
Molecular Profiling of Docetaxel-Resistant Prostate Cancer Cells Identifies Multiple Mechanisms of Therapeutic Resistance

 , ,
, ,  ,
,  , and
, and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Resistant Cell Lines Development
2.2. Gene Expression Analysis and Microarray Profiling
2.3. Cytotoxicity Assay
2.4. Real-Time Cell Proliferation Monitoring
2.5. Cell Cycle Analysis by Flow Cytometry
2.6. Protein Extraction, Western Blotting, and Nanocapillary Electrophoresis
2.7. Immunostaining
2.8. Cytogenetic Analysis
2.9. Fluorescence In Situ Hybridization
3. Results
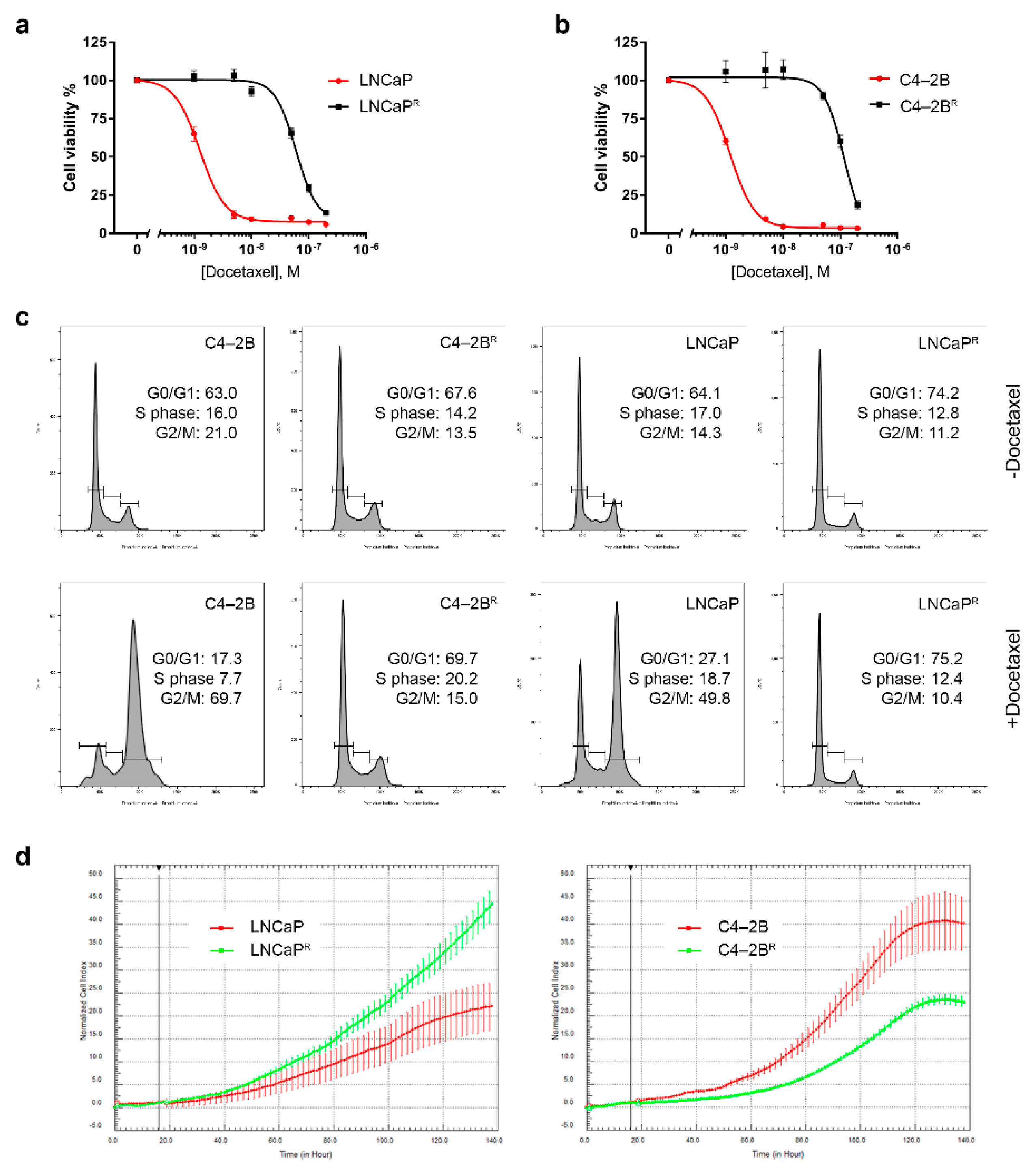
3.1. Establishment of Docetaxel-Resistant PCa Cell Lines
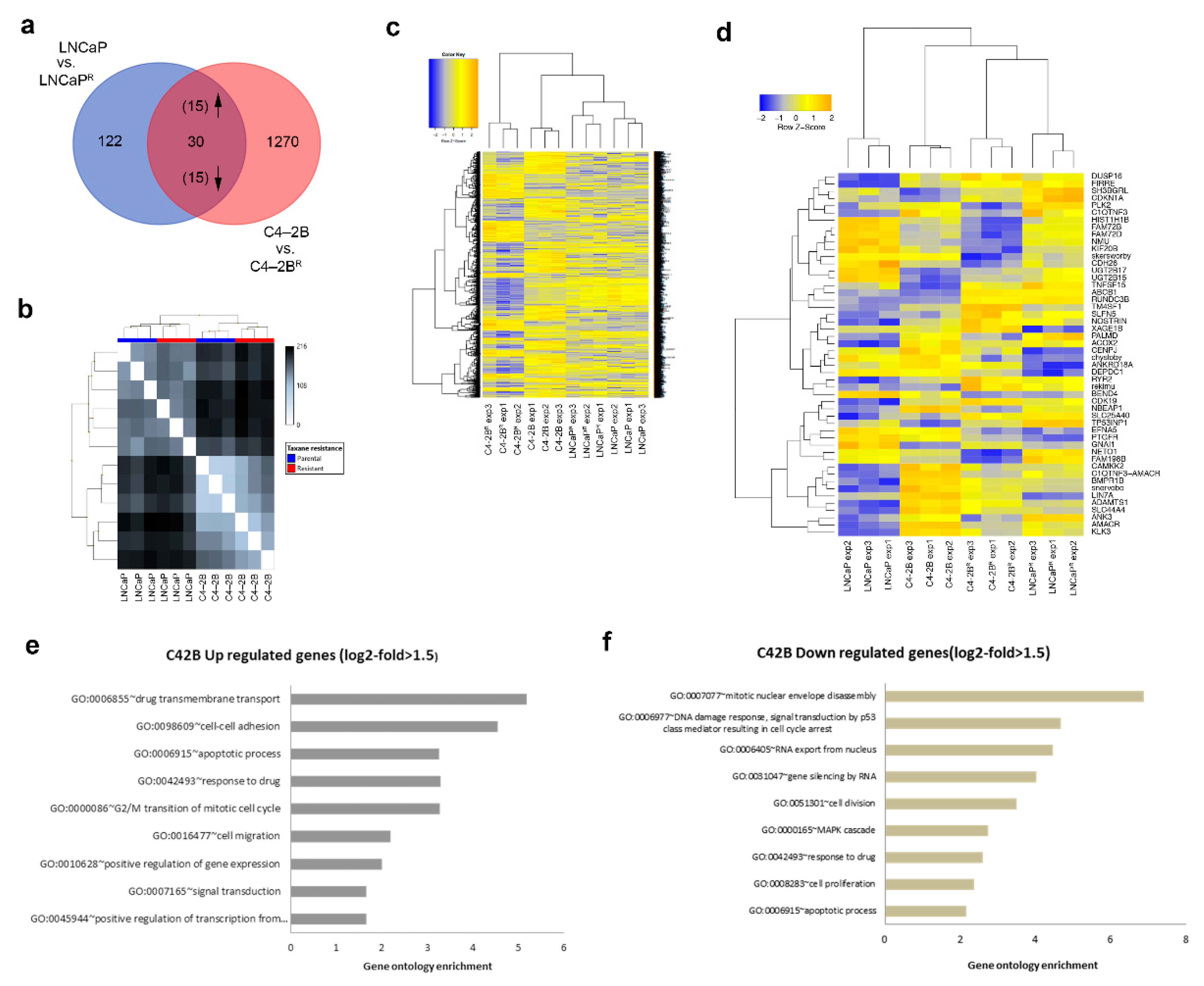
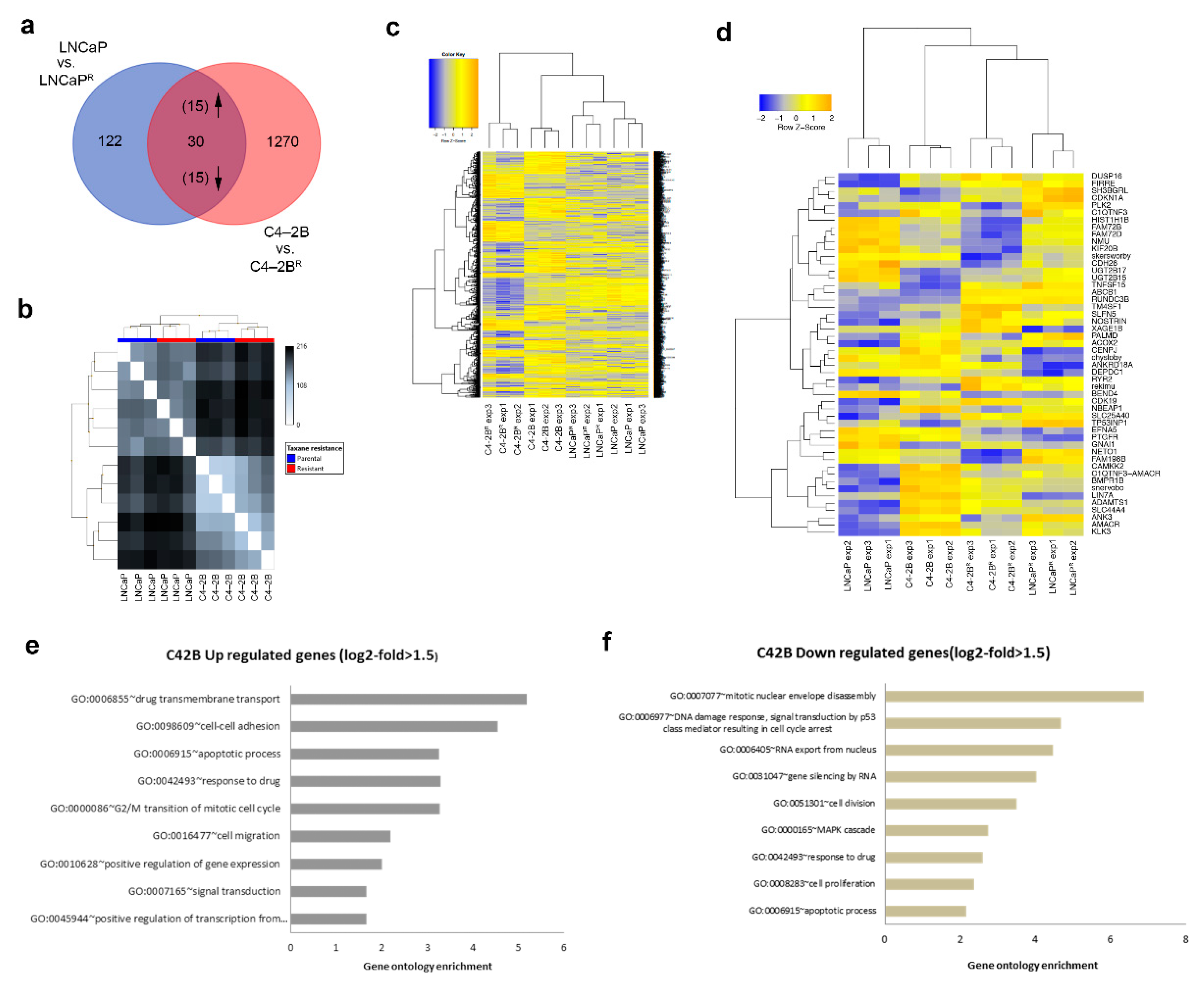
3.2. Identification of Differentially Expressed Genes in Docetaxel-Resistant Cell Lines
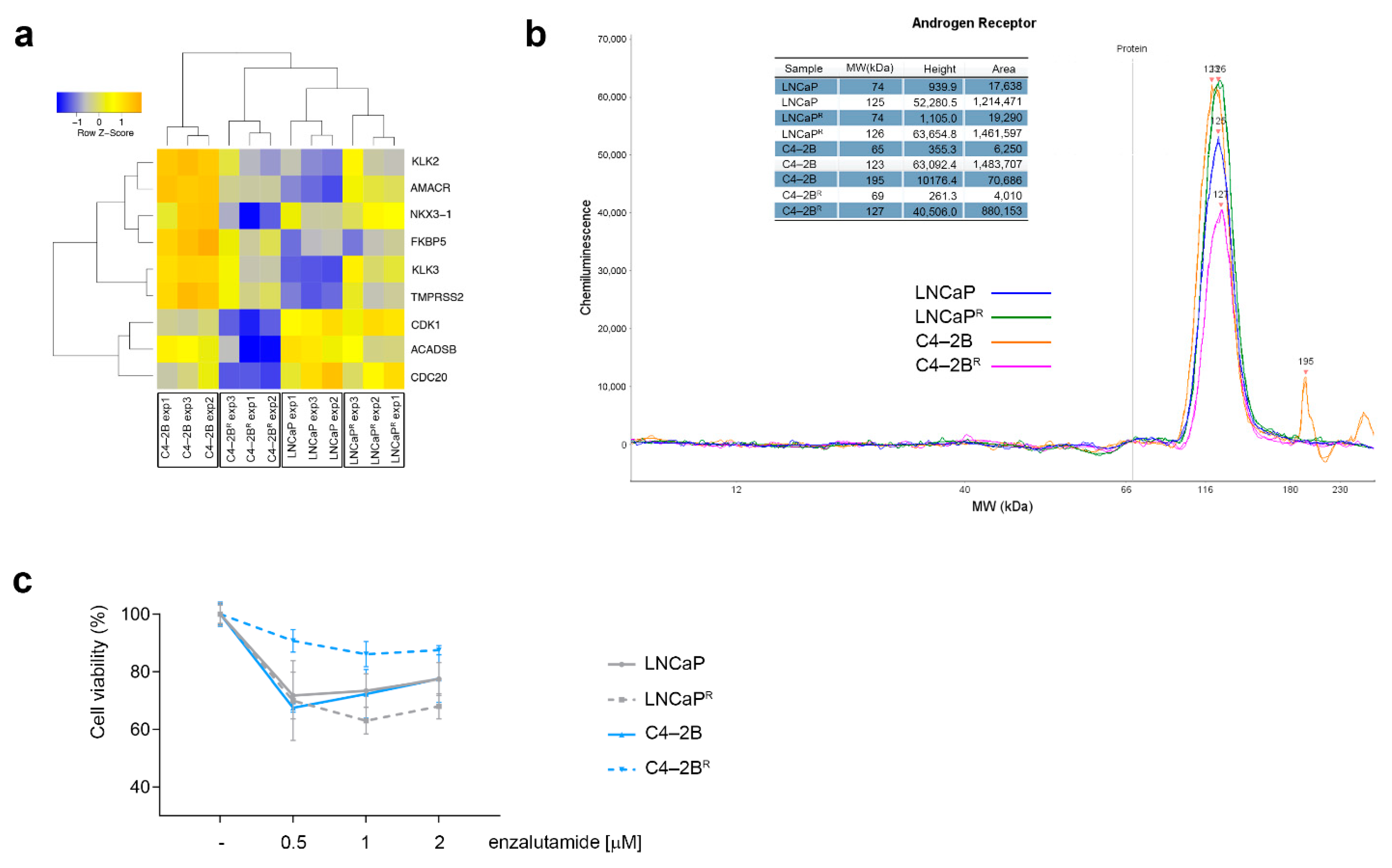
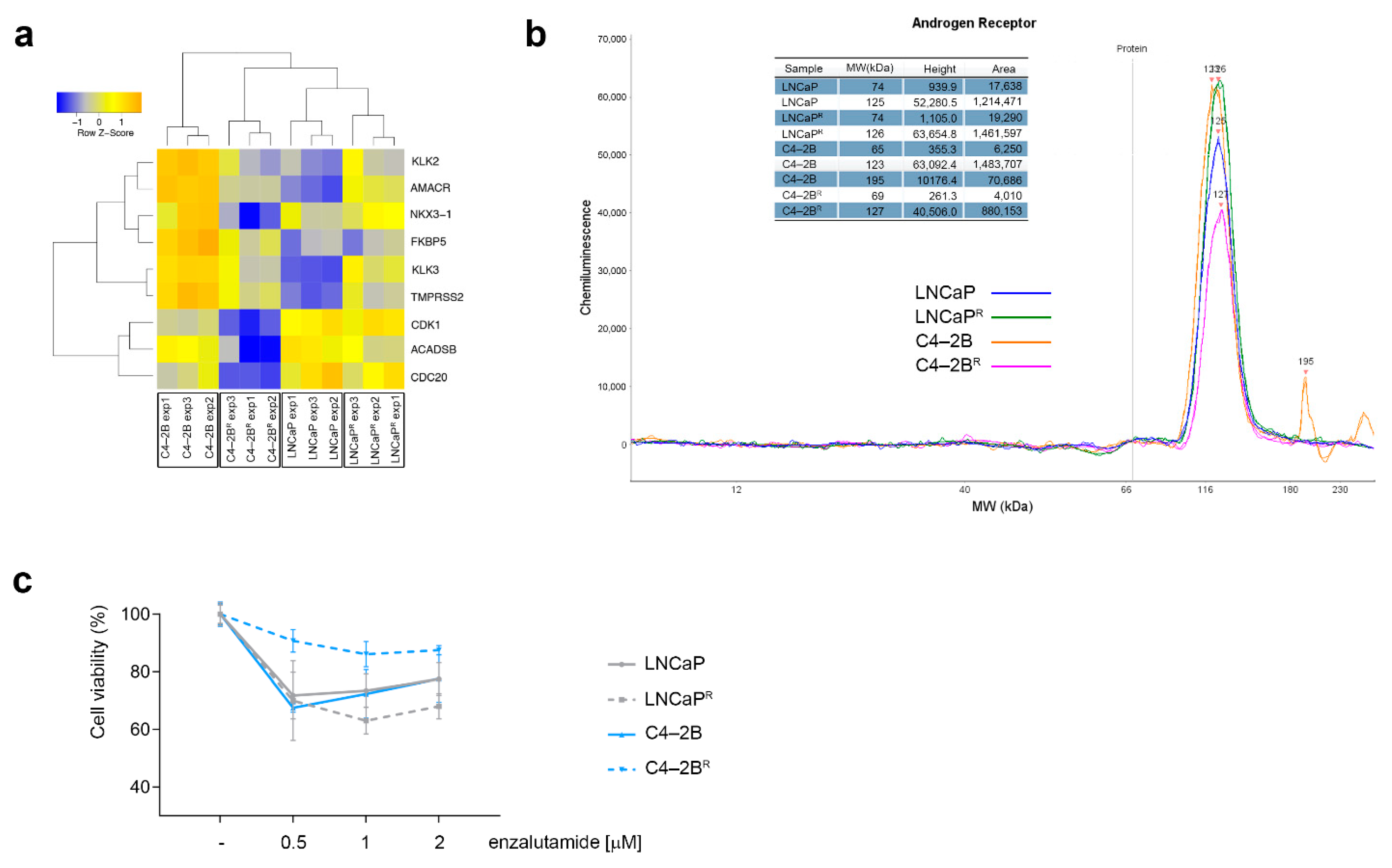
3.3. Differential Androgen Receptor Signaling Behavior in Docetaxel-Resistant C4-2BR and LNCaPR Cell Lines
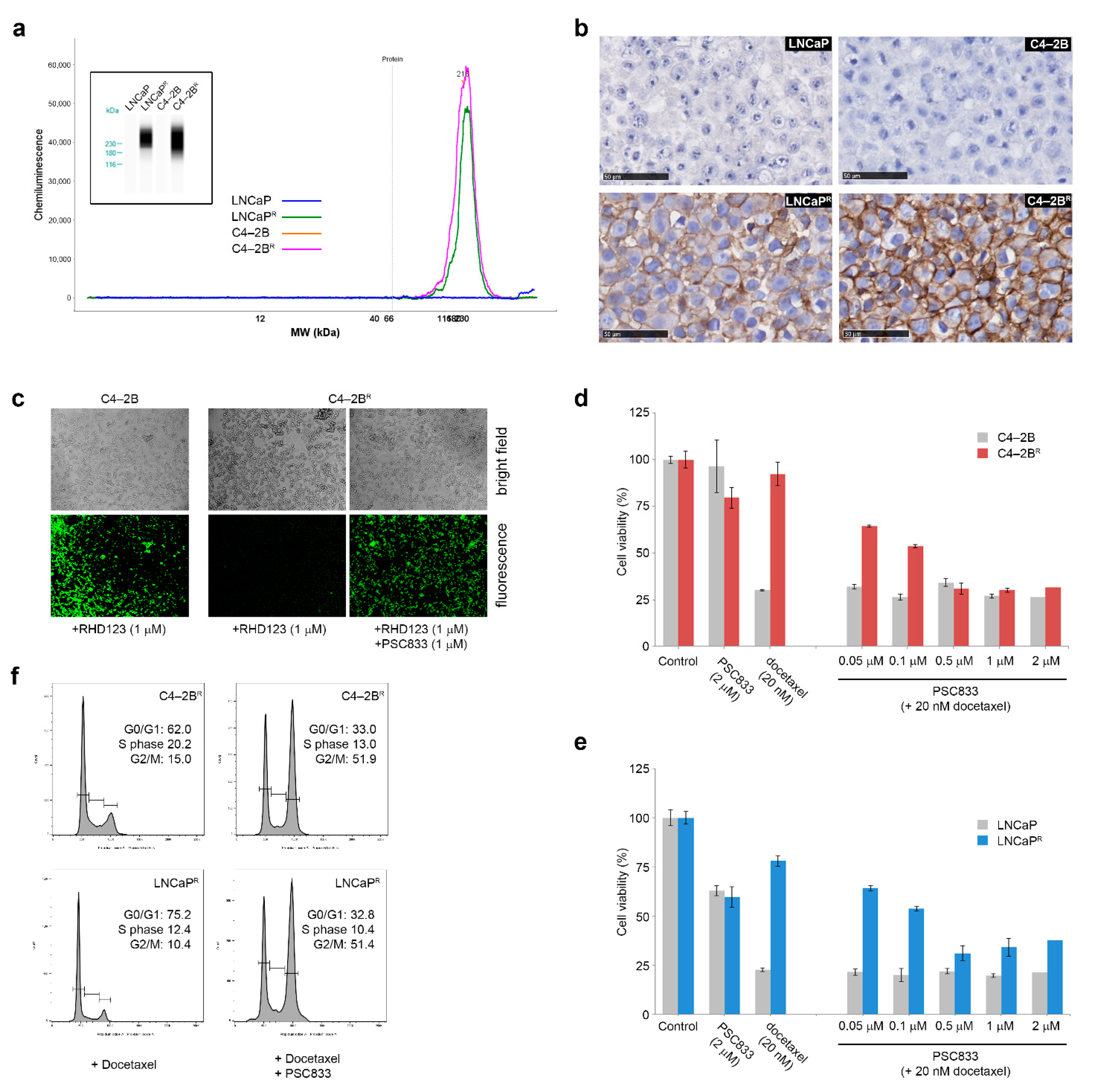
3.4. Increased ABCB1 Expression Is a Common Feature of the Docetaxel-Resistant Sub-Lines
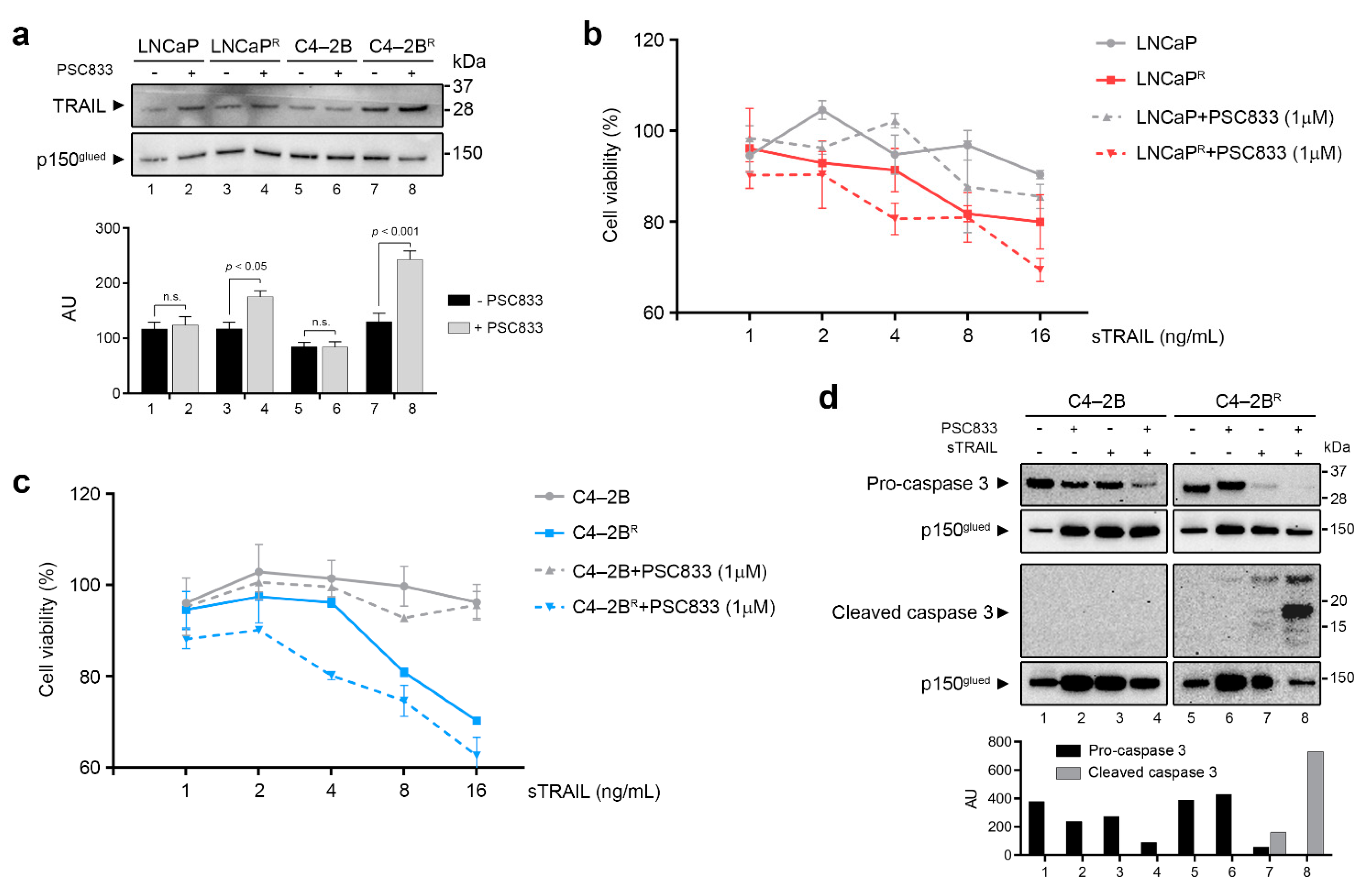
3.5. Modulation of Cell Death Mechanisms May Contribute to Multifactorial Docetaxel Resistance
4. Discussion
4.1. Modulation of AR Signaling
4.2. Up-Regulation of the ABCB1 Drug Efflux Pump
4.3. Modulation of Anti-Apoptotic Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pernar, C.H.; Ebot, E.M.; Wilson, K.M.; Mucci, L.A. The Epidemiology of Prostate Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a030361. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Attard, G.; Reid, A.H.; Yap, T.A.; Raynaud, F.; Dowsett, M.; Settatree, S.; Barrett, M.; Parker, C.; Martins, V.; Folkerd, E.; et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J. Clin. Oncol. 2008, 26, 4563–4571. [Google Scholar] [CrossRef]
- Scher, H.I.; Beer, T.M.; Higano, C.S.; Anand, A.; Taplin, M.E.; Efstathiou, E.; Rathkopf, D.; Shelkey, J.; Yu, E.Y.; Alumkal, J.; et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: A phase 1-2 study. Lancet 2010, 375, 1437–1446. [Google Scholar] [CrossRef] [Green Version]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Theodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [Green Version]
- Kyriakopoulos, C.E.; Chen, Y.H.; Carducci, M.A.; Liu, G.; Jarrard, D.F.; Hahn, N.M.; Shevrin, D.H.; Dreicer, R.; Hussain, M.; Eisenberger, M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer: Long-Term Survival Analysis of the Randomized Phase III E3805 CHAARTED Trial. J. Clin. Oncol. 2018, 36, 1080–1087. [Google Scholar] [CrossRef] [Green Version]
- Gravis, G.; Fizazi, K.; Joly, F.; Oudard, S.; Priou, F.; Esterni, B.; Latorzeff, I.; Delva, R.; Krakowski, I.; Laguerre, B.; et al. Androgen-deprivation therapy alone or with docetaxel in non-castrate metastatic prostate cancer (GETUG-AFU 15): A randomised, open-label, phase 3 trial. Lancet Oncol. 2013, 14, 149–158. [Google Scholar] [CrossRef]
- Parker, C.; Gillessen, S.; Heidenreich, A.; Horwich, A.; Committee, E.G. Cancer of the prostate: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26, v69–v77. [Google Scholar] [CrossRef]
- Petrylak, D.P.; Tangen, C.M.; Hussain, M.H.; Lara, P.N., Jr.; Jones, J.A.; Taplin, M.E.; Burch, P.A.; Berry, D.; Moinpour, C.; Kohli, M.; et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N. Engl. J. Med. 2004, 351, 1513–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, C.; Mendoza, P.; Contreras, H.R.; Vergara, J.; McCubrey, J.A.; Huidobro, C.; Castellon, E.A. Expression of multidrug resistance proteins in prostate cancer is related with cell sensitivity to chemotherapeutic drugs. Prostate 2009, 69, 1448–1459. [Google Scholar] [CrossRef]
- Shman, T.V.; Fedasenka, U.U.; Savitski, V.P.; Aleinikova, O.V. CD34+ leukemic subpopulation predominantly displays lower spontaneous apoptosis and has higher expression levels of Bcl-2 and MDR1 genes than CD34- cells in childhood AML. Ann. Hematol. 2008, 87, 353–360. [Google Scholar] [CrossRef]
- Wang, Q.; He, W.Y.; Zeng, Y.Z.; Hossain, A.; Gou, X. Inhibiting autophagy overcomes docetaxel resistance in castration-resistant prostate cancer cells. Int. Urol. Nephrol. 2018, 50, 675–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, T.; Ushio, K.; Nishiwaki, M.; Kouno, J.; Araki, H.; Hikichi, Y.; Hattori, M.; Imai, Y.; Yamaoka, M. A mutation in beta-tubulin and a sustained dependence on androgen receptor signalling in a newly established docetaxel-resistant prostate cancer cell line. Cell Biol. Int. 2010, 34, 177–184. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Nakazawa, M.; Nadal, R.; Paller, C.J.; Denmeade, S.R.; Carducci, M.A.; et al. Androgen Receptor Splice Variant 7 and Efficacy of Taxane Chemotherapy in Patients With Metastatic Castration-Resistant Prostate Cancer. J. Urol. 2015, 1, 582–591. [Google Scholar] [CrossRef] [Green Version]
- Komura, K.; Jeong, S.H.; Hinohara, K.; Qu, F.; Wang, X.; Hiraki, M.; Azuma, H.; Lee, G.S.; Kantoff, P.W.; Sweeney, C.J. Resistance to docetaxel in prostate cancer is associated with androgen receptor activation and loss of KDM5D expression. Proc. Natl. Acad. Sci. USA 2016, 113, 6259–6264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horoszewicz, J.S.; Leong, S.S.; Chu, T.M.; Wajsman, Z.L.; Friedman, M.; Papsidero, L.; Kim, U.; Chai, L.S.; Kakati, S.; Arya, S.K.; et al. The LNCaP cell line--a new model for studies on human prostatic carcinoma. Prog. Clin. Biol. Res. 1980, 37, 115–132. [Google Scholar]
- Thalmann, G.N.; Anezinis, P.E.; Chang, S.M.; Zhau, H.E.; Kim, E.E.; Hopwood, V.L.; Pathak, S.; von Eschenbach, A.C.; Chung, L.W. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 1994, 54, 2577–2581. [Google Scholar]
- Wu, H.C.; Hsieh, J.T.; Gleave, M.E.; Brown, N.M.; Pathak, S.; Chung, L.W. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: Role of bone stromal cells. Int. J. Cancer 1994, 57, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Thalmann, G.N.; Sikes, R.A.; Wu, T.T.; Degeorges, A.; Chang, S.M.; Ozen, M.; Pathak, S.; Chung, L.W. LNCaP progression model of human prostate cancer: Androgen-independence and osseous metastasis. Prostate 2000, 44, 91–103. [Google Scholar] [CrossRef]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [Green Version]
- Chauffaille Mde, L.; Coutinho, V.; Yamamoto, M.; Kerbauy, J. Combined method for simultaneous morphology, immunophenotype and karyotype (MAC) in leukemias. Sao Paulo Med. J. 1997, 115, 1336–1342. [Google Scholar] [CrossRef] [Green Version]
- Reed, K.; Hembruff, S.L.; Laberge, M.L.; Villeneuve, D.J.; Cote, G.B.; Parissenti, A.M. Hypermethylation of the ABCB1 downstream gene promoter accompanies ABCB1 gene amplification and increased expression in docetaxel-resistant MCF-7 breast tumor cells. Epigenetics 2008, 3, 270–280. [Google Scholar] [CrossRef] [Green Version]
- Krongrad, A.; Wilson, C.M.; Wilson, J.D.; Allman, D.R.; McPhaul, M.J. Androgen increases androgen receptor protein while decreasing receptor mRNA in LNCaP cells. Mol. Cell. Endocrinol. 1991, 76, 79–88. [Google Scholar] [CrossRef]
- Manin, M.; Baron, S.; Goossens, K.; Beaudoin, C.; Jean, C.; Veyssiere, G.; Verhoeven, G.; Morel, L. Androgen receptor expression is regulated by the phosphoinositide 3-kinase/Akt pathway in normal and tumoral epithelial cells. Biochem. J. 2002, 366, 729–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.Q.; Heldman, M.R.; Herrmann, M.A.; Kedei, N.; Woo, W.; Blumberg, P.M.; Goldsmith, P.K. Absolute quantitation of endogenous proteins with precision and accuracy using a capillary Western system. Anal. Biochem. 2013, 442, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juranka, P.F.; Zastawny, R.L.; Ling, V. P-glycoprotein: Multidrug-resistance and a superfamily of membrane-associated transport proteins. FASEB J. 1989, 3, 2583–2592. [Google Scholar] [CrossRef]
- Gheuens, E.E.; van Bockstaele, D.R.; van der Keur, M.; Tanke, H.J.; van Oosterom, A.T.; De Bruijn, E.A. Flow cytometric double labeling technique for screening of multidrug resistance. J. Int. Soc. Anal. Cytol. 1991, 12, 636–644. [Google Scholar] [CrossRef]
- Smith, A.J.; Mayer, U.; Schinkel, A.H.; Borst, P. Availability of PSC833, a substrate and inhibitor of P-glycoproteins, in various concentrations of serum. J. Natl. Cancer Inst. 1998, 90, 1161–1166. [Google Scholar] [CrossRef]
- Callen, D.F.; Baker, E.; Simmers, R.N.; Seshadri, R.; Roninson, I.B. Localization of the human multiple drug resistance gene, MDR1, to 7q21.1. Hum. Genet. 1987, 77, 142–144. [Google Scholar] [CrossRef]
- Slovak, M.L.; Hoeltge, G.A.; Trent, J.M. Cytogenetic alterations associated with the acquisition of doxorubicin resistance: Possible significance of chromosome 7 alterations. Cancer Res. 1987, 47, 6646–6652. [Google Scholar]
- Nieuwint, A.W.; Baas, F.; Wiegant, J.; Joenje, H. Cytogenetic alterations associated with P-glycoprotein- and non-P-glycoprotein-mediated multidrug resistance in SW-1573 human lung tumor cell lines. Cancer Res. 1992, 52, 4361–4371. [Google Scholar]
- Kim, I.W.; Han, N.; Kim, M.G.; Kim, T.; Oh, J.M. Copy number variability analysis of pharmacogenes in patients with lymphoma, leukemia, hepatocellular, and lung carcinoma using The Cancer Genome Atlas data. Pharm. Genom. 2015, 25, 1–7. [Google Scholar] [CrossRef]
- Chekhun, V.F.; Lukyanova, N.Y.; Kovalchuk, O.; Tryndyak, V.P.; Pogribny, I.P. Epigenetic profiling of multidrug-resistant human MCF-7 breast adenocarcinoma cells reveals novel hyper- and hypomethylated targets. Mol. Cancer Ther. 2007, 6, 1089–1098. [Google Scholar] [CrossRef] [Green Version]
- Kearney, L. Molecular cytogenetics. Best Pract. Res. Clin. Haematol. 2001, 14, 645–669. [Google Scholar] [CrossRef] [PubMed]
- Gillet, J.P.; Gottesman, M.M. Mechanisms of multidrug resistance in cancer. Methods Mol. Biol. 2010, 596, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Abe, S.; Kurata, M.; Hasegawa, M.; Yamamoto, K.; Inoue, M.; Takemura, T.; Suzuki, K.; Kitagawa, M. IAP family protein expression correlates with poor outcome of multiple myeloma patients in association with chemotherapy-induced overexpression of multidrug resistance genes. Am. J. Hematol. 2006, 81, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Amort, M.; Nachbauer, B.; Tuzlak, S.; Kieser, A.; Schepers, A.; Villunger, A.; Polacek, N. Expression of the vault RNA protects cells from undergoing apoptosis. Nat. Commun. 2015, 6, 7030. [Google Scholar] [CrossRef] [Green Version]
- Horos, R.; Buscher, M.; Kleinendorst, R.; Alleaume, A.M.; Tarafder, A.K.; Schwarzl, T.; Dziuba, D.; Tischer, C.; Zielonka, E.M.; Adak, A.; et al. The Small Non-coding Vault RNA1-1 Acts as a Riboregulator of Autophagy. Cell 2019, 176, 1054–1067.e12. [Google Scholar] [CrossRef] [Green Version]
- Souza, P.S.; Madigan, J.P.; Gillet, J.P.; Kapoor, K.; Ambudkar, S.V.; Maia, R.C.; Gottesman, M.M.; Fung, K.L. Expression of the multidrug transporter P-glycoprotein is inversely related to that of apoptosis-associated endogenous TRAIL. Exp. Cell Res. 2015, 336, 318–328. [Google Scholar] [CrossRef] [Green Version]
- Galski, H.; Oved-Gelber, T.; Simanovsky, M.; Lazarovici, P.; Gottesman, M.M.; Nagler, A. P-glycoprotein-dependent resistance of cancer cells toward the extrinsic TRAIL apoptosis signaling pathway. Biochem. Pharmacol. 2013, 86, 584–596. [Google Scholar] [CrossRef] [Green Version]
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.; Parker, C.C.; Russell, J.M.; Attard, G.; et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016, 387, 1163–1177. [Google Scholar] [CrossRef] [Green Version]
- Kelly, W.K.; Halabi, S.; Carducci, M.; George, D.; Mahoney, J.F.; Stadler, W.M.; Morris, M.; Kantoff, P.; Monk, J.P.; Kaplan, E.; et al. Randomized, double-blind, placebo-controlled phase III trial comparing docetaxel and prednisone with or without bevacizumab in men with metastatic castration-resistant prostate cancer: CALGB 90401. J. Clin. Oncol. 2012, 30, 1534–1540. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.J.; Dutta, S.; Kelly, W.K.; Russell, C.; Small, E.J.; Morris, M.J.; Taplin, M.E.; Halabi, S. From The Alliance for Clinical Trials in Oncology Genitourinary, C. Androgen decline and survival during docetaxel therapy in metastatic castration resistant prostate cancer (mCRPC). Prostate Cancer Prostatic Dis. 2020, 23, 66–73. [Google Scholar] [CrossRef]
- Burmeister, D.W.; Smith, E.H.; Cristel, R.T.; McKay, S.D.; Shi, H.; Arthur, G.L.; Davis, J.W.; Taylor, K.H. The expression of RUNDC3B is associated with promoter methylation in lymphoid malignancies. Hematol. Oncol. 2017, 35, 25–33. [Google Scholar] [CrossRef]
- Balaguer, T.M.; Gomez-Martinez, A.; Garcia-Morales, P.; Lacueva, J.; Calpena, R.; Reverte, L.R.; Riquelme, N.L.; Martinez-Lacaci, I.; Ferragut, J.A.; Saceda, M. Dual regulation of P-glycoprotein expression by trichostatin A in cancer cell lines. BMC Mol. Biol. 2012, 13, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raguz, S.; De Bella, M.T.; Slade, M.J.; Higgins, C.F.; Coombes, R.C.; Yague, E. Expression of RPIP9 (Rap2 interacting protein 9) is activated in breast carcinoma and correlates with a poor prognosis. Int. J. Cancer 2005, 117, 934–941. [Google Scholar] [CrossRef]
- Scherer, S.W.; Cheung, J.; MacDonald, J.R.; Osborne, L.R.; Nakabayashi, K.; Herbrick, J.A.; Carson, A.R.; Parker-Katiraee, L.; Skaug, J.; Khaja, R.; et al. Human chromosome 7: DNA sequence and biology. Science 2003, 300, 767–772. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; OuYang, H.; An, X.; Liu, S. Vault RNAs partially induces drug resistance of human tumor cells MCF-7 by binding to the RNA/DNA-binding protein PSF and inducing oncogene GAGE6. PLoS ONE 2018, 13, e0191325. [Google Scholar] [CrossRef] [Green Version]
- Kickhoefer, V.A.; Rajavel, K.S.; Scheffer, G.L.; Dalton, W.S.; Scheper, R.J.; Rome, L.H. Vaults are up-regulated in multidrug-resistant cancer cell lines. J. Biol. Chem. 1998, 273, 8971–8974. [Google Scholar] [CrossRef] [Green Version]
- Steiner, E.; Holzmann, K.; Elbling, L.; Micksche, M.; Berger, W. Cellular functions of vaults and their involvement in multidrug resistance. Curr. Drug Targets 2006, 7, 923–934. [Google Scholar] [CrossRef]
- Kitazono, M.; Sumizawa, T.; Takebayashi, Y.; Chen, Z.S.; Furukawa, T.; Nagayama, S.; Tani, A.; Takao, S.; Aikou, T.; Akiyama, S. Multidrug resistance and the lung resistance-related protein in human colon carcinoma SW-620 cells. J. Natl. Cancer Inst. 1999, 91, 1647–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, S.J.; An, H.J.; Oh, Y.S.; Choi, H.R.; Ha, M.K.; Park, S.C. On the role of major vault protein in the resistance of senescent human diploid fibroblasts to apoptosis. Cell Death Differ. 2008, 15, 1673–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misawa, A.; Takayama, K.; Urano, T.; Inoue, S. Androgen-induced Long Noncoding RNA (lncRNA) SOCS2-AS1 Promotes Cell Growth and Inhibits Apoptosis in Prostate Cancer Cells. J. Biol. Chem. 2016, 291, 17861–17880. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.; Wang, J.; Xu, H.; Yang, X. Resistance to docetaxel-induced apoptosis in prostate cancer cells by p38/p53/p21 signaling. Prostate 2011, 71, 1158–1166. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Log2 Ratio | mRNA Accession | Probe Set ID | |
|---|---|---|---|---|
| LNCaPR vs. LNCaP | C4-2BR vs. C4-2B | |||
| NOSTRIN | 4.1 | 2.71 | NM_001171631 | TC0900010056.hg.1 |
| REKIMU | 4.17 | 11.1 | rekimu.aAug10-unspliced | TC0400010553.hg.1 |
| TNFSF15 | 4.31 | 8.42 | NM_001204344 | TC0600007847.hg.1 |
| FIRRE | 17.9 | 2.36 | NR_026975 | TC1000010727.hg.1 |
| ABCB1 | 10.3 | 20.1 | NM_000927 | TC0700011706.hg.1 |
| SH3BGRL | 7.9 | 1.8 | NM_003022 | TC0500013296.hg.1 |
| RUNDC3B | 174.8 | 107.4 | NM_001134405 | TC0400008183.hg.1 |
| SLC25A40 | 3.97 | 2.28 | NM_018843 | TC1200012199.hg.1 |
| TP53INP1 | 3.92 | 2.54 | NM_001135733 | TC1300008371.hg.1 |
| RYR2 | 3.29 | 2.54 | NM_001035 | TC0500013298.hg.1 |
| TM4SF1 | 3.5 | 41 | NM_014220 | TC0600012839.hg.1 |
| CDK19 | 3.7 | 2.62 | NM_001300960 | TC0300011309.hg.1 |
| CDKN1A | 9.65 | 3.25 | NM_000389 | TC2100007821.hg.1 |
| SLFN5 | 3.8 | 5.59 | NM_144975 | TC2000007943.hg.1 |
| DUSP16 | 2.95 | 2.17 | NM_030640 | TC0500013297.hg.1 |
| Gene Symbol | Log2 Ratio | mRNA Accession | Probe Set ID | |
|---|---|---|---|---|
| LNCaPR vs. LNCaP | C42BR vs. C42B | |||
| FAM72B | 0.23 | 0.32 | NM_001100910 | TC0X00010826.hg.1 |
| PTGFR | 0.04 | 0.21 | NM_000959 | TC1500008627.hg.1 |
| CENPJ | 0.38 | 0.45 | NM_018451 | TC0500011648.hg.1 |
| KIF20B | 0.31 | 0.42 | NM_001284259 | TC1000008406.hg.1 |
| LIN7A | 0.24 | 0.09 | NM_004664 | TC1900008607.hg.1 |
| CDH26 | 0.17 | 0.44 | NM_021810 | TSUnmapped00000374.hg.1 |
| BEND4 | 0.14 | 0.31 | NM_001159547 | TC0100014543.hg.1 |
| FAM72D | 0.31 | 0.31 | NM_207418 | TC0700008181.hg.1 |
| DEPDC1 | 0.04 | 0.24 | NM_001114120 | TC0100015509.hg.1 |
| HIST1H1B | 0.21 | 0.24 | NM_005322 | TC0600011232.hg.1 |
| EFNA5 | 0.1 | 0.18 | NM_001962 | TC0100009723.hg.1 |
| CHYSLOBY | 0.17 | 0.22 | chysloby.aAug10-unspliced | TC0400012245.hg.1 |
| SKERSWORBY | 0.24 | 0.06 | skersworby.aAug10-unspliced | TC1800009011.hg.1 |
| NMU | 0.21 | 0.52 | NM_001292045 | TC1200011385.hg.1 |
| ANKRD18A | 0.14 | 0.44 | NM_147195 | TC0500011649.hg.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lima, T.S.; Iglesias-Gato, D.; Souza, L.D.O.; Stenvang, J.; Lima, D.S.; Røder, M.A.; Brasso, K.; Moreira, J.M.A. Molecular Profiling of Docetaxel-Resistant Prostate Cancer Cells Identifies Multiple Mechanisms of Therapeutic Resistance. Cancers 2021, 13, 1290. https://doi.org/10.3390/cancers13061290
Lima TS, Iglesias-Gato D, Souza LDO, Stenvang J, Lima DS, Røder MA, Brasso K, Moreira JMA. Molecular Profiling of Docetaxel-Resistant Prostate Cancer Cells Identifies Multiple Mechanisms of Therapeutic Resistance. Cancers. 2021; 13(6):1290. https://doi.org/10.3390/cancers13061290
Chicago/Turabian StyleLima, Thiago S., Diego Iglesias-Gato, Luciano D. O. Souza, Jan Stenvang, Diego S. Lima, Martin A. Røder, Klaus Brasso, and José M. A. Moreira. 2021. "Molecular Profiling of Docetaxel-Resistant Prostate Cancer Cells Identifies Multiple Mechanisms of Therapeutic Resistance" Cancers 13, no. 6: 1290. https://doi.org/10.3390/cancers13061290
APA StyleLima, T. S., Iglesias-Gato, D., Souza, L. D. O., Stenvang, J., Lima, D. S., Røder, M. A., Brasso, K., & Moreira, J. M. A. (2021). Molecular Profiling of Docetaxel-Resistant Prostate Cancer Cells Identifies Multiple Mechanisms of Therapeutic Resistance. Cancers, 13(6), 1290. https://doi.org/10.3390/cancers13061290