
p300 Serine 89: A Critical Signaling Integrator and Its Effects on Intestinal Homeostasis and Repair

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Isolation of Crypt Cells from Ileum
2.3. Co-Immunoprecipitation
2.4. Western Blotting
2.5. RT-qPCR and PCR
2.6. 16S rRNA Gene Sequencing of Microbiota
2.7. Intestinal Organoids
2.8. Genomic Analysis
2.9. Proteomic Analysis
2.10. Ingenuity Pathway Analysis of Proteomic Data
2.11. DSS-induced Colitis Mouse Model
2.12. Data Analysis
3. Results
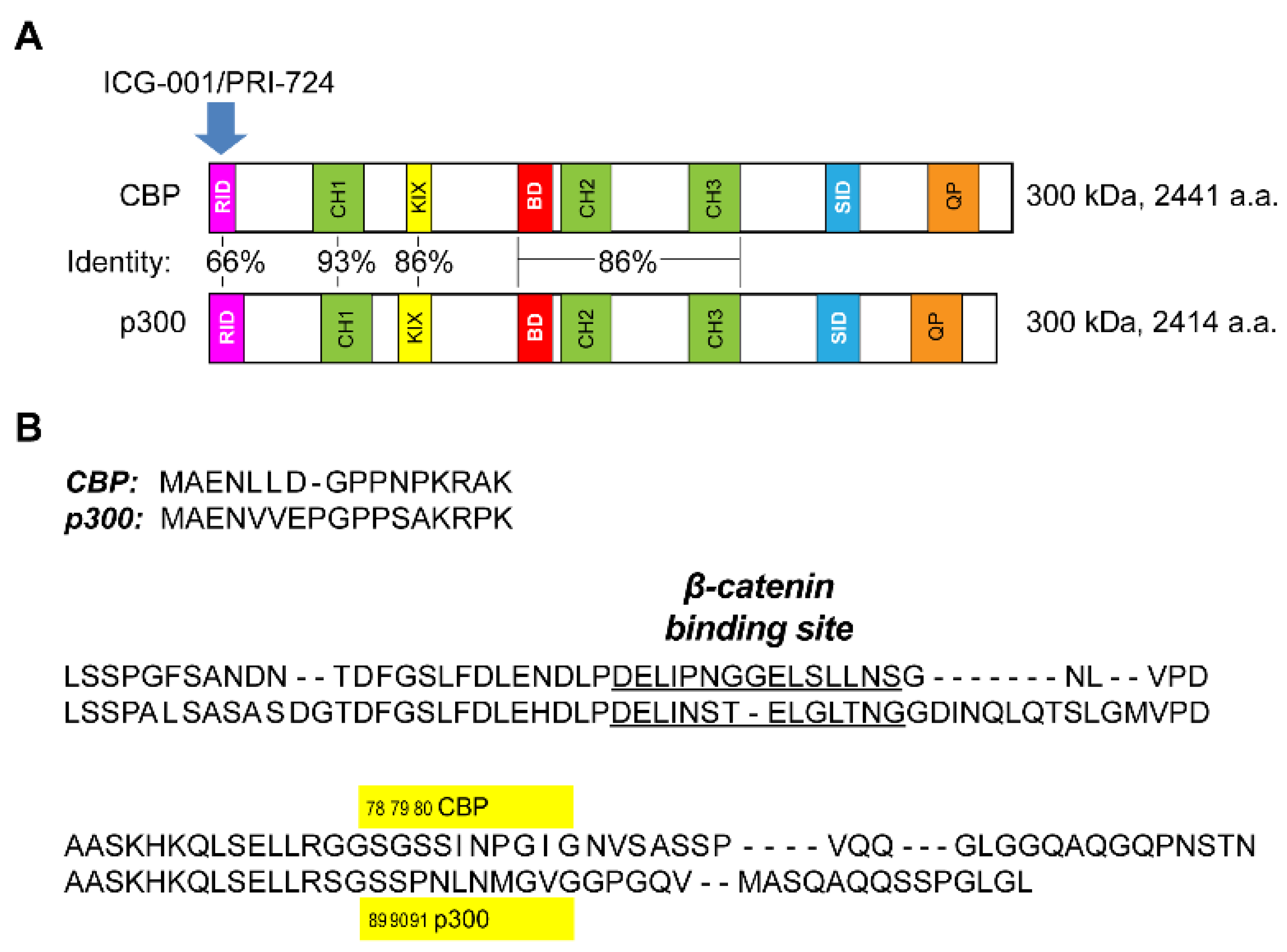
3.1. Generation of p300 S89A Point Mutant Knock-in Mice
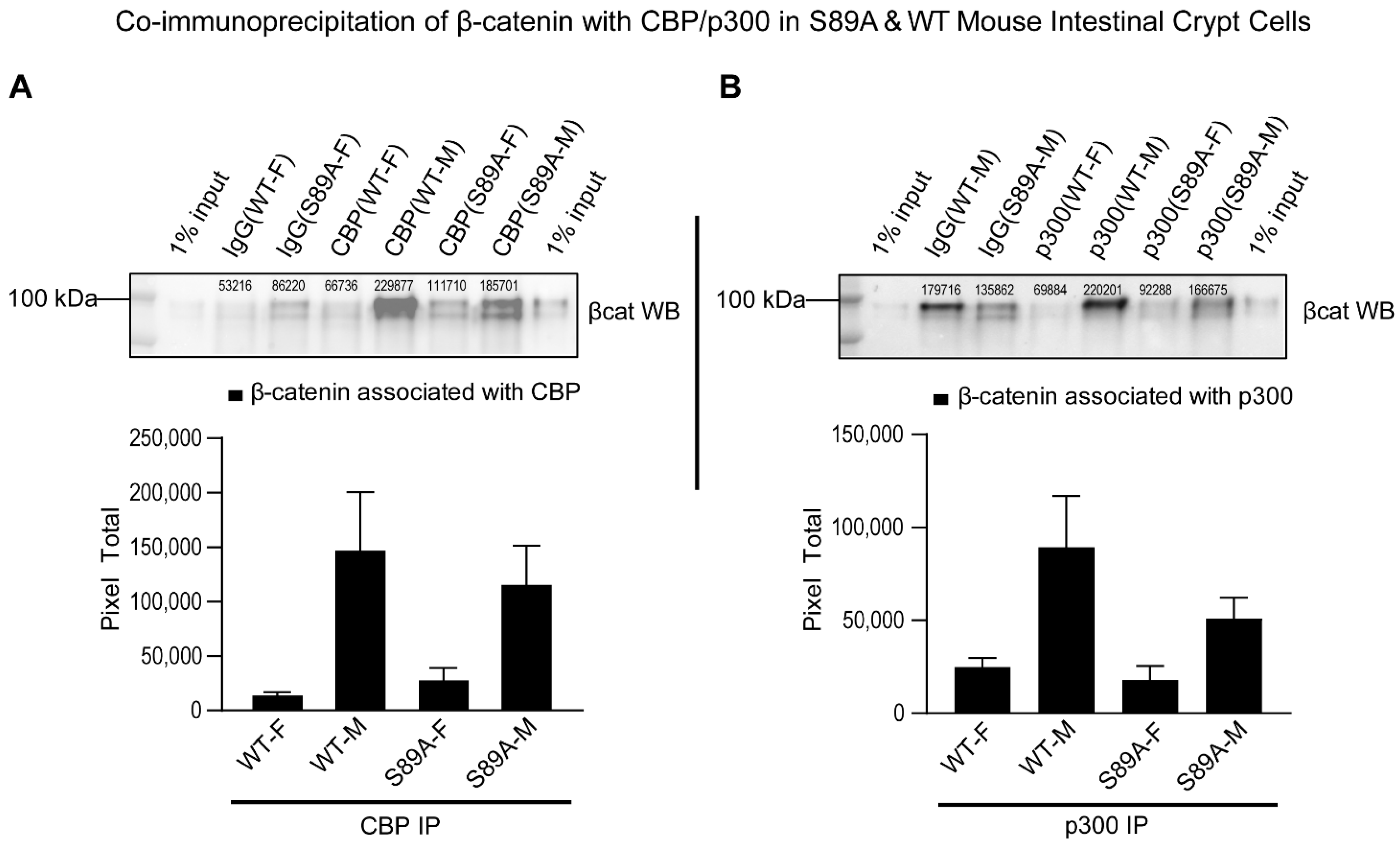
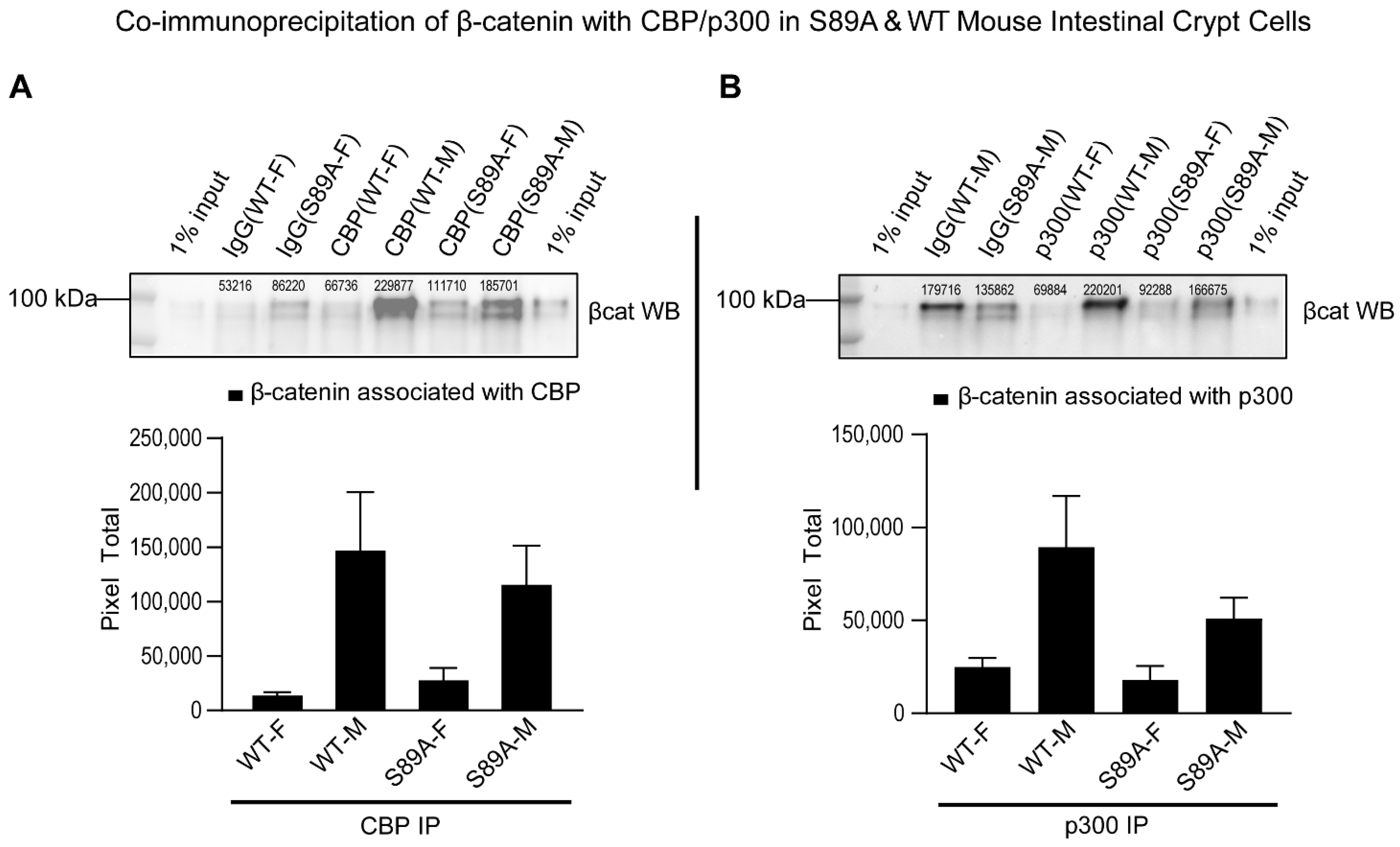
3.2. p300 S89A Mice and Differential β-Catenin Kat3 Coactivator Usage
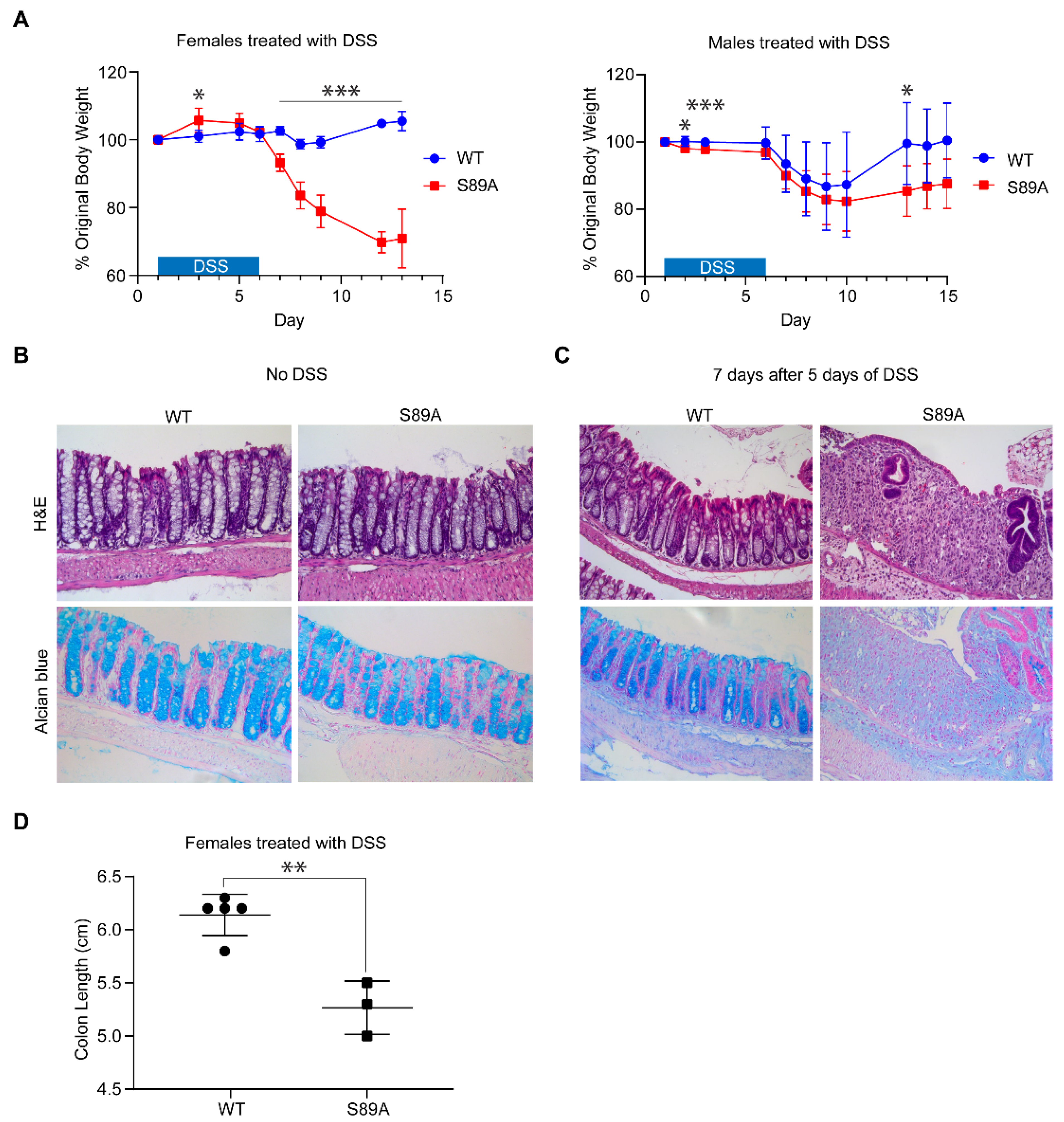
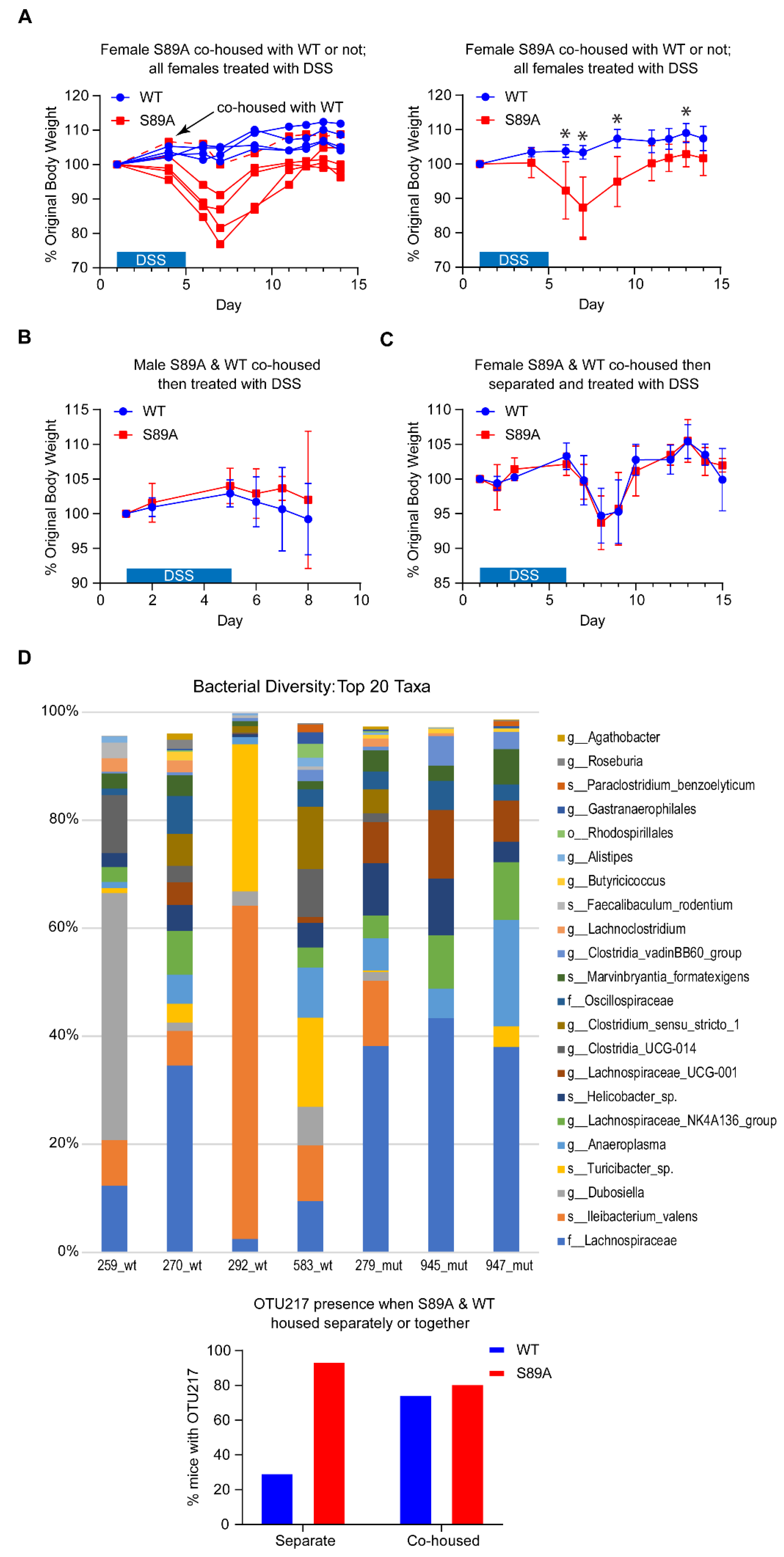
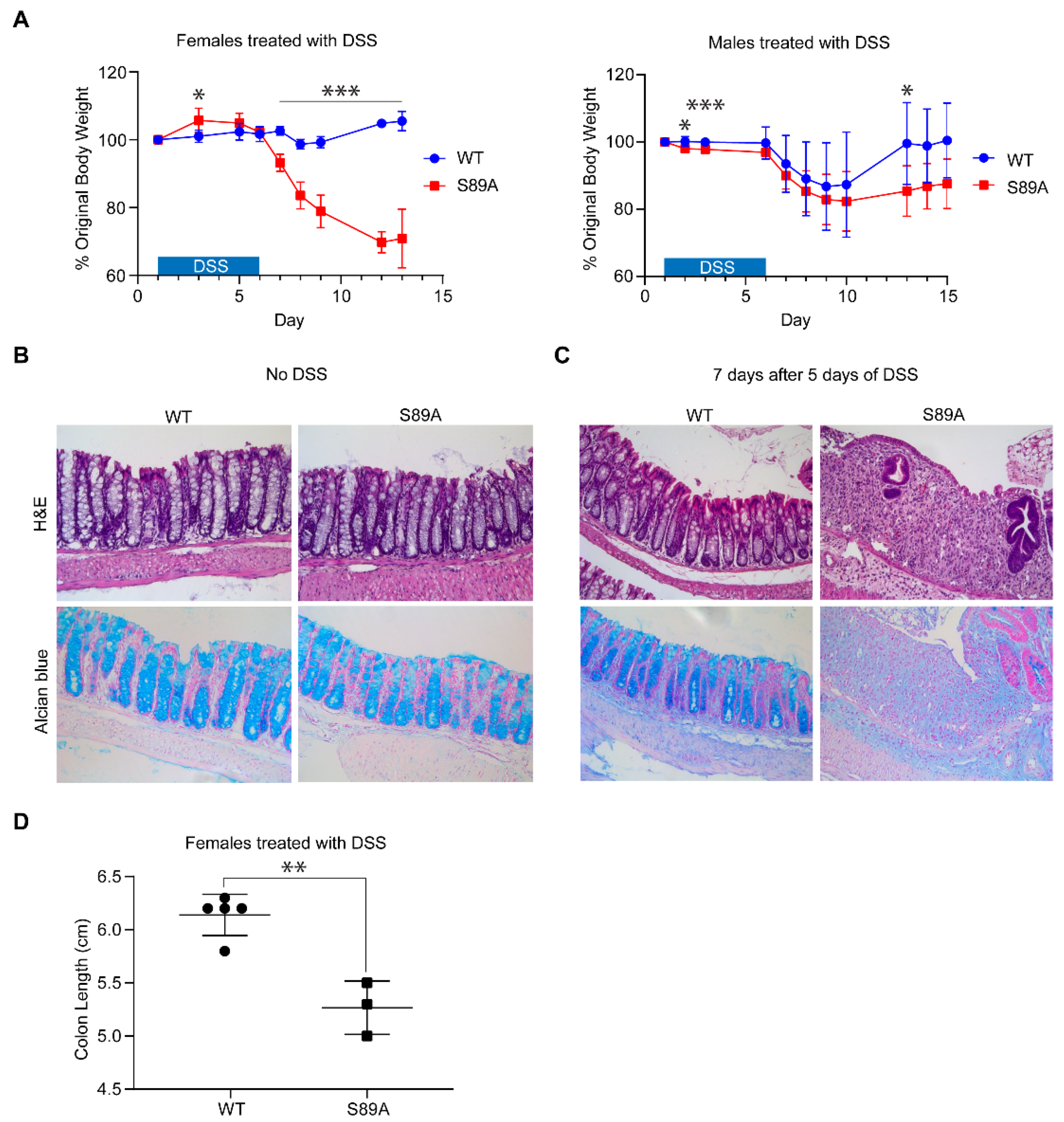
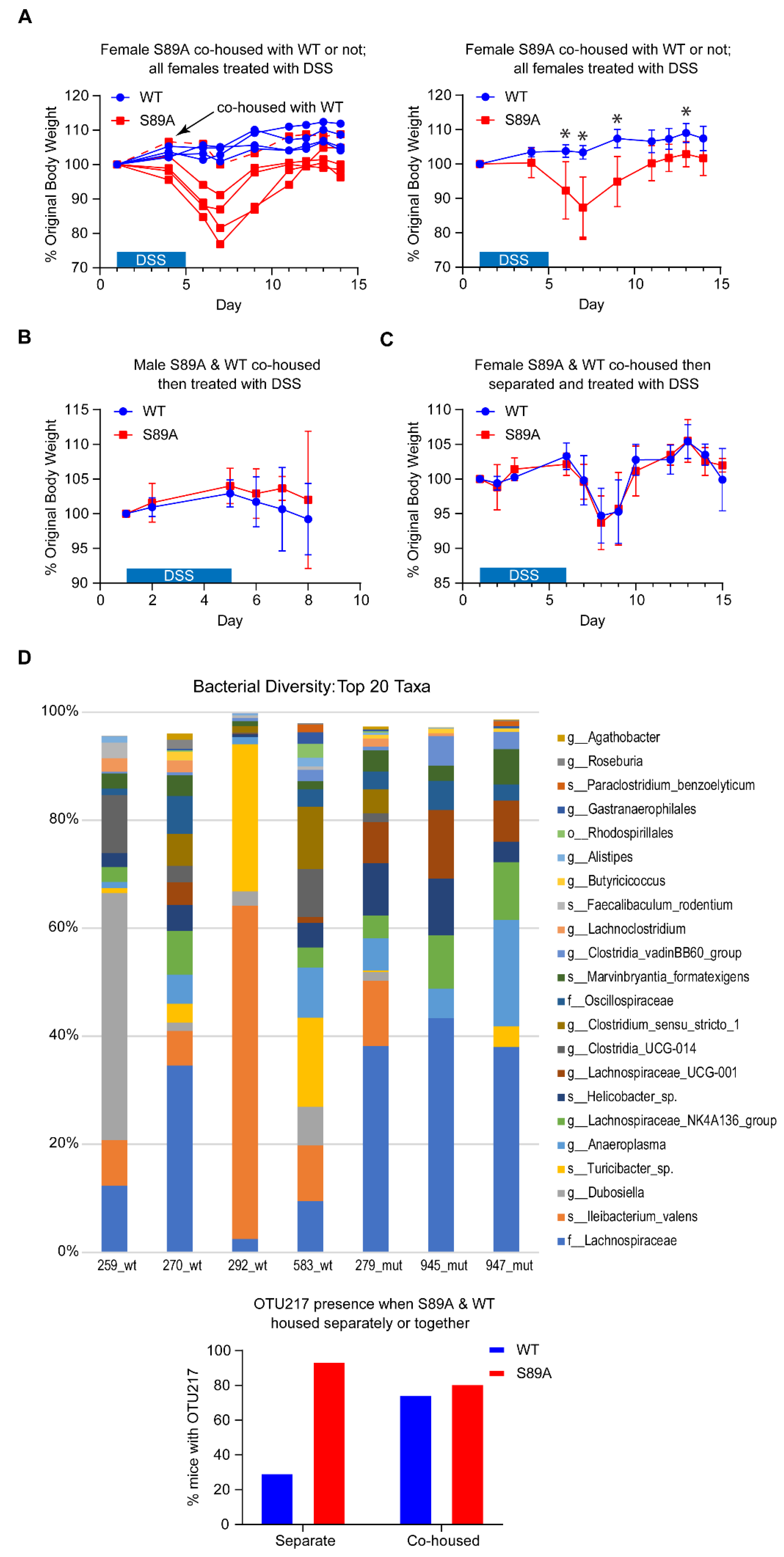
3.3. p300 S89A Mice Are Extremely Sensitive to Intestinal Insult
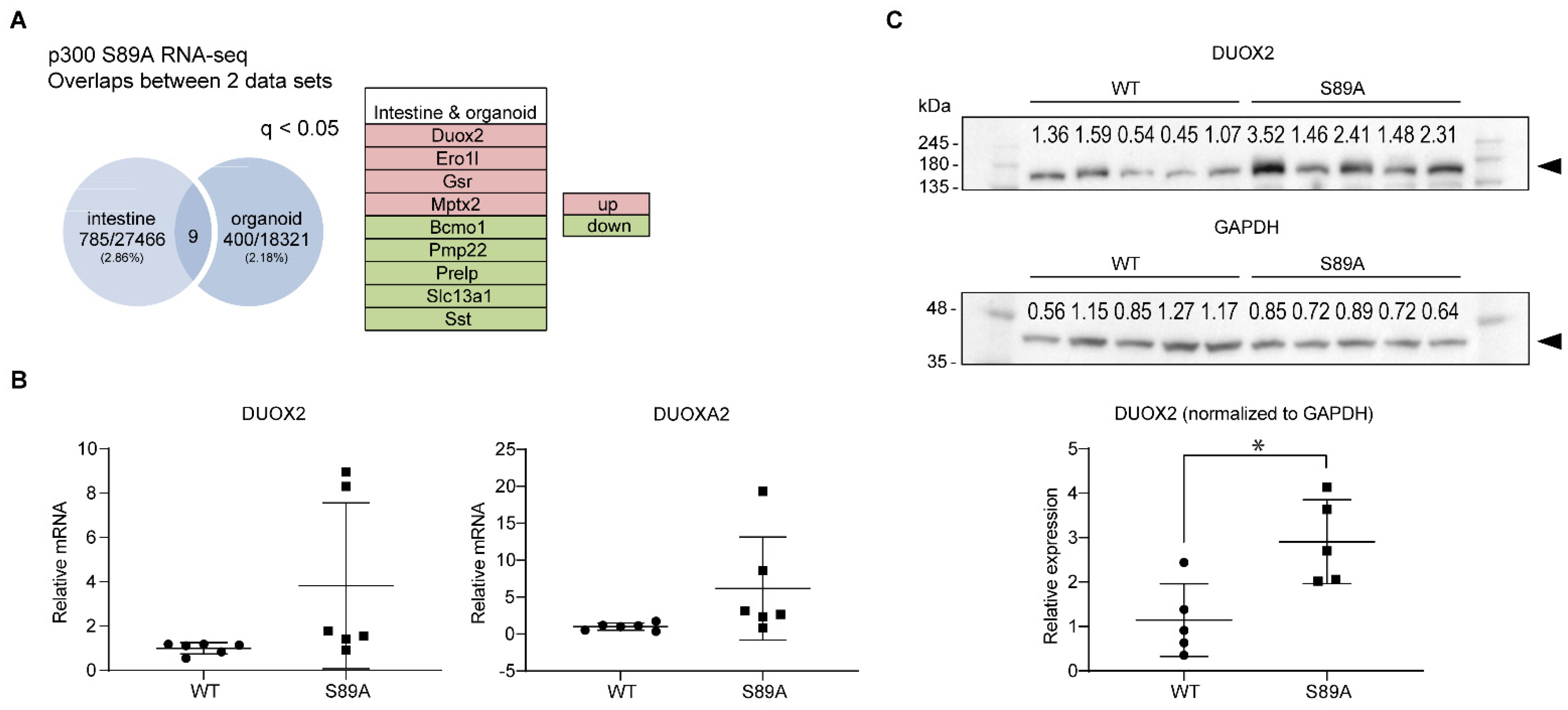
3.4. Genomic Analysis
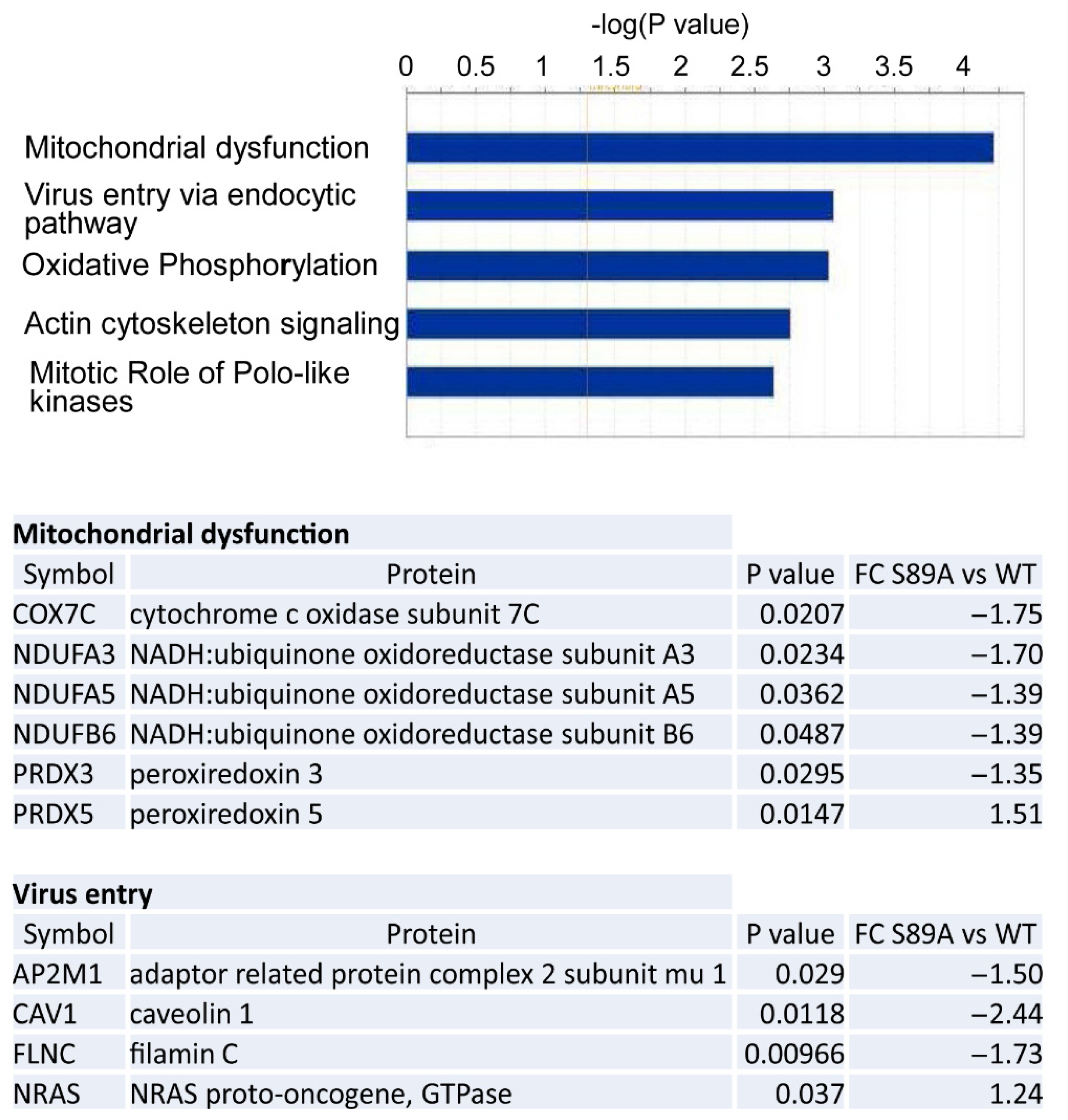
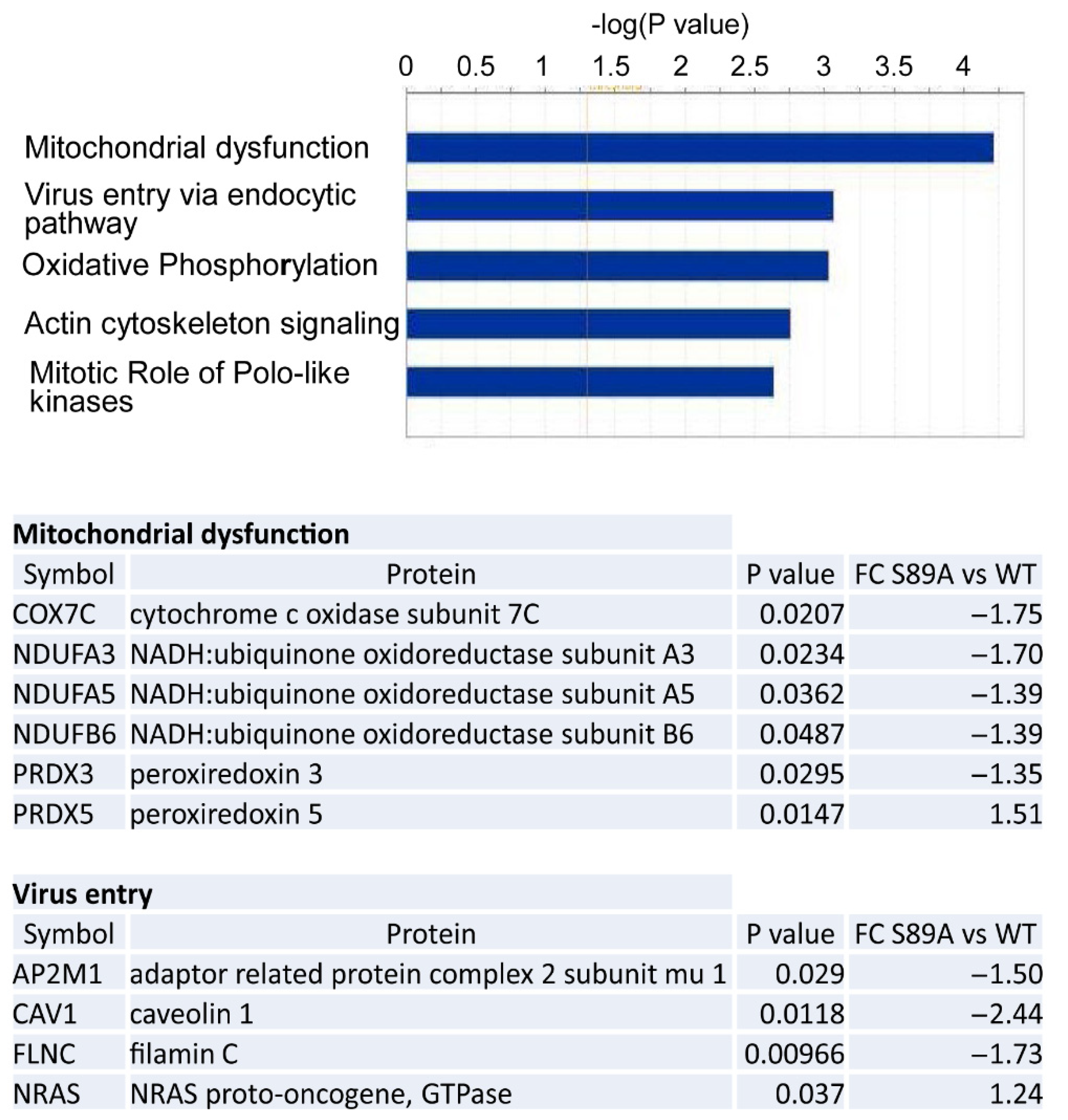
3.5. Proteomic Analysis
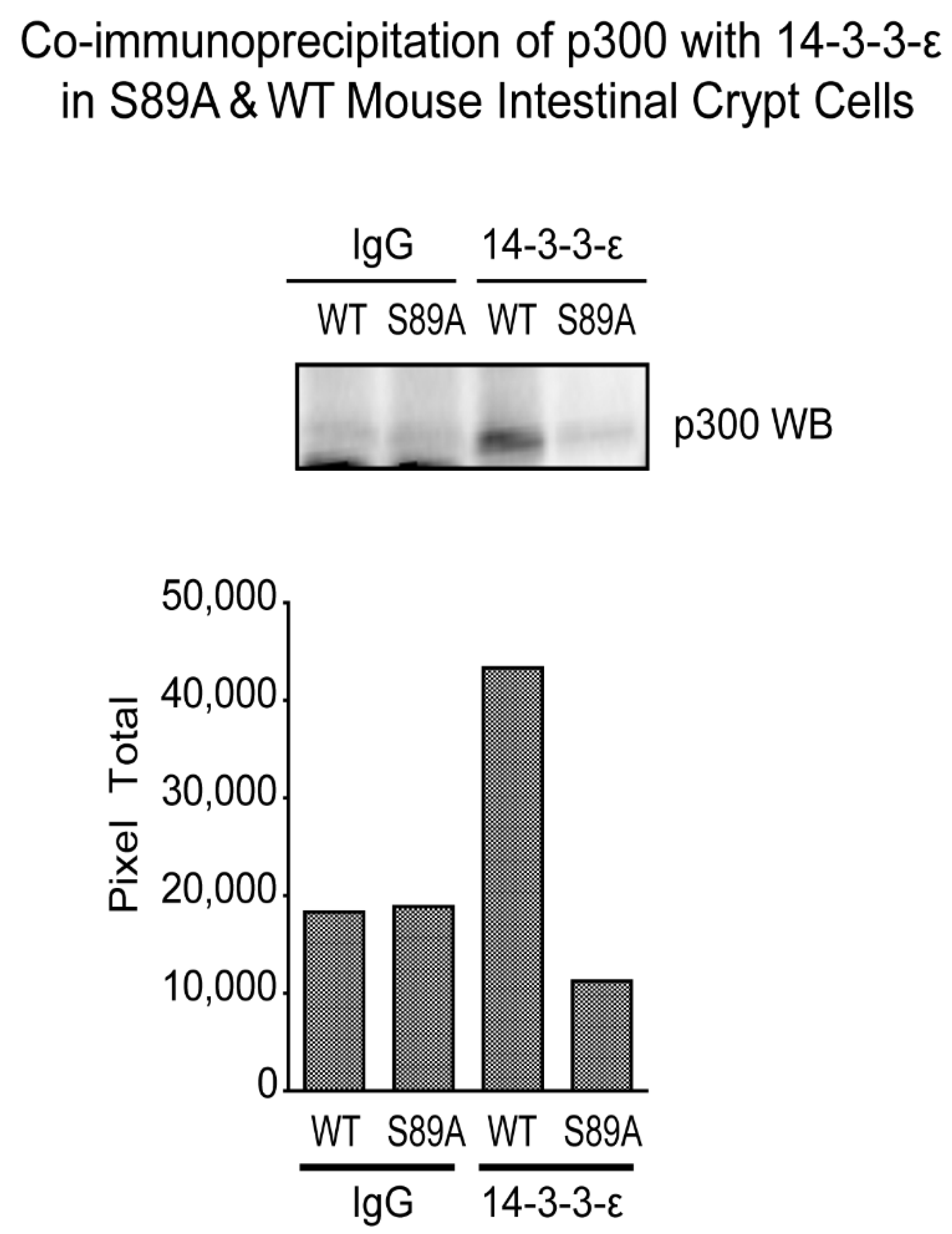
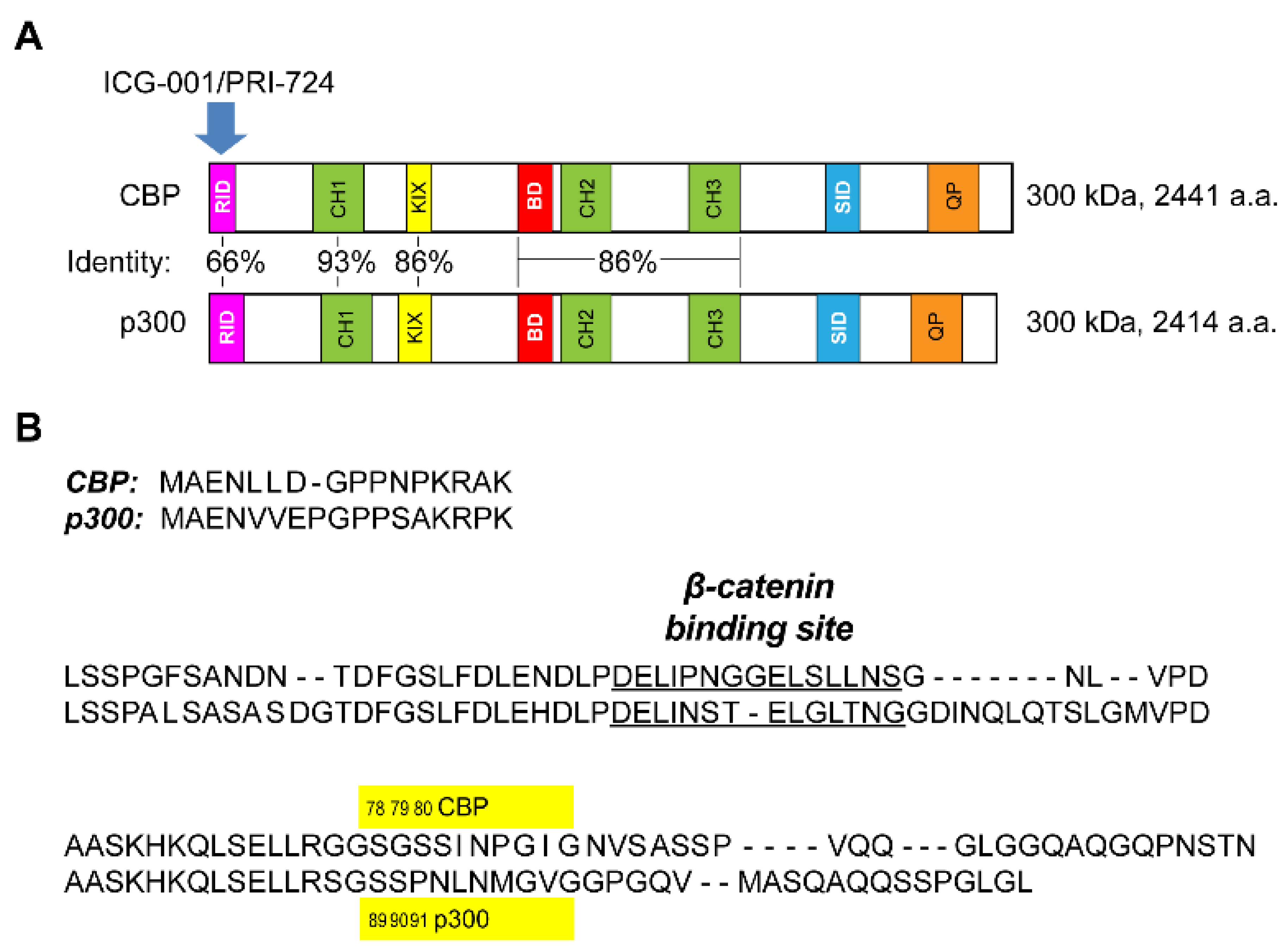
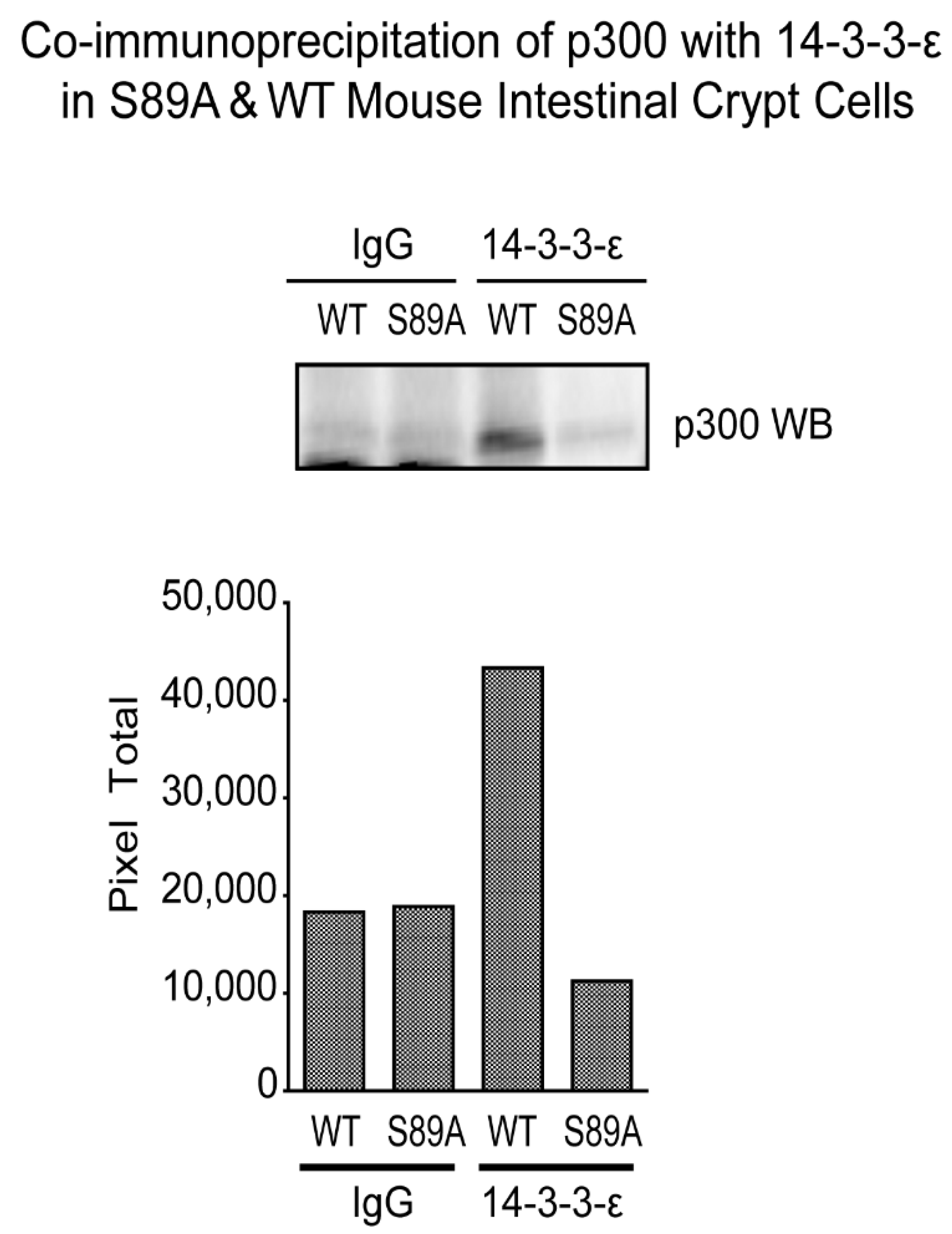
3.6. p300 S89A Is a Part of a 14-3-3 Binding Motif
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thomas, P.D.; Kahn, M. Kat3 coactivators in somatic stem cells and cancer stem cells: Biological roles, evolution, and pharmacologic manipulation. Cell Biol. Toxicol. 2016, 32, 61–81. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M. Wnt Signaling in Stem Cells and Cancer Stem Cells: A Tale of Two Coactivators. Prog. Mol. Biol. Transl. Sci. 2018, 153, 209–244. [Google Scholar] [CrossRef]
- Trosko, J.E.; Kang, K.-S. Evolution of Energy Metabolism, Stem Cells and Cancer Stem Cells: How the Warburg and Barker Hypotheses Might Be Linked. Int. J. Stem Cells 2012, 5, 39–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arany, Z.; Sellers, W.R.; Livingston, D.M.; Eckner, R. E1A-associated p300 and CREB-associated CBP belong to a conserved family of coactivators. Cell 1994, 77, 799–800. [Google Scholar] [CrossRef]
- Eckner, R.; Ewen, M.E.; Newsome, D.; Gerdes, M.; DeCaprio, J.A.; Lawrence, J.B.; Livingston, D.M. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 1994, 8, 869–884. [Google Scholar] [CrossRef] [Green Version]
- Kung, A.L.; Rebel, V.I.; Bronson, R.T.; Ch’Ng, L.-E.; Sieff, C.A.; Livingston, D.M.; Yao, T.-P. Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genome Res. 2000, 14, 272–277. [Google Scholar]
- Yamauchi, T.; Oike, Y.; Kamon, J.; Waki, H.; Komeda, K.; Tsuchida, A.; Date, Y.; Li, M.-X.; Miki, H.; Akanuma, Y.; et al. Increased insulin sensitivity despite lipodystrophy in Crebbp heterozygous mice. Nat. Genet. 2002, 30, 221–226. [Google Scholar] [CrossRef]
- Roth, J.-F.; Shikama, N.; Henzen, C.; Desbaillets, I.; Lutz, W.; Marino, S.; Wittwer, J.; Schorle, H.; Gassmann, M.; Eckner, R. Differential role of p300 and CBP acetyltransferase during myogenesis: p300 acts upstream of MyoD and Myf5. EMBO J. 2003, 22, 5186–5196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, M.; Lai, K.K.Y.; Wu, K.; Nguyen, C.; Lin, D.P.; Murali, R.; Kahn, M. Nuclear receptor/Wnt beta-catenin interactions are regulated via differential CBP/p300 coactivator usage. PLoS ONE 2018, 13, e0200714. [Google Scholar] [CrossRef]
- Ma, H.; Nguyen, C.; Lee, K.-S.; Kahn, M. Differential roles for the coactivators CBP and p300 on TCF/β-catenin-mediated survivin gene expression. Oncogene 2005, 24, 3619–3631. [Google Scholar] [CrossRef] [Green Version]
- Moon, R.T. Wnt/ -Catenin Pathway. Sci. Signal. 2005, 2005, cm1. [Google Scholar] [CrossRef]
- Teo, J.-L.; Kahn, M. The Wnt signaling pathway in cellular proliferation and differentiation: A tale of two coactivators. Adv. Drug Deliv. Rev. 2010, 62, 1149–1155. [Google Scholar] [CrossRef]
- McMillan, M.; Kahn, M. Investigating Wnt signaling: A chemogenomic safari. Drug Discov. Today 2005, 10, 1467–1474. [Google Scholar] [CrossRef]
- Emami, K.H.; Nguyen, C.; Ma, H.; Kim, D.H.; Jeong, K.W.; Eguchi, M.; Moon, R.T.; Teo, J.-L.; Oh, S.W.; Kim, H.Y.; et al. A small molecule inhibitor of -catenin/CREB-binding protein transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 12682–12687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higuchi, Y.; Nguyen, C.; Yasuda, S.-Y.; McMillan, M.; Hasegawa, K.; Kahn, M. Specific Direct Small Molecule p300/?-Catenin Antagonists Maintain Stem Cell Potency. Curr. Mol. Pharmacol. 2016, 9, 272–279. [Google Scholar] [CrossRef]
- Torchia, J.; Rose, D.W.; Inostroza, J.; Kamei, Y.; Westin, S.; Glass, C.K.; Rosenfeld, M.G. The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nat. Cell Biol. 1997, 387, 677–684. [Google Scholar] [CrossRef]
- Dillard, A.C.; Lane, M.A. Retinol decreases β-catenin protein levels in retinoic acid-resistant colon cancer cell lines. Mol. Carcinog. 2007, 46, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Easwaran, V.; Pishvaian, M.; Salimuddin; Byers, S. Cross-regulation of beta-catenin-LEF/TCF and retinoid signaling pathways. Curr. Biol. 1999, 9, 1415–1418. [Google Scholar] [CrossRef] [Green Version]
- Larriba, M.J.; Martín-Villar, E.; Garcia, J.M.; Pereira, F.; Peña, C.; De Herreros, A.G.; Bonilla, F.; Munoz, A. Snail2 cooperates with Snail1 in the repression of vitamin D receptor in colon cancer. Carcinogen 2009, 30, 1459–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyabayashi, T.; Teo, J.-L.; Yamamoto, M.; McMillan, M.; Nguyen, C.; Kahn, M. Wnt/beta-catenin/CBP signaling maintains long-term murine embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. USA 2007, 104, 5668–5673. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.W.; Gambee, J.E. Phosphorylation of p300 at Serine 89 by Protein Kinase C. J. Biol. Chem. 2000, 275, 40946–40951. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.W.; Soh, J.W.; Weinstein, I.B. Inhibition of histone acetyltransferase function of p300 by PKCdelta. Biochim. Biophys. Acta 2002, 1592, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Hong, Y.H.; Shen, X.-Q.; Frankowski, C.; Camp, H.S.; Leff, T. Regulation of Transcription by AMP-activated Protein Kinase. J. Biol. Chem. 2001, 276, 38341–38344. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Dentin, R.; Chen, D.; Hedrick, S.; Ravnskjaer, K.; Schenk, S.; Milne, J.; Meyers, D.J.; Cole, P.; Iii, J.Y.; et al. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nat. Cell Biol. 2008, 456, 269–273. [Google Scholar] [CrossRef]
- Gusterson, R.J.; Yuan, L.; Latchman, D.S. Distinct serine residues in CBP and p300 are necessary for their activation by phenylephrine. Int. J. Biochem. Cell Biol. 2004, 36, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Rieger, M.E.; Zhou, B.; Solomon, N.; Sunohara, M.; Li, C.; Nguyen, C.; Liu, Y.; Pan, J.-H.; Minoo, P.; Crandall, E.D.; et al. p300/β-Catenin Interactions Regulate Adult Progenitor Cell Differentiation Downstream of WNT5a/Protein Kinase C (PKC). J. Biol. Chem. 2016, 291, 6569–6582. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.K.; Horvath, B.; Xie, H.; Wu, X.; Lewis, B.L.; Pai, R.K.; Plesec, T.; Patil, D.T.; Gordon, I.O.; Wang, Y.; et al. Risk for Colorectal Neoplasia in Patients with Inflammatory Bowel Disease and Mucosa Indefinite for Dysplasia. Inflamm. Bowel Dis. 2015, 21, 378–384. [Google Scholar] [CrossRef]
- Chen, R.; Rabinovitch, P.S.; Crispin, D.A.; Emond, M.J.; Bronner, M.P.; Brentnall, T.A. The initiation of colon cancer in a chronic inflammatory setting. Carcinogen 2005, 26, 1513–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, T.-P.; Oh, S.P.; Fuchs, M.; Zhou, N.-D.; Ch’Ng, L.-E.; Newsome, D.; Bronson, R.T.; Li, E.; Livingston, D.M.; Eckner, R. Gene Dosage–Dependent Embryonic Development and Proliferation Defects in Mice Lacking the Transcriptional Integrator p300. Cell 1998, 93, 361–372. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, K.; Ackerman, S.; Dow, L.; Lowe, S. Isolation, Culture, and Maintenance of Mouse Intestinal Stem Cells. Bio Protocol 2016, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Clevers, H. Primary Mouse Small Intestinal Epithelial Cell Cultures. Adv. Struct. Saf. Stud. 2012, 945, 319–328. [Google Scholar] [CrossRef]
- Ring, A.; Nguyen, C.; Smbatyan, G.; Tripathy, D.; Yu, M.; Press, M.; Kahn, M.; Lang, J.E. CBP/β-Catenin/FOXM1 Is a Novel Therapeutic Target in Triple Negative Breast Cancer. Cancers 2018, 10, 525. [Google Scholar] [CrossRef] [Green Version]
- Okuda, S.; Watanabe, Y.; Moriya, Y.; Kawano, S.; Yamamoto, T.; Matsumoto, M.; Takami, T.; Kobayashi, D.; Araki, N.; Yoshizawa, A.C.; et al. jPOSTrepo: An international standard data repository for proteomes. Nucleic Acids Res. 2017, 45, D1107–D1111. [Google Scholar] [CrossRef] [Green Version]
- Whittem, C.G.; Williams, A.D.; Williams, C.S. Murine Colitis Modeling using Dextran Sulfate Sodium (DSS). J. Vis. Exp. 2010, e1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory Bowel Disease. Annu. Rev. Immunol. 2010, 28, 573–621. [Google Scholar] [CrossRef] [Green Version]
- Sartor, R.B.; Wu, G.D. Roles for Intestinal Bacteria, Viruses, and Fungi in Pathogenesis of Inflammatory Bowel Diseases and Therapeutic Approaches. Gastroenterology 2017, 152, 327–339.e4. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-A.; Cho, K.-C.; Kim, B.; Jang, I.-H.; Nam, K.; Kwon, Y.E.; Kim, M.; Hyeon, D.Y.; Hwang, D.; Seol, J.-H.; et al. Inflammation-Modulated Metabolic Reprogramming Is Required for DUOX-Dependent Gut Immunity in Drosophila. Cell Host Microbe 2018, 23, 338–352.e5. [Google Scholar] [CrossRef] [Green Version]
- Rigoni, A.; Poulsom, R.; Jeffery, R.; Mehta, S.; Lewis, A.; Yau, C.; Giannoulatou, E.; Feakins, R.; Lindsay, J.O.; Colombo, M.P.; et al. Separation of Dual Oxidase 2 and Lactoperoxidase Expression in Intestinal Crypts and Species Differences May Limit Hydrogen Peroxide Scavenging During Mucosal Healing in Mice and Humans. Inflamm. Bowel Dis. 2017, 24, 136–148. [Google Scholar] [CrossRef] [Green Version]
- Thompson, C.A.; Wojta, K.; Pulakanti, K.; Rao, S.; Dawson, P.; Battle, M.A. GATA4 Is Sufficient to Establish Jejunal Versus Ileal Identity in the Small Intestine. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 422–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cash, H.L.; Whitham, C.V.; Behrendt, C.L.; Hooper, L.V. Symbiotic Bacteria Direct Expression of an Intestinal Bactericidal Lectin. Science 2006, 313, 1126–1130. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Dassopoulos, T.; Cope, L.; Maitra, A.; Brant, S.R.; Harris, M.L.; Bayless, T.M.; Parmigiani, G.; Chakravarti, S. Genome-wide gene expression differences in Crohnʼs disease and ulcerative colitis from endoscopic pinch biopsies: Insights into distinctive pathogenesis. Inflamm. Bowel Dis. 2007, 13, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Varga, J. The transcriptional coactivator and acetyltransferase p300 in fibroblast biology and fibrosis. J. Cell. Physiol. 2007, 213, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Kemper, J.K.; Xiao, Z.; Ponugoti, B.; Miao, J.; Fang, S.; Kanamaluru, D.; Tsang, S.; Wu, S.-Y.; Chiang, C.-M.; Veenstra, T.D. FXR Acetylation Is Normally Dynamically Regulated by p300 and SIRT1 but Constitutively Elevated in Metabolic Disease States. Cell Metab. 2009, 10, 392–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawazoe, Y.; Sekimoto, T.; Araki, M.; Takagi, K.; Araki, K.; Yamamura, K.-I. Region-specific gastrointestinal Hox code during murine embryonal gut development. Dev. Growth Differ. 2002, 44, 77–84. [Google Scholar] [CrossRef]
- Giles, E.M.; Sanders, T.J.; McCarthy, N.E.; Lung, J.; Pathak, M.; Macdonald, T.T.; Lindsay, J.O.; Stagg, A.J. Regulation of human intestinal T-cell responses by type 1 interferon-STAT1 signaling is disrupted in inflammatory bowel disease. Mucosal Immunol. 2016, 10, 184–193. [Google Scholar] [CrossRef] [Green Version]
- Eckhardt, E.R.M.; Witta, J.; Zhong, J.; Arsenescu, R.; Arsenescu, V.; Wang, Y.; Ghoshal, S.; De Beer, M.C.; De Beer, F.C.; De Villiers, W.J.S. Intestinal Epithelial Serum Amyloid A Modulates Bacterial Growth In Vitro and Pro-Inflammatory Responses in Mouse Experimental Colitis. BMC Gastroenterol. 2010, 10, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, M.; Yoshitane, H.; Du, N.-H.; Okano, T.; Fukada, Y. Preferential Inhibition of BMAL2-CLOCK Activity by PER2 Reemphasizes Its Negative Role and a Positive Role of BMAL2 in the Circadian Transcription. J. Biol. Chem. 2009, 284, 25149–25159. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Hida, A.; McGuinness, O.P.; Wasserman, D.H.; Yamazaki, S.; Johnson, C.H. Circadian Clock Gene Bmal1 Is Not Essential; Functional Replacement with its Paralog, Bmal2. Curr. Biol. 2010, 20, 316–321. [Google Scholar] [CrossRef] [Green Version]
- Etchegaray, J.-P.; Lee, C.; Wade, P.A.; Reppert, S.M. Rhythmic histone acetylation underlies transcription in the mammalian circadian clock. Nat. Cell Biol. 2002, 421, 177–182. [Google Scholar] [CrossRef]
- Zhong, X.; Yu, J.; Frazier, K.; Weng, X.; Li, Y.; Cham, C.M.; Dolan, K.; Zhu, X.; Hubert, N.; Tao, Y.; et al. Circadian Clock Regulation of Hepatic Lipid Metabolism by Modulation of m6A mRNA Methylation. Cell Rep. 2018, 25, 1816–1828.e4. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Downes, M.; Yu, R.T.; Bookout, A.L.; He, W.; Straume, M.; Mangelsdorf, D.J.; Evans, R.M. Nuclear Receptor Expression Links the Circadian Clock to Metabolism. Cell 2006, 126, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Venkatachalam, A.B.; Sawler, D.L.; Wright, J.M. Tissue-specific transcriptional modulation of fatty acid-binding protein genes, fabp2, fabp3 and fabp6, by fatty acids and the peroxisome proliferator, clofibrate, in zebrafish (Danio rerio). Gene 2013, 520, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Yang, L.; Wang, Z.; Huang, W. Bile acid nuclear receptor FXR and digestive system diseases. Acta Pharm. Sin. B 2015, 5, 135–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohkuro, M.; Kim, J.-D.; Kuboi, Y.; Hayashi, Y.; Mizukami, H.; Kobayashi-Kuramochi, H.; Muramoto, K.; Shirato, M.; Michikawa-Tanaka, F.; Moriya, J.; et al. Calreticulin and integrin alpha dissociation induces anti-inflammatory programming in animal models of inflammatory bowel disease. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Krysko, D.V.; Ravichandran, K.S.; Vandenabeele, P. Macrophages regulate the clearance of living cells by calreticulin. Nat. Commun. 2018, 9, 4644. [Google Scholar] [CrossRef] [Green Version]
- Pennington, K.L.; Chan, T.Y.; Torres, M.P.; Andersen, J.L. The dynamic and stress-adaptive signaling hub of 14-3-3: Emerging mechanisms of regulation and context-dependent protein–protein interactions. Oncogene 2018, 37, 5587–5604. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.; Crowther, S.; Stafford, M.J.; Campbell, D.G.; Toth, R.; Mackintosh, C. Bioinformatic and experimental survey of 14-3-3-binding sites. Biochem. J. 2010, 427, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Teo, J.-L.; Ma, H.; Nguyen, C.; Lam, C.; Kahn, M. Specific inhibition of CBP/ -catenin interaction rescues defects in neuronal differentiation caused by a presenilin-1 mutation. Proc. Natl. Acad. Sci. USA 2005, 102, 12171–12176. [Google Scholar] [CrossRef] [Green Version]
- Kahn, M. Symmetric division versus asymmetric division: A tale of two coactivators. Futur. Med. Chem. 2011, 3, 1745–1763. [Google Scholar] [CrossRef]
- Zhang, J.J.; Vinkemeier, U.; Gu, W.; Chakravarti, D.; Horvath, C.M.; Darnell, J.E. Two contact regions between Stat1 and CBP/p300 in interferon signaling. Proc. Natl. Acad. Sci. USA 1996, 93, 15092–15096. [Google Scholar] [CrossRef] [Green Version]
- Horvai, A.E.; Xu, L.; Korzus, E.; Brard, G.; Kalafus, D.; Mullen, T.-M.; Rose, D.W.; Rosenfeld, M.G.; Glass, C.K. Nuclear integration of JAK/STAT and Ras/AP-1 signaling by CBP and p300. Proc. Natl. Acad. Sci. USA 1997, 94, 1074–1079. [Google Scholar] [CrossRef] [Green Version]
- Kurokawa, R.; Kalafus, D.; Ogliastro, M.-H.; Kioussi, C.; Xu, L.; Torchia, J.; Rosenfeld, M.G.; Glass, C.K. Differential Use of CREB Binding Protein-Coactivator Complexes. Science 1998, 279, 700–703. [Google Scholar] [CrossRef]
- Metwaly, A.; Dunkel, A.; Waldschmitt, N.; Raj, A.C.D.; Lagkouvardos, I.; Corraliza, A.M.; Mayorgas, A.; Martinez-Medina, M.; Reiter, S.; Schloter, M.; et al. Integrated microbiota and metabolite profiles link Crohn’s disease to sulfur metabolism. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.; Turnbaugh, P.J. Deconstructing Mechanisms of Diet-Microbiome-Immune Interactions. Immunology 2020, 53, 264–276. [Google Scholar] [CrossRef]
- Hernandez, J.B.; Chang, C.; Leblanc, M.; Grimm, D.; Le Lay, J.; Kaestner, K.H.; Zheng, Y.; Montminy, M. The CREB/CRTC2 pathway modulates autoimmune disease by promoting Th17 differentiation. Nat. Commun. 2015, 6, 7216. [Google Scholar] [CrossRef] [Green Version]
- Mazmanian, S.K.; Liu, C.H.; Tzianabos, A.O.; Kasper, D.L. An Immunomodulatory Molecule of Symbiotic Bacteria Directs Maturation of the Host Immune System. Cell 2005, 122, 107–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markovich, D. Physiological Roles and Regulation of Mammalian Sulfate Transporters. Physiol. Rev. 2001, 81, 1499–1533. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.R.T.; Cox, I.J.; Walker, D.G.; North, B.V.; Patel, V.M.; Marshall, S.E.; Jewell, D.P.; Ghosh, S.; Thomas, H.J.W.; Teare, J.P.; et al. Characterization of Inflammatory Bowel Disease With Urinary Metabolic Profiling. Am. J. Gastroenterol. 2009, 104, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the Regulation of Innate and Adaptive Immunity. Immunology 2015, 42, 406–417. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Pearce, E.L. Mitochondrial Dynamics at the Interface of Immune Cell Metabolism and Function. Trends Immunol. 2018, 39, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Litvak, Y.; Byndloss, M.X.; Bäumler, A.J. Colonocyte metabolism shapes the gut microbiota. Science 2018, 362, eaat9076. [Google Scholar] [CrossRef] [Green Version]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chávez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Furuta, G.T.; Turner, J.R.; Taylor, C.T.; Hershberg, R.M.; Comerford, K.; Narravula, S.; Podolsky, D.K.; Colgan, S.P. Hypoxia-Inducible Factor 1–Dependent Induction of Intestinal Trefoil Factor Protects Barrier Function during Hypoxia. J. Exp. Med. 2001, 193, 1027–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-Y.; Cevallos, S.A.; Byndloss, M.X.; Tiffany, C.R.; Olsan, E.E.; Butler, B.P.; Young, B.M.; Rogers, A.W.; Nguyen, H.; Kim, K.; et al. High-Fat Diet and Antibiotics Cooperatively Impair Mitochondrial Bioenergetics to Trigger Dysbiosis that Exacerbates Pre-inflammatory Bowel Disease. Cell Host Microbe 2020, 28, 273–284.e6. [Google Scholar] [CrossRef] [PubMed]
- Rigottier-Gois, L. Dysbiosis in inflammatory bowel diseases: The oxygen hypothesis. ISME J. 2013, 7, 1256–1261. [Google Scholar] [CrossRef]
- Rousseaux, C.; El-Jamal, N.; Fumery, M.; Dubuquoy, C.; Romano, O.; Chatelain, D.; Langlois, A.; Bertin, B.; Buob, D.; Colombel, J.F.; et al. The 5-aminosalicylic acid antineoplastic effect in the intestine is mediated by PPARγ. Carcinogen 2013, 34, 2580–2586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, K.K.Y.; Hu, X.; Chosa, K.; Nguyen, C.; Lin, D.P.; Lai, K.K.; Kato, N.; Higuchi, Y.; Highlander, S.K.; Melendez, E.; et al. p300 Serine 89: A Critical Signaling Integrator and Its Effects on Intestinal Homeostasis and Repair. Cancers 2021, 13, 1288. https://doi.org/10.3390/cancers13061288
Lai KKY, Hu X, Chosa K, Nguyen C, Lin DP, Lai KK, Kato N, Higuchi Y, Highlander SK, Melendez E, et al. p300 Serine 89: A Critical Signaling Integrator and Its Effects on Intestinal Homeostasis and Repair. Cancers. 2021; 13(6):1288. https://doi.org/10.3390/cancers13061288
Chicago/Turabian StyleLai, Keane K. Y., Xiaohui Hu, Keisuke Chosa, Cu Nguyen, David P. Lin, Keith K. Lai, Nobuo Kato, Yusuke Higuchi, Sarah K. Highlander, Elizabeth Melendez, and et al. 2021. "p300 Serine 89: A Critical Signaling Integrator and Its Effects on Intestinal Homeostasis and Repair" Cancers 13, no. 6: 1288. https://doi.org/10.3390/cancers13061288
APA StyleLai, K. K. Y., Hu, X., Chosa, K., Nguyen, C., Lin, D. P., Lai, K. K., Kato, N., Higuchi, Y., Highlander, S. K., Melendez, E., Eriguchi, Y., Fueger, P. T., Ouellette, A. J., Chimge, N.-O., Ono, M., & Kahn, M. (2021). p300 Serine 89: A Critical Signaling Integrator and Its Effects on Intestinal Homeostasis and Repair. Cancers, 13(6), 1288. https://doi.org/10.3390/cancers13061288