
Molecular Mechanism of Autophagy and Its Regulation by Cannabinoids in Cancer



Abstract
:Simple Summary
Abstract
1. Introduction
2. Molecular Mechanism of Autophagy
3. Regulation of Autophagy
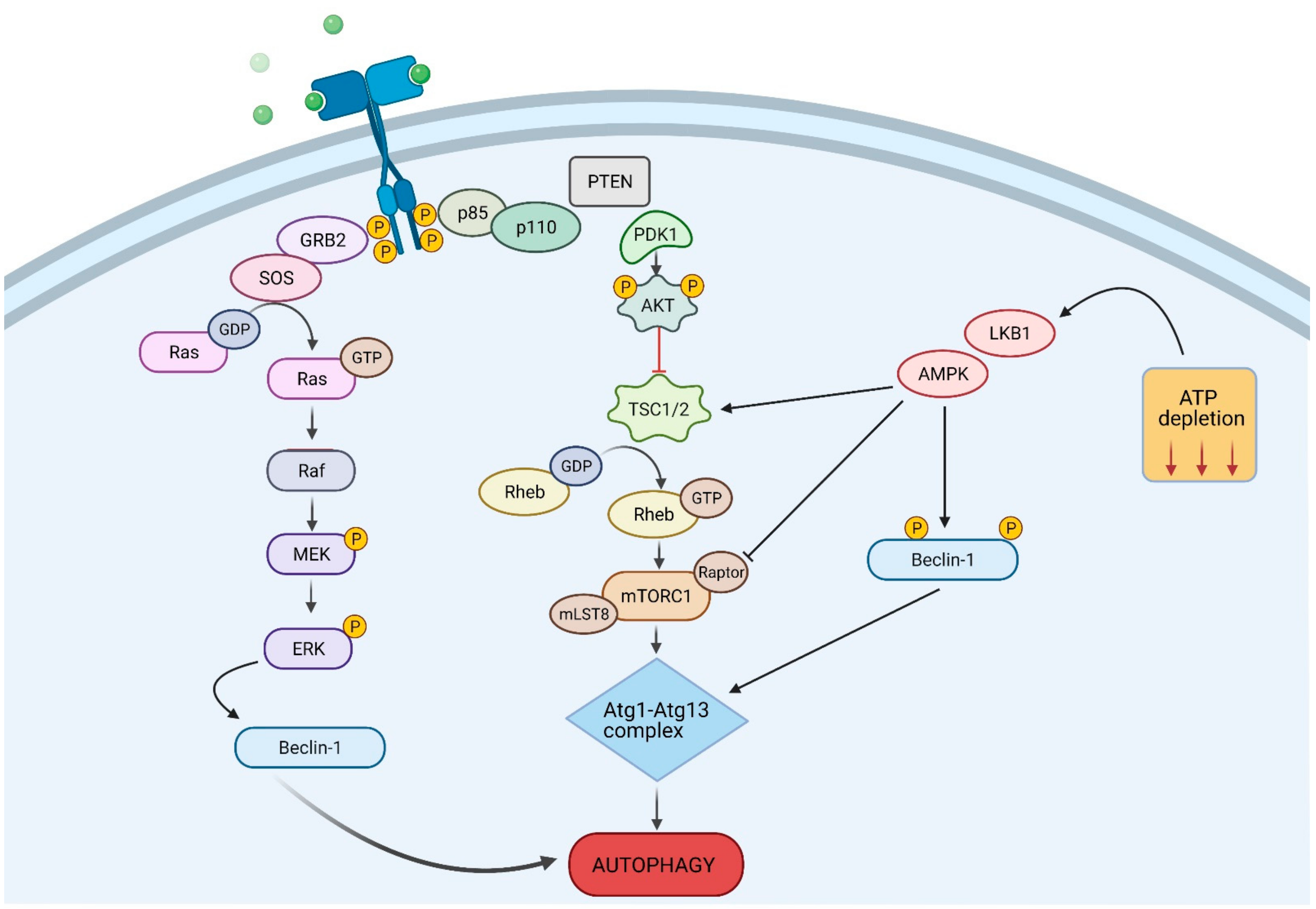
4. Autophagy Signalling Pathways
5. The Role of Autophagy in Cancer
5.1. Autophagy and its Tumour Suppressive Role
5.2. Autophagy and Its Tumour Promoter Role
5.3. Autophagy as a Therapeutic Target in Cancer
6. The Endocannabinoid System
7. Anticancer Properties of Cannabinoids
7.1. Cannabinoids Modulating Cell Cycle Checkpoint
7.2. Role of Cannabinoids in Invasion, Metastasis and Angiogenesis
7.3. Tumour-Immune Interactions
7.4. Cannabinoid-Induced Cell Death Mechanisms
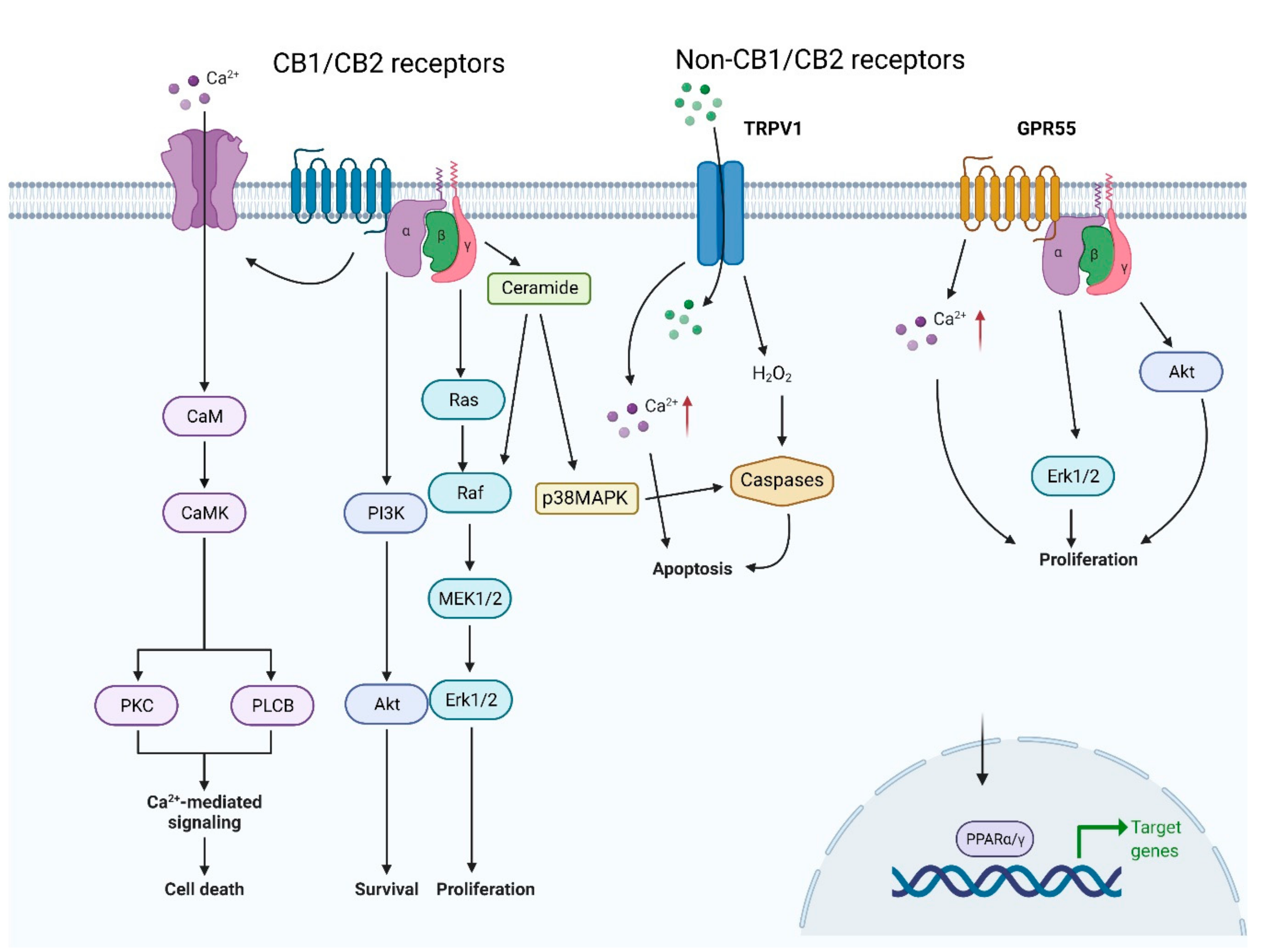
8. Cannabinoid-Induced Autophagy
9. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Activating transcription factor 4 | ATF4 |
| adenosine monophosphate-activated protein kinase | AMPK |
| arachidonyl-2′-chloroethylamide | ACEA |
| arachidonoyl cyclopropramide | APCA |
| 2-arachidonoylglycerol | 2-AG |
| autophagy-related | Atg |
| bax interacting factor-1 | Bif-1 |
| B-cell lymphoma 2 | Bcl2 |
| Bcl-2 homology 3 domain | BH3 |
| BECN1-regulated autophagy protein 1 | Ambra1 |
| Camptothecin | CPT |
| Cannabidiol | CBD |
| cannabinoid receptor 1 | CB1 |
| cannabinoid receptor 2 | CB2 |
| cdc25 homologue A | Cdc25A |
| CCAAT/enhancer-binding protein homologous protein | CHOP |
| cell division cycle | Cdc2 |
| chloroquine | CQ |
| central nervous system | CNS |
| extracellular matrix, cyclin dependent kinases | CDKs |
| cyclin-dependent kinase 1 | CDK1 |
| cyclin-dependent kinase 2 | Cdk2 |
| cyclic-AMP | cAMP |
| ECM; eukaryotic translation initiation factor 2, subunit 1 alpha | eIF2α |
| Endoplasmic reticulum | ER |
| extracellular-regulated kinase | ERK |
| 5–Fluorouracil, fatty acid amide hydrolase | FAAH; 5FU |
| genetically engineered mouse models | GEMMs |
| glioblastoma multiforme | GBM |
| G-protein coupled receptor | GPCR |
| G-protein coupled receptor 55 | GPR55 |
| Hydroxychloroquine | HCQ |
| hypoxia-induce factor-1 alpha | HIF-1α |
| intercellular adhesion molecule 1 | ICAM-1 |
| jun N-terminal kinase | JNK |
| lymphocyte function-associated antigen 1 | LFA-1 |
| mammalian target of rapamycin | mTOR |
| mammalian target of rapamycin complex 1 | mTORC1 |
| 3-methyladenine | 3-MA |
| microtubule-associated protein light chain 3 | LC3B |
| mitogen-activated protein kinases | MAPKs |
| monoacylglycerol lipase | MAGL |
| multiple myeloma | MM |
| peroxisome proliferator-activated receptors | PPARs |
| phosphoinositide 3-kinase | P13K |
| N-arachidonoylethanolamine | AEA |
| non-small-cell lung cancer | NSCLC |
| N-oleoylethanolamine | OEA |
| N-palmitoylethanolamine | PEA |
| protein kinase B | AKT |
| pseudokinase tribbles homologue-3 | TRIB3 |
| reactive oxygen species | ROS |
| retinoblastoma | Rb |
| sequestosome-1 | SQSTM1 |
| soluble N-ethylmaleimide-sensitive factor attachment protein receptor | SNARE |
| stress-modulated protein p8 | p8 |
| Δ9-tetrahydrocannabinol | THC |
| transient receptor potential vanilloid type 1 | TRPV1 |
| transient receptor potential vanilloid type 2 | TRPV2 |
| tuberous sclerosis protein 2 | TSC2 |
| UV radiation resistance-associated gene | UVRAG |
| vascular-endothelial growth factor | VEGF |
References
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codogno, P.; Meijer, A.J. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ. 2005, 12, 1509–1518. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef] [Green Version]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L. The dual role of autophagy in cancer. Curr. Opin. Pharmacol. 2011, 11, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, L.; Amaral, C.; Teixeira, N.; Correia-da-Silva, G.; Fonseca, B.M. Cannabinoid-induced autophagy: Protective or death role? Prostaglandins Other Lipid Mediat. 2016, 122, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Velasco, G.; Sánchez, C.; Guzmán, M. Anticancer mechanisms of cannabinoids. Curr. Oncol. 2016, 23, S23–S32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Śledziński, P.; Zeyland, J.; Słomski, R.; Nowak, A. The current state and future perspectives of cannabinoids in cancer biology. Cancer Med. 2018, 7, 765–775. [Google Scholar] [CrossRef]
- Bielawiec, P.; Harasim-Symbor, E.; Chabowski, A. Phytocannabinoids: Useful Drugs for the Treatment of Obesity? Special Focus on Cannabidiol. Front. Endocrinol. 2020, 11, 114. [Google Scholar] [CrossRef] [PubMed]
- Biazik, J.; Ylä-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.L. Ultrastructural relationship of the phagophore with surrounding organelles. Autophagy 2015, 11, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Sugita, H.; Yoshimori, T.; Ohsumi, Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 1998, 273, 33889–33892. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Wild, P.; McEwan, D.G.; Dikic, I. The LC3 interactome at a glance. J. Cell Sci. 2014, 127, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, L.; Hou, C.; Lai, Y.; Long, J.; Liu, J.; Zhong, Q.; Diao, J. SNARE-mediated membrane fusion in autophagy. Semin. Cell Dev. Biol. 2016, 60, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian Autophagy: How Does It Work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Paolo, G.; de Camilli, P. Phosphoinositides in cell regulation and membrane dynamics. Nature 2006, 443, 651–657. [Google Scholar] [CrossRef]
- Cebollero, E.; van der Vaart, A.; Reggiori, F. Understanding phosphatidylinositol-3-phosphate dynamics during autophagosome biogenesis. Autophagy 2012, 8, 1868–1870. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, U.; Strauss, P. Autophagic vacuoles in heart muscle and liver. A comparative morphometric study including circadian variations in meal-fed rats. J. Mol. Cell Cardiol. 1981, 13, 37–49. [Google Scholar] [CrossRef]
- Pfeifer, U.; Warmuth-Metz, M. Inhibition by insulin of cellular autophagy in proximal tubular cells of rat kidney. Am. J. Physiol. 1983, 244, E109–E114. [Google Scholar] [CrossRef]
- Mortimore, G.E.; Hutson, N.J.; Surmacz, C.A. Quantitative correlation between proteolysis and macro- and microautophagy in mouse hepatocytes during starvation and refeeding. Proc. Natl. Acad. Sci. USA 1983, 80, 2179–2183. [Google Scholar] [CrossRef] [Green Version]
- Mortimore, G.E.; Ward, W.F. Behavior of the lysosomal system during organ perfusion. An inquiry into the mechanism of hepatic proteolysis. Front. Biol. 1976, 45, 157–184. [Google Scholar]
- Deter, R.L.; Baudhuin, P.; de Duve, C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J. Cell Biol. 1976, 35, C11–C16. [Google Scholar] [CrossRef] [Green Version]
- Lum, J.J.; DeBerardinis, R.J.; Thompson, C.B. Autophagy in metazoans: Cell survival in the land of plenty. Nat. Rev. Mol. Cell Biol. 2005, 6, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Ohsumi, Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998, 273, 3963–3966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Karantza-Wadsworth, V. Role and regulation of autophagy in cancer. Biochim. Biophys. Acta 2009, 1793, 1516–1523. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, H.; Kondo, Y.; Fujiwara, K.; Kanzawa, T.; Aoki, H.; Mills, G.B.; Kondo, S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005, 65, 3336–3346. [Google Scholar] [CrossRef] [Green Version]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.; Park, R.; Kim, H.; Namkoong, S.; Jo, D.; Huh, Y.H.; Jang, I.S.; Lee, J.I.; Park, J. AMPK contributes to autophagosome maturation and lysosomal fusion. Sci. Rep. 2018, 8, 12637. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; She, H.; Zhang, T.; Xu, H.; Cheng, L.; Yepes, M.; Zhao, Y.; Mao, Z. p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1. J. Cell Biol. 2018, 217, 315–328. [Google Scholar] [CrossRef] [Green Version]
- Kenific, C.M.; Thorburn, A.; Debnath, J. Autophagy and metastasis: Another double-edged sword. Curr. Opin. Cell Biol. 2010, 22, 241–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notte, A.; Leclere, L.; Michiels, C. Autophagy as a mediator of chemotherapy-induced cell death in cancer. Biochem. Pharmacol. 2011, 82, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Langley, R.R.; Fidler, I.J. The seed and soil hypothesis revisited—The role of tumor-stroma interactions in metastasis to different organs. Int. J. Cancer 2011, 128, 2527–2535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Shen, Y.; Li, D.D.; Wang, L.L.; Deng, R.; Zhu, X.F. Decreased expression of autophagy-related proteins in malignant epithelial ovarian cancer. Autophagy 2008, 4, 1067–1068. [Google Scholar] [CrossRef] [Green Version]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Eng. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wechman, S.L.; Pradhan, A.K.; DeSalle, R.; Das, S.K.; Emdad, L.; Sarkar, D.; Fisher, P.B. New Insights Into Beclin-1: Evolution and Pan-Malignancy Inhibitor Activity. Adv. Cancer Res. 2018, 137, 77–114. [Google Scholar] [CrossRef]
- Miracco, C.; Cosci, E.; Oliveri, G.; Luzi, P.; Pacenti, L.; Monciatti, I.; Mannucci, S.; de Nisi, M.C.; Toscano, M.; Malagnino, V.; et al. Protein and mRNA expression of autophagy gene Beclin 1 in human brain tumours. Int. J. Oncol. 2007, 30, 429–436. [Google Scholar] [PubMed]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 2006, 8, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mulé, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef]
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar] [CrossRef] [Green Version]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Vicencio, J.M.; Criollo, A.; Maiuri, M.C.; Kroemer, G. Anti- and pro-tumor functions of autophagy. Biochim. Biophys. Acta 2009, 1793, 1524–1532. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.J.; Zhang, X.; Rodriguez-Velez, A.; Evans, T.D.; Razani, B. p62/SQSTM1 and Selective Autophagy in Cardiometabolic Diseases. Antioxid. Redox Signal. 2019, 31, 458–471. [Google Scholar] [CrossRef]
- Moscat, J.; Diaz-Meco, M.T. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009, 137, 1001–1004. [Google Scholar] [CrossRef] [Green Version]
- Duran, A.; Linares, J.F.; Galvez, A.S.; Wikenheiser, K.; Flores, J.M.; Diaz-Meco, M.T.; Moscat, J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 2008, 13, 343–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef]
- Todoric, J.; Antonucci, L.; di Caro, G.; Li, N.; Wu, X.; Lytle, N.K.; Dhar, D.; Banerjee, S.; Fagman, J.B.; Browne, C.D.; et al. Stress-Activated NRF2-MDM2 Cascade Controls Neoplastic Progression in Pancreas. Cancer Cell 2017, 32, 824–839.e828. [Google Scholar] [CrossRef] [Green Version]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valencia, T.; Kim, J.Y.; Abu-Baker, S.; Moscat-Pardos, J.; Ahn, C.S.; Reina-Campos, M.; Duran, A.; Castilla, E.A.; Metallo, C.M.; Diaz-Meco, M.T.; et al. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell 2014, 26, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, A.; Hernandez, E.D.; Reina-Campos, M.; Castilla, E.A.; Subramaniam, S.; Raghunandan, S.; Roberts, L.R.; Kisseleva, T.; Karin, M.; Diaz-Meco, M.T.; et al. p62/SQSTM1 by Binding to Vitamin D Receptor Inhibits Hepatic Stellate Cell Activity, Fibrosis, and Liver Cancer. Cancer Cell 2016, 30, 595–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Duran, A.; Reina-Campos, M.; Valencia, T.; Castilla, E.A.; Müller, T.D.; Tschöp, M.H.; Moscat, J.; Diaz-Meco, M.T. Adipocyte p62/SQSTM1 Suppresses Tumorigenesis through Opposite Regulations of Metabolism in Adipose Tissue and Tumor. Cancer Cell 2018, 33, 770–784.e776. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Zhang, J.; Zhao, J.; Ma, N.; Kim, S.W.; Qiao, S.; Ma, X. Autophagy: The Last Defense against Cellular Nutritional Stress. Adv. Nutr. 2018, 9, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Daskalaki, I.; Gkikas, I.; Tavernarakis, N. Hypoxia and Selective Autophagy in Cancer Development and Therapy. Front. Cell Dev. Biol. 2018, 6, 104. [Google Scholar] [CrossRef] [Green Version]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Mazure, N.M.; Pouysségur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef]
- Ngabire, D.; Kim, G.D. Autophagy and Inflammatory Response in the Tumor Microenvironment. Int. J. Mol. Sci. 2017, 18, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.J.; Yoon, J.H.; Ryu, J.H. Mitophagy: A balance regulator of NLRP3 inflammasome activation. BMB Rep. 2016, 49, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 2007, 7, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef]
- Yang, A.; Rajeshkumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef] [Green Version]
- Elgendy, M.; Sheridan, C.; Brumatti, G.; Martin, S.J. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol. Cell 2011, 42, 23–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Koh, J.Y.; Price, S.; White, E.; Mehnert, J.M. Atg7 Overcomes Senescence and Promotes Growth of BrafV600E-Driven Melanoma. Cancer Discov. 2015, 5, 410–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; He, Z.; von Rütte, T.; Yousefi, S.; Hunger, R.E.; Simon, H.U. Down-regulation of autophagy-related protein 5 (ATG5) contributes to the pathogenesis of early-stage cutaneous melanoma. Sci. Transl. Med. 2013, 5, 202ra123. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 1994, 124, 619–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanharanta, S.; Massagué, J. Origins of metastatic traits. Cancer Cell 2013, 24, 410–421. [Google Scholar] [CrossRef] [Green Version]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.F.; Shi, Y.H.; Ding, Z.B.; Ke, A.W.; Gu, C.Y.; Hui, B.; Zhou, J.; Qiu, S.J.; Dai, Z.; Fan, J. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy 2013, 9, 2056–2068. [Google Scholar] [CrossRef] [Green Version]
- Avivar-Valderas, A.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Bardeesy, N.; Debnath, J.; Aguirre-Ghiso, J.A. Regulation of autophagy during ECM detachment is linked to a selective inhibition of mTORC1 by PERK. Oncogene 2013, 32, 4932–4940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalano, M.; D’Alessandro, G.; Lepore, F.; Corazzari, M.; Caldarola, S.; Valacca, C.; Faienza, F.; Esposito, V.; Limatola, C.; Cecconi, F.; et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol. Oncol. 2015, 9, 1612–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gugnoni, M.; Sancisi, V.; Gandolfi, G.; Manzotti, G.; Ragazzi, M.; Giordano, D.; Tamagnini, I.; Tigano, M.; Frasoldati, A.; Piana, S.; et al. Cadherin-6 promotes EMT and cancer metastasis by restraining autophagy. Oncogene 2017, 36, 667–677. [Google Scholar] [CrossRef]
- Morel, E.; Mehrpour, M.; Botti, J.; Dupont, N.; Hamaï, A.; Nascimbeni, A.C.; Codogno, P. Autophagy: A Druggable Process. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 375–398. [Google Scholar] [CrossRef]
- Malet-Martino, M.; Jolimaitre, P.; Martino, R. The prodrugs of 5-fluorouracil. Curr. Med. Chem. Anticancer Agents 2002, 2, 267–310. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Huang, S.; Wu, T.T.; Foster, N.R.; Sinicrope, F.A. Prognostic impact of Beclin 1, p62/sequestosome 1 and LC3 protein expression in colon carcinomas from patients receiving 5-fluorouracil as adjuvant chemotherapy. Cancer Biol. Ther. 2013, 14, 100–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.; Tang, J.; Liang, Y.; Jin, R.; Cai, X. Suppression of autophagy by chloroquine sensitizes 5-fluorouracil-mediated cell death in gallbladder carcinoma cells. Cell Biosci. 2014, 4, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, X.; Kong, N.; Wang, X.; Fang, Y.; Hu, X.; Xu, Y.; Chen, W.; Wang, K.; Li, D.; Jin, W.; et al. JNK confers 5-fluorouracil resistance in p53-deficient and mutant p53-expressing colon cancer cells by inducing survival autophagy. Sci. Rep. 2014, 4, 4694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, J.A.; Peixoto, A.; Neves, M.; Gaiteiro, C.; Reis, C.A.; Assaraf, Y.G.; Santos, L.L. Mechanisms of cisplatin resistance and targeting of cancer stem cells: Adding glycosylation to the equation. Drug Resist. Updat. 2016, 24, 34–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wu, G.S. Role of autophagy in cisplatin resistance in ovarian cancer cells. J. Biol. Chem. 2014, 289, 17163–17173. [Google Scholar] [CrossRef] [Green Version]
- Bao, L.; Jaramillo, M.C.; Zhang, Z.; Zheng, Y.; Yao, M.; Zhang, D.D.; Yi, X. Induction of autophagy contributes to cisplatin resistance in human ovarian cancer cells. Mol. Med. Rep. 2015, 11, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Yang, Y.; Liu, Q.; Wang, J. Inhibition of autophagy by 3-MA potentiates cisplatin-induced apoptosis in esophageal squamous cell carcinoma cells. Med. Oncol. 2011, 28, 105–111. [Google Scholar] [CrossRef]
- Zhu, L.; Du, H.; Shi, M.; Chen, Z.; Hang, J. ATG7 deficiency promote apoptotic death induced by Cisplatin in human esophageal squamous cell carcinoma cells. Bull. Cancer 2013, 100, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.; Thorburn, A. Targeting autophagy during cancer therapy to improve clinical outcomes. Pharmacol. Ther. 2011, 131, 130–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abedin, M.J.; Wang, D.; McDonnell, M.A.; Lehmann, U.; Kelekar, A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007, 14, 500–510. [Google Scholar] [CrossRef]
- O’Donovan, T.R.; O’Sullivan, G.C.; McKenna, S.L. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 2011, 7, 509–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwitkowski, V.E.; Prowell, T.M.; Ibrahim, A.; Farrell, A.T.; Justice, R.; Mitchell, S.S.; Sridhara, R.; Pazdur, R. FDA approval summary: Temsirolimus as treatment for advanced renal cell carcinoma. Oncologist 2010, 15, 428–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manic, G.; Obrist, F.; Kroemer, G.; Vitale, I.; Galluzzi, L. Chloroquine and hydroxychloroquine for cancer therapy. Mol. Cell Oncol. 2014, 1, e29911. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Lin, J.F.; Wen, S.I.; Yang, S.C.; Tsai, T.F.; Chen, H.E.; Chou, K.Y.; Hwang, T.I. Chloroquine and hydroxychloroquine inhibit bladder cancer cell growth by targeting basal autophagy and enhancing apoptosis. Kaohsiung J. Med. Sci. 2017, 33, 215–223. [Google Scholar] [CrossRef]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Sasaki, K.; Tsuno, N.H.; Sunami, E.; Tsurita, G.; Kawai, K.; Okaji, Y.; Nishikawa, T.; Shuno, Y.; Hongo, K.; Hiyoshi, M.; et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 2010, 10, 370. [Google Scholar] [CrossRef] [Green Version]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B.; et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Barnard, R.A.; Wittenburg, L.A.; Amaravadi, R.K.; Gustafson, D.L.; Thorburn, A.; Thamm, D.H. Phase I clinical trial and pharmacodynamic evaluation of combination hydroxychloroquine and doxorubicin treatment in pet dogs treated for spontaneously occurring lymphoma. Autophagy 2014, 10, 1415–1425. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, B. SAR405, a PIK3C3/Vps34 inhibitor that prevents autophagy and synergizes with MTOR inhibition in tumor cells. Autophagy 2015, 11, 725–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronan, B.; Flamand, O.; Vescovi, L.; Dureuil, C.; Durand, L.; Fassy, F.; Bachelot, M.F.; Lamberton, A.; Mathieu, M.; Bertrand, T.; et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat. Chem. Biol. 2014, 10, 1013–1019. [Google Scholar] [CrossRef]
- Shao, S.; Li, S.; Qin, Y.; Wang, X.; Yang, Y.; Bai, H.; Zhou, L.; Zhao, C.; Wang, C. Spautin-1, a novel autophagy inhibitor, enhances imatinib-induced apoptosis in chronic myeloid leukemia. Int. J. Oncol. 2014, 44, 1661–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maccarrone, M.; Bab, I.; Bíró, T.; Cabral, G.A.; Dey, S.K.; di Marzo, V.; Konje, J.C.; Kunos, G.; Mechoulam, R.; Pacher, P.; et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol. Sci. 2015, 36, 277–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanus, L.O.; Mechoulam, R. Novel natural and synthetic ligands of the endocannabinoid system. Curr. Med. Chem. 2010, 17, 1341–1359. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R.; et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Verme, J.L.; Fu, J.; Astarita, G.; la Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Gaoni, Y.; Mechoulam, R. The isolation and structure of delta-1-tetrahydrocannabinol and other neutral cannabinoids from hashish. J. Am. Chem. Soc. 1971, 93, 217–224. [Google Scholar] [CrossRef] [PubMed]
- ElSohly, M.A.; Radwan, M.M.; Gul, W.; Chandra, S.; Galal, A. Phytochemistry of Cannabis sativa L. Prog. Chem. Org. Nat. Prod. 2017, 103, 1–36. [Google Scholar] [PubMed]
- Morales, P.; Jagerovic, N. Antitumor Cannabinoid Chemotypes: Structural Insights. Front. Pharmacol. 2019, 10, 621. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.; di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB1 and CB2. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef] [Green Version]
- Chiocchetti, R.; Galiazzo, G.; Tagliavia, C.; Stanzani, A.; Giancola, F.; Menchetti, M.; Militerno, G.; Bernardini, C.; Forni, M.; Mandrioli, L. Cellular Distribution of Canonical and Putative Cannabinoid Receptors in Canine Cervical Dorsal Root Ganglia. Front. Vet. Sci. 2019, 6, 313. [Google Scholar] [CrossRef]
- Howlett, A.C. Cannabinoid inhibition of adenylate cyclase: Relative activity of constituents and metabolites of marihuana. Neuropharmacology 1987, 26, 507–512. [Google Scholar] [CrossRef]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Hanus, L.; Gopher, A.; Almog, S.; Mechoulam, R. Two new unsaturated fatty acid ethanolamides in brain that bind to the cannabinoid receptor. J. Med. Chem. 1993, 36, 3032–3034. [Google Scholar] [CrossRef]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Ferro, R.; Adamska, A.; Lattanzio, R.; Mavrommati, I.; Edling, C.E.; Arifin, S.A.; Fyffe, C.A.; Sala, G.; Sacchetto, L.; Chiorino, G.; et al. GPR55 signalling promotes proliferation of pancreatic cancer cells and tumour growth in mice, and its inhibition increases effects of gemcitabine. Oncogene 2018, 37, 6368–6382. [Google Scholar] [CrossRef] [PubMed]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Dinh, T.P.; Carpenter, D.; Leslie, F.M.; Freund, T.F.; Katona, I.; Sensi, S.L.; Kathuria, S.; Piomelli, D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc. Natl. Acad. Sci. USA 2002, 99, 10819–10824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tramèr, M.R.; Carroll, D.; Campbell, F.A.; Reynolds, D.J.; Moore, R.A.; McQuay, H.J. Cannabinoids for control of chemotherapy induced nausea and vomiting: Quantitative systematic review. BMJ 2001, 323, 16–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Han, L.; Zhang, X.; Li, L.; Jiang, C.; Qiu, Y.; Huang, R.; Xie, B.; Lin, Z.; Ren, J.; et al. Alteration of endocannabinoid system in human gliomas. J. Neurochem. 2012, 120, 842–849. [Google Scholar] [CrossRef]
- Xu, X.; Liu, Y.; Huang, S.; Liu, G.; Xie, C.; Zhou, J.; Fan, W.; Li, Q.; Wang, Q.; Zhong, D.; et al. Overexpression of cannabinoid receptors CB1 and CB2 correlates with improved prognosis of patients with hepatocellular carcinoma. Cancer Genet. Cytogenet. 2006, 171, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Ramer, R.; Schwarz, R.; Hinz, B. Modulation of the Endocannabinoid System as a Potential Anticancer Strategy. Front. Pharmacol. 2019, 10, 430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalski, C.W.; Oti, F.E.; Erkan, M.; Sauliunaite, D.; Bergmann, F.; Pacher, P.; Batkai, S.; Müller, M.W.; Giese, N.A.; Friess, H.; et al. Cannabinoids in pancreatic cancer: Correlation with survival and pain. Int. J. Cancer 2008, 122, 742–750. [Google Scholar] [CrossRef] [Green Version]
- Vitale, I.; Shema, E.; Loi, S.; Galluzzi, L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat. Med. 2021, 27, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Todoric, J.; Karin, M. The Fire within: Cell-Autonomous Mechanisms in Inflammation-Driven Cancer. Cancer Cell 2019, 35, 714–720. [Google Scholar] [CrossRef]
- Śledziński, P.; Nowak-Terpiłowska, A.; Zeyland, J. Cannabinoids in Medicine: Cancer, Immunity, and Microbial Diseases. Int. J. Mol. Sci. 2021, 22, 263. [Google Scholar] [CrossRef]
- Kienzl, M.; Kargl, J.; Schicho, R. The Immune Endocannabinoid System of the Tumor Microenvironment. Int. J. Mol. Sci. 2020, 21, 8929. [Google Scholar] [CrossRef] [PubMed]
- Bar-Sela, G.; Cohen, I.; Campisi-Pinto, S.; Lewitus, G.M.; Oz-Ari, L.; Jehassi, A.; Peer, A.; Turgeman, I.; Vernicova, O.; Berman, P.; et al. Cannabis Consumption Used by Cancer Patients during Immunotherapy Correlates with Poor Clinical Outcome. Cancers 2020, 12, 2447. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Carracedo, A.; Salanueva, I.J.; Hernández-Tiedra, S.; Lorente, M.; Egia, A.; Vázquez, P.; Blázquez, C.; Torres, S.; García, S.; et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Investig. 2009, 119, 1359–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, J.L.; Hill, D.S.; McKee, C.S.; Hernandez-Tiedra, S.; Lorente, M.; Lopez-Valero, I.; Anagnostou, M.E.; Babatunde, F.; Corazzari, M.; Redfern, C.P.F.; et al. Exploiting cannabinoid-induced cytotoxic autophagy to drive melanoma cell death. J. Investig. Dermatol. 2015, 135, 1629–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qamri, Z.; Preet, A.; Nasser, M.W.; Bass, C.E.; Leone, G.; Barsky, S.H.; Ganju, R.K. Synthetic cannabinoid receptor agonists inhibit tumor growth and metastasis of breast cancer. Mol. Cancer Ther. 2009, 8, 3117–3129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vara, D.; Salazar, M.; Olea-Herrero, N.; Guzmán, M.; Velasco, G.; Díaz-Laviada, I. Anti-tumoral action of cannabinoids on hepatocellular carcinoma: Role of AMPK-dependent activation of autophagy. Cell Death Differ. 2011, 18, 1099–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Hinz, B.; Ramer, R. Anti-tumour actions of cannabinoids. Br. J. Pharmacol. 2019, 176, 1384–1394. [Google Scholar] [CrossRef]
- Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J.; Cui, Q. The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.; Hokinson, D.; Park, S.; Elvira, R.; Kusuma, F.; Lee, J.M.; Yun, M.; Lee, S.G.; Han, J. ER Stress Induces Cell Cycle Arrest at the G2/M Phase Through eIF2α Phosphorylation and GADD45α. Int. J. Mol. Sci. 2019, 20, 6309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caffarel, M.M.; Sarrió, D.; Palacios, J.; Guzmán, M.; Sánchez, C. Delta9-tetrahydrocannabinol inhibits cell cycle progression in human breast cancer cells through Cdc2 regulation. Cancer Res. 2006, 66, 6615–6621. [Google Scholar] [CrossRef] [Green Version]
- Al Bitar, S.; Gali-Muhtasib, H. The Role of the Cyclin Dependent Kinase Inhibitor p21(cip1/waf1) in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics. Cancers 2019, 11, 1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blázquez, C.; Carracedo, A.; Barrado, L.; Real, P.J.; Fernández-Luna, J.L.; Velasco, G.; Malumbres, M.; Guzmán, M. Cannabinoid receptors as novel targets for the treatment of melanoma. FASEB J. 2006, 20, 2633–2635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasco, G.; Sánchez, C.; Guzmán, M. Towards the use of cannabinoids as antitumour agents. Nat. Rev. Cancer 2012, 12, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, D.J.; Marnett, L.J. Cannabinoids, endocannabinoids, and cancer. Cancer Metastasis Rev. 2011, 30, 599–612. [Google Scholar] [CrossRef]
- Katayama, Y.; Uchino, J.; Chihara, Y.; Tamiya, N.; Kaneko, Y.; Yamada, T.; Takayama, K. Tumor Neovascularization and Developments in Therapeutics. Cancers 2019, 11, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef]
- Klauber, N.; Parangi, S.; Flynn, E.; Hamel, E.; D’Amato, R.J. Inhibition of angiogenesis and breast cancer in mice by the microtubule inhibitors 2-methoxyestradiol and taxol. Cancer Res. 1997, 57, 81–86. [Google Scholar]
- Stanley, G.; Harvey, K.; Slivova, V.; Jiang, J.; Sliva, D. Ganoderma lucidum suppresses angiogenesis through the inhibition of secretion of VEGF and TGF-beta1 from prostate cancer cells. Biochem. Biophys. Res. Commun. 2005, 330, 46–52. [Google Scholar] [CrossRef]
- Haustein, M.; Ramer, R.; Linnebacher, M.; Manda, K.; Hinz, B. Cannabinoids increase lung cancer cell lysis by lymphokine-activated killer cells via upregulation of ICAM-1. Biochem. Pharmacol. 2014, 92, 312–325. [Google Scholar] [CrossRef]
- Scheau, C.; Badarau, I.A.; Mihai, L.G.; Scheau, A.E.; Costache, D.O.; Constantin, C.; Calina, D.; Caruntu, C.; Costache, R.S.; Caruntu, A. Cannabinoids in the Pathophysiology of Skin Inflammation. Molecules 2020, 25, 652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.H.; Iskandar, K.B.; Yadav, S.K.; Hirpara, J.L.; Loh, T.; Pervaiz, S. Simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation. PLoS ONE 2010, 5, e9996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munson, A.E.; Harris, L.S.; Friedman, M.A.; Dewey, W.L.; Carchman, R.A. Antineoplastic activity of cannabinoids. J. Natl. Cancer Inst. 1975, 55, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Calvaruso, G.; Pellerito, O.; Notaro, A.; Giuliano, M. Cannabinoid-associated cell death mechanisms in tumor models (review). Int. J. Oncol. 2012, 41, 407–413. [Google Scholar] [CrossRef] [Green Version]
- Bifulco, M.; Malfitano, A.M.; Pisanti, S.; Laezza, C. Endocannabinoids in endocrine and related tumours. Endocr. Relat. Cancer 2008, 15, 391–408. [Google Scholar] [CrossRef]
- Maccarrone, M.; Lorenzon, T.; Bari, M.; Melino, G.; Finazzi-Agro, A. Anandamide induces apoptosis in human cells via vanilloid receptors. Evidence for a protective role of cannabinoid receptors. J. Biol. Chem. 2000, 275, 31938–31945. [Google Scholar] [CrossRef] [Green Version]
- Demuth, D.G.; Molleman, A. Cannabinoid signalling. Life Sci. 2006, 78, 549–563. [Google Scholar] [CrossRef]
- Cianchi, F.; Papucci, L.; Schiavone, N.; Lulli, M.; Magnelli, L.; Vinci, M.C.; Messerini, L.; Manera, C.; Ronconi, E.; Romagnani, P.; et al. Cannabinoid receptor activation induces apoptosis through tumor necrosis factor alpha-mediated ceramide de novo synthesis in colon cancer cells. Clin. Cancer Res. 2008, 14, 7691–7700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carracedo, A.; Gironella, M.; Lorente, M.; Garcia, S.; Guzmán, M.; Velasco, G.; Iovanna, J.L. Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress-related genes. Cancer Res. 2006, 66, 6748–6755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carracedo, A.; Lorente, M.; Egia, A.; Blázquez, C.; García, S.; Giroux, V.; Malicet, C.; Villuendas, R.; Gironella, M.; González-Feria, L.; et al. The stress-regulated protein p8 mediates cannabinoid-induced apoptosis of tumor cells. Cancer Cell 2006, 9, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Galve-Roperh, I.; Sánchez, C.; Cortés, M.L.; del Pulgar, T.G.; Izquierdo, M.; Guzmán, M. Anti-tumoral action of cannabinoids: Involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat. Med. 2000, 6, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Del Pulgar, T.G.; Velasco, G.; Sánchez, C.; Haro, A.; Guzmán, M. De novo-synthesized ceramide is involved in cannabinoid-induced apoptosis. Biochem. J. 2002, 363, 183–188. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Lorente, M.; García-Taboada, E.; Hernández-Tiedra, S.; Davila, D.; Francis, S.E.; Guzmán, M.; Kiss-Toth, E.; Velasco, G. The pseudokinase tribbles homologue-3 plays a crucial role in cannabinoid anticancer action. Biochim. Biophys. Acta 2013, 1831, 1573–1578. [Google Scholar] [CrossRef] [PubMed]
- Bachari, A.; Piva, T.J.; Salami, S.A.; Jamshidi, N.; Mantri, N. Roles of Cannabinoids in Melanoma: Evidence from In Vivo Studies. Int. J. Mol. Sci. 2020, 21, 6040. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, C.; Ruiz-Calvo, A.; Bajo-Grañeras, R.; Baufreton, J.M.; Resel, E.; Varilh, M.; Zottola, A.C.P.; Mariani, Y.; Cannich, A.; Rodríguez-Navarro, J.A.; et al. Cannabinoid-induced motor dysfunction via autophagy inhibition. Autophagy 2020, 16, 2289–2291. [Google Scholar] [CrossRef]
- Nabissi, M.; Morelli, M.B.; Offidani, M.; Amantini, C.; Gentili, S.; Soriani, A.; Cardinali, C.; Leoni, P.; Santoni, G. Cannabinoids synergize with carfilzomib, reducing multiple myeloma cells viability and migration. Oncotarget 2016, 7, 77543–77557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morelli, M.B.; Offidani, M.; Alesiani, F.; Discepoli, G.; Liberati, S.; Olivieri, A.; Santoni, M.; Santoni, G.; Leoni, P.; Nabissi, M. The effects of cannabidiol and its synergism with bortezomib in multiple myeloma cell lines. A role for transient receptor potential vanilloid type-2. Int. J. Cancer 2014, 134, 2534–2546. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Rozenfeld, R.; Wu, D.; Devi, L.A.; Zhang, Z.; Cederbaum, A. Cannabidiol protects liver from binge alcohol-induced steatosis by mechanisms including inhibition of oxidative stress and increase in autophagy. Free Radic. Biol. Med. 2014, 68, 260–267. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, A.; Kuzontkoski, P.M.; Groopman, J.E.; Prasad, A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol. Cancer Ther. 2011, 10, 1161–1172. [Google Scholar] [CrossRef] [Green Version]
- Emery, S.M.; Alotaibi, M.R.; Tao, Q.; Selley, D.E.; Lichtman, A.H.; Gewirtz, D.A. Combined antiproliferative effects of the aminoalkylindole WIN55,212–2 and radiation in breast cancer cells. J. Pharmacol. Exp. Ther. 2014, 348, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Schoeman, R.; Beukes, N.; Frost, C. Cannabinoid Combination Induces Cytoplasmic Vacuolation in MCF-7 Breast Cancer Cells. Molecules 2020, 25, 4682. [Google Scholar] [CrossRef]
- Ivanov, V.N.; Grabham, P.W.; Wu, C.-C.; Hei, T.K. Inhibition of autophagic flux differently modulates cannabidiol-induced death in 2D and 3D glioblastoma cell cultures. Sci. Rep. 2020, 10, 2687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.; Huang, Y.; Zhang, Y.; Wang, C.; Wu, H.; Tian, X.; Liu, Y.; Hou, B.; Liang, Y.; Rong, H.; et al. Cannabinoid receptor 2-selective agonist JWH015 attenuates bone cancer pain through the amelioration of impaired autophagy flux induced by inflammatory mediators in the spinal cord. Mol. Med. Rep. 2019, 20, 5100–5110. [Google Scholar] [CrossRef] [Green Version]
- Murase, R.; Kawamura, R.; Singer, E.; Pakdel, A.; Sarma, P.; Judkins, J.; Elwakeel, E.; Dayal, S.; Martinez-Martinez, E.; Amere, M.; et al. Targeting multiple cannabinoid anti-tumour pathways with a resorcinol derivative leads to inhibition of advanced stages of breast cancer. Br. J. Pharmacol. 2014, 171, 4464–4477. [Google Scholar] [CrossRef] [Green Version]
- Pellerito, O.; Notaro, A.; Sabella, S.; de Blasio, A.; Vento, R.; Calvaruso, G.; Giuliano, M. WIN induces apoptotic cell death in human colon cancer cells through a block of autophagic flux dependent on PPARγ down-regulation. Apoptosis 2014, 19, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Notaro, A.; Sabella, S.; Pellerito, O.; di Fiore, R.; de Blasio, A.; Vento, R.; Calvaruso, G.; Giuliano, M. Involvement of PAR-4 in cannabinoid-dependent sensitization of osteosarcoma cells to TRAIL-induced apoptosis. Int. J. Biol. Sci. 2014, 10, 466–478. [Google Scholar] [CrossRef] [Green Version]
- Dando, I.; Donadelli, M.; Costanzo, C.; Pozza, E.D.; D’Alessandro, A.; Zolla, L.; Palmieri, M. Cannabinoids inhibit energetic metabolism and induce AMPK-dependent autophagy in pancreatic cancer cells. Cell Death Dis. 2013, 4, e664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellert-Miklaszewska, A.; Ciechomska, I.A.; Kaminska, B. Synthetic Cannabinoids Induce Autophagy and Mitochondrial Apoptotic Pathways in Human Glioblastoma Cells Independently of Deficiency in TP53 or PTEN Tumor Suppressors. Cancers 2021, 13, 419. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy revisited: A conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef] [Green Version]
- Saftig, P.; Beertsen, W.; Eskelinen, E.L. LAMP-2: A control step for phagosome and autophagosome maturation. Autophagy 2008, 4, 510–512. [Google Scholar] [CrossRef] [Green Version]
- Kraft, C.; Deplazes, A.; Sohrmann, M.; Peter, M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat. Cell Biol. 2008, 10, 602–610. [Google Scholar] [CrossRef]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N.; et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Mochida, K.; Oikawa, Y.; Kimura, Y.; Kirisako, H.; Hirano, H.; Ohsumi, Y.; Nakatogawa, H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015, 522, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends Cell Biol. 2016, 26, 6–16. [Google Scholar] [CrossRef]
- Mancias, J.D.; Kimmelman, A.C. Mechanisms of Selective Autophagy in Normal Physiology and Cancer. J. Mol. Biol. 2016, 428, 1659–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, P.; Casari, I.; Falasca, M. Therapeutic potential of cannabinoids in combination cancer therapy. Adv. Biol. Regul. 2021, 79, 100774. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cannabinoid Classification | Cannabinoid | Diseases | Proposed Mechanism Mediating Autophagy | Reference |
|---|---|---|---|---|
| Phytocannabinoid | THC | Glioma |
| Salazar et al., (2009) [140] |
| THC + CBD | Melanoma |
| Bachari et al., (2020) [180] | |
| THC |
| Blázquez et al., (2006) [181] | ||
| THC + CBD | Multiple Myeloma |
| Nabissi et al., (2016) [182] | |
| THC + CBD + Carfilzomib (Immuno-proteasome inhibitor) |
| |||
| THC + CBD + Bortezomib (Proteasome Inhibitor) |
| Morelli et al., (2014) [183] | ||
| CBD | Liver Steatosis |
| Yang et al., (2014) [184] | |
| CBD | Breast Cancer |
| Shrivastava et al., (2011) [185] Emery et al., (2014) [186] | |
| THC |
| Schoeman et al., (2020) [187] | ||
| CBN | ||||
| CBG | ||||
| THC | Hepatocellular Carcinoma |
| Vara et al., (2011) [131] | |
| CBD | Pancreatic Cancer |
| Dando et al., (2013) [169] | |
| CBD | Glioblastoma Multiforme |
| Ivanov et al., (2020) [188] | |
| Synthetic Cannabinoid | JWH-015 | Bone Cancer |
| Mao et al., (2019) [189] |
| JWH-015 | Breast Cancer |
| Emery et al., (2014) [186] | |
| CP55 940 |
| Murase et al., (2014) [190] | ||
| O-1663 | ||||
| JWH-015 | Hepatocellular Carcinoma |
| Vara et al., (2011) [149] | |
| WIN55,212-2 | Colon Cancer Osteosarcoma |
| Pellerito et al., (2014) [191] Notaro et al., (2014) [192] | |
| WIN55,212-2 | Melanoma |
| Blázquez et al., (2006) [181] | |
| arachidonoyl cyclopropamide | Pancreatic Cancer |
| Dando et al., (2013) [193] | |
| GW405833 | ||||
| WIN55,212-2 JWH133 | Glioblastoma |
| Ellert-Miklaszewska et al., (2021) [194] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, X.C.; Werner, E.; Falasca, M. Molecular Mechanism of Autophagy and Its Regulation by Cannabinoids in Cancer. Cancers 2021, 13, 1211. https://doi.org/10.3390/cancers13061211
Lee XC, Werner E, Falasca M. Molecular Mechanism of Autophagy and Its Regulation by Cannabinoids in Cancer. Cancers. 2021; 13(6):1211. https://doi.org/10.3390/cancers13061211
Chicago/Turabian StyleLee, Xin Chien, Evelyn Werner, and Marco Falasca. 2021. "Molecular Mechanism of Autophagy and Its Regulation by Cannabinoids in Cancer" Cancers 13, no. 6: 1211. https://doi.org/10.3390/cancers13061211