Signaling Attenuates Antitumor Effects of Imatinib in Gastrointestinal Stromal Tumors. Cancer Discov. 2015, 5, 438–451. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Corless, C.L.; Demetri, G.D.; Blanke, C.D.; Von Mehren, M.; Joensuu, H.; McGreevey, L.S.; Chen, C.-J.; Abbeele, A.D.V.D.; Druker, B.J.; et al. Kinase Mutations and Imatinib Response in Patients With Metastatic Gastrointestinal Stromal Tumor. J. Clin. Oncol. 2003, 21, 4342–4349. [Google Scholar] [CrossRef]
- Debiec-Rychter, M.; Sciot, R.; Le Cesne, A.; Schlemmer, M.; Hohenberger, P.; van Oosterom, A.T.; Blay, J.-Y.; Leyvraz, S.; Stul, M.; Casali, P.G.; et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur. J. Cancer 2006, 42, 1093–1103. [Google Scholar] [CrossRef]
- Serrano, C.; George, S. Gastrointestinal Stromal Tumor: Challenges and Opportunities for a New Decade. Clin. Cancer Res. 2020, 26, 5078–5085. [Google Scholar] [CrossRef]
- Vincenzi, B.; Nannini, M.; Badalamenti, G.; Grignani, G.; Fumagalli, E.; Gasperoni, S.; D’Ambrosio, L.; Incorvaia, L.; Stellato, M.; Ceruso, M.S.; et al. Imatinib rechallenge in patients with advanced gastrointestinal stromal tumors following progression with imatinib, sunitinib and regorafenib. Ther. Adv. Med Oncol. 2018, 10. [Google Scholar] [CrossRef]
- Casali, P.G.; Zalcberg, J.; Le Cesne, A.; Reichardt, P.; Blay, J.-Y.; Lindner, L.H.; Judson, I.R.; Schöffski, P.; Leyvraz, S.; Italiano, A.; et al. Ten-Year Progression-Free and Overall Survival in Patients With Unresectable or Metastatic GI Stromal Tumors: Long-Term Analysis of the European Organisation for Research and Treatment of Cancer, Italian Sarcoma Group, and Australasian Gastrointestinal Trials Group Intergroup Phase III Randomized Trial on Imatinib at Two Dose Levels. J. Clin. Oncol. 2017, 35, 1713–1720. [Google Scholar] [CrossRef]
- Nannini, M.; Nigro, M.C.; Vincenzi, B.; Fumagalli, E.; Grignani, G.; D’Ambrosio, L.; Badalamenti, G.; Incorvaia, L.; Bracci, R.; Gasperoni, S.; et al. Personalization of regorafenib treatment in metastatic gastrointestinal stromal tumours in real-life clinical practice. Ther. Adv. Med Oncol. 2017, 9, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, H.; Wardelmann, E.; Sihto, H.; Eriksson, M.; Hall, K.S.; Reichardt, A.; Hartmann, J.T.; Pink, D.; Cameron, S.; Hohenberger, P.; et al. Effect of KIT and PDGFRA Mutations on Survival in Patients with Gastrointestinal Stromal Tumors Treated With Adjuvant Imatinib: An Exploratory Analysis of a Randomized Clinical Trial. JAMA Oncol. 2017, 3, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, A.; Rutkowski, P.; Schöffski, P.; Ray-Coquard, I.; Hostein, I.; Schildhaus, H.-U.; Le Cesne, A.; Bylina, E.; Limon, J.; Blay, J.-Y.; et al. Tumor Genotype Is an Independent Prognostic Factor in Primary Gastrointestinal Stromal Tumors of Gastric Origin: A European Multicenter Analysis Based on ConticaGIST. Clin. Cancer Res. 2014, 20, 6105–6116. [Google Scholar] [CrossRef] [PubMed]
- Martin-Broto, J.; Gutierrez, A.; Garcia-Del-Muro, X.; Lopez-Guerrero, J.; Martinez-Trufero, J.; De Sande, L.; Lainez, N.; Maurel, J.; De Juan, A.; Losa, F.; et al. Prognostic time dependence of deletions affecting codons 557 and/or 558 of KIT gene for relapse-free survival (RFS) in localized GIST: A Spanish Group for Sarcoma Research (GEIS) Study. Ann. Oncol. 2010, 21, 1552–1557. [Google Scholar] [CrossRef]
- Patrikidou, A.; Domont, J.; Chabaud, S.; Ray-Coquard, I.; Coindre, J.-M.; Bui-Nguyen, B.; Adenis, A.; Rios, M.; Bertucci, F.; Duffaud, F.; et al. Long-term outcome of molecular subgroups of GIST patients treated with standard-dose imatinib in the BFR14 trial of the French Sarcoma Group. Eur. J. Cancer 2016, 52, 173–180. [Google Scholar] [CrossRef]
- Lasota, J.; Dobosz, A.J.V.; Wasag, B.; Woźniak, A.; Kraszewska, E.; Michej, W.; Ptaszynski, K.; Rutkowski, P.; Sarlomo-Rikala, M.; Steigen, S.E.; et al. Presence of homozygous KIT exon 11 mutations is strongly associated with malignant clinical behavior in gastrointestinal stromal tumors. Lab. Investig. 2007, 87, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Bachet, J.B.; Hostein, I.; Le Cesne, A.; Brahimi, S.; Beauchet, A.; Tabone-Eglinger, S.; Subra, F.; Bui, B.; Duffaud, F.; Terrier, P.; et al. Prognosis and predictive value of KIT exon 11 deletion in GISTs. Br. J. Cancer 2009, 101, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, P.; Demetri, G.D.; Gelderblom, H.; Rutkowski, P.; Im, S.-A.; Gupta, S.; Kang, Y.-K.; Schöffski, P.; Schuette, J.; Soulières, D.; et al. Correlation of KIT and PDGFRA mutational status with clinical benefit in patients with gastrointestinal stromal tumor treated with sunitinib in a worldwide treatment-use trial. BMC Cancer 2016, 16. [Google Scholar] [CrossRef]
- Badalamenti, G.; Rodolico, V.; Fulfaro, F.; Cascio, S.; Cipolla, C.; Cicero, G.; Incorvaia, L.; Sanfilippo, M.; Intrivici, C.; Sandonato, L.; et al. Gastrointestinal stromal tumors (GISTs): Focus on histopathological diagnosis and biomolecular features. Ann. Oncol. 2007, 18, vi136–vi140. [Google Scholar] [CrossRef]
- Casali, P.; Abecassis, N.; Bauer, S.; Biagini, R.; Bielack, S.; Bonvalot, S.; Boukovinas, I.; Bovee, J.; Brodowicz, T.; Broto, J.; et al. Gastrointestinal stromal tumours: ESMO–EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv68–iv78. [Google Scholar] [CrossRef]
- Agaram, N.P.; Besmer, P.; Wong, G.C.; Guo, T.; Socci, N.D.; Maki, R.G.; DeSantis, D.; Brennan, M.F.; Singer, S.; DeMatteo, R.P.; et al. Pathologic and Molecular Heterogeneity in Imatinib-Stable or Imatinib-Responsive Gastrointestinal Stromal Tumors. Clin. Cancer Res. 2007, 13, 170–181. [Google Scholar] [CrossRef]
- Kim, T.S.; Cavnar, M.J.; Cohen, N.A.; Sorenson, E.C.; Greer, J.B.; Seifert, A.M.; Crawley, M.H.; Green, B.L.; Popow, R.; Pillarsetty, N.; et al. Increased KIT Inhibition Enhances Therapeutic Efficacy in Gastrointestinal Stromal Tumor. Clin. Cancer Res. 2014, 20, 2350–2362. [Google Scholar] [CrossRef] [PubMed]
- Liegl, B.; Kepten, I.; Le, C.; Zhu, M.; Demetri, G.D.; Heinrich, M.C.; Fletcher, C.D.M.; Corless, C.L.; Fletcher, J.A. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J. Pathol. 2008, 216, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST). Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: A meta-analysis of 1640 patients. J. Clin. Oncol. 2010, 28, 1247–1253. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Besmer, P.; Guo, T.; Arkun, K.; Hom, G.; Koryotowski, B.; Leversha, M.A.; Jeffrey, P.D.; DeSantis, D.; Singer, S.; et al. Acquired Resistance to Imatinib in Gastrointestinal Stromal Tumor Occurs Through Secondary Gene Mutation. Clin. Cancer Res. 2005, 11, 4182–4190. [Google Scholar] [CrossRef]
- Szucs, Z.; Thway, K.; Fisher, C.; Bulusu, R.; Constantinidou, A.; Benson, C.; Van Der Graaf, W.T.A.; Jones, R.L. Molecular subtypes of gastrointestinal stromal tumors and their prognostic and therapeutic implications. Future Oncol. 2017, 13, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Duensing, A.; Demetri, G.D.; Fletcher, J.A. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene 2007, 26, 7560–7568. [Google Scholar] [CrossRef]
- Ma, Y.; Cunningham, M.E.; Wang, X.; Ghosh, I.; Regan, L.; Longley, B.J. Inhibition of Spontaneous Receptor Phosphorylation by Residues in a Putative α-Helix in the KIT Intracellular Juxtamembrane Region. J. Biol. Chem. 1999, 274, 13399–13402. [Google Scholar] [CrossRef] [PubMed]
- Judson, I.; Ma, P.; Peng, B.; Verweij, J.; Racine, A.; Di Paola, E.D.; Van Glabbeke, M.; Dimitrijevic, S.; Scurr, M.; Dumez, H.; et al. Imatinib pharmacokinetics in patients with gastrointestinal stromal tumour: A retrospective population pharmacokinetic study over time. EORTC Soft Tissue and Bone Sarcoma Group. Cancer Chemother. Pharmacol. 2004, 55, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, A.; Bal, M.; Swami, R.; Shetty, O.; Bose, S.; Pai, T.; Gurav, M.; Gupta, S.; Ostwal, V. Early outcomes of exon 11 mutants in GIST treated with standard dose Imatinib. Ann. Transl. Med. 2017, 5, 134. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).