
Epithelial Mutant p53 Promotes Resistance to Anti-PD-1-Mediated Oral Cancer Immunoprevention in Carcinogen-Induced Mouse Models

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mouse Models
2.2. Immunoprevention Preclinical Studies
2.3. Histology and Immunohistochemistry Analyses
2.4. RNA and DNA Purification
2.5. Nanostring Analysis
2.6. p53 Sequencing
2.7. Statistical Analyses
3. Results
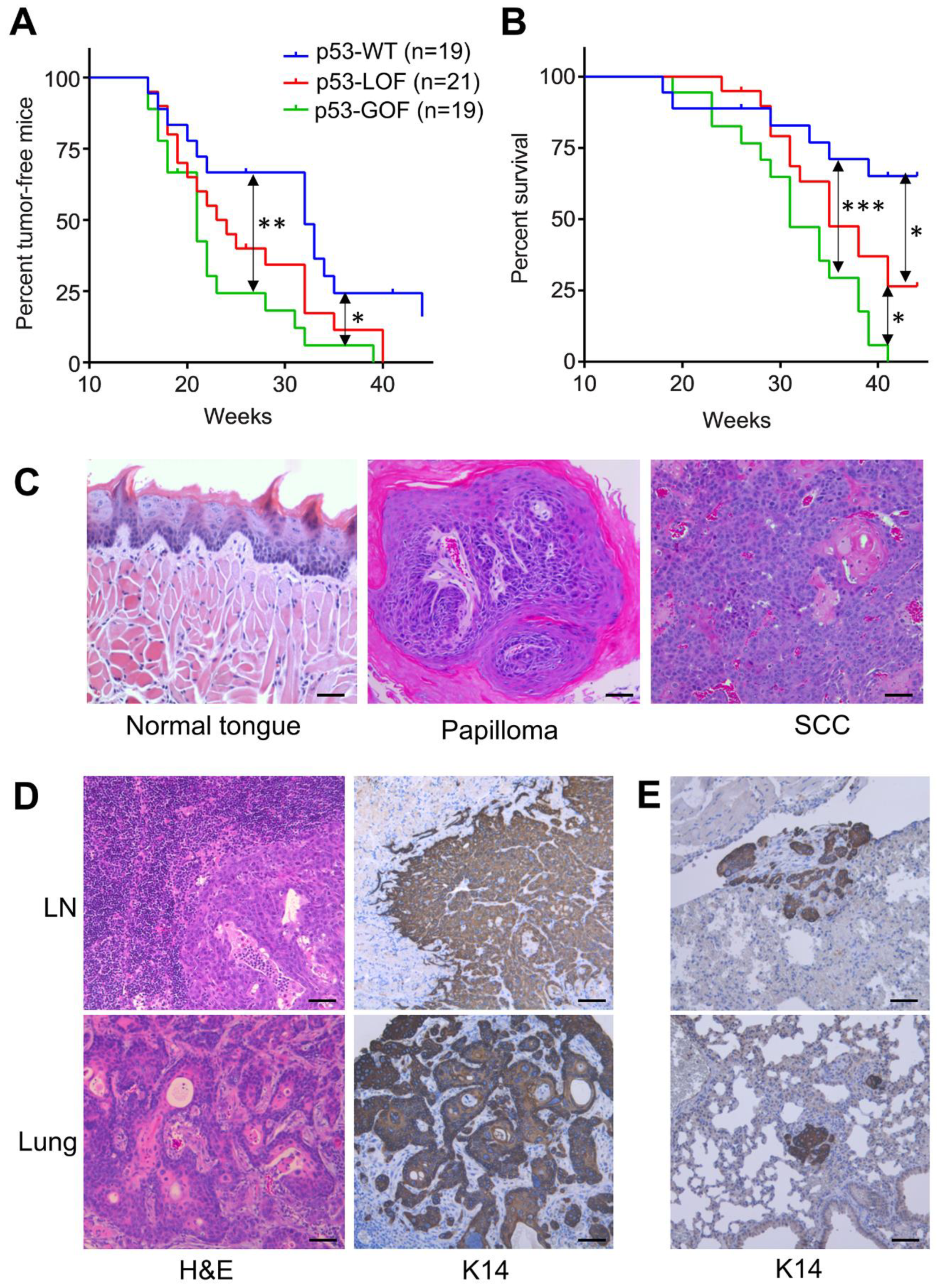
3.1. Mutant p53R172H Promotes Metastasis in 4NQO-Induced Oral Tumors
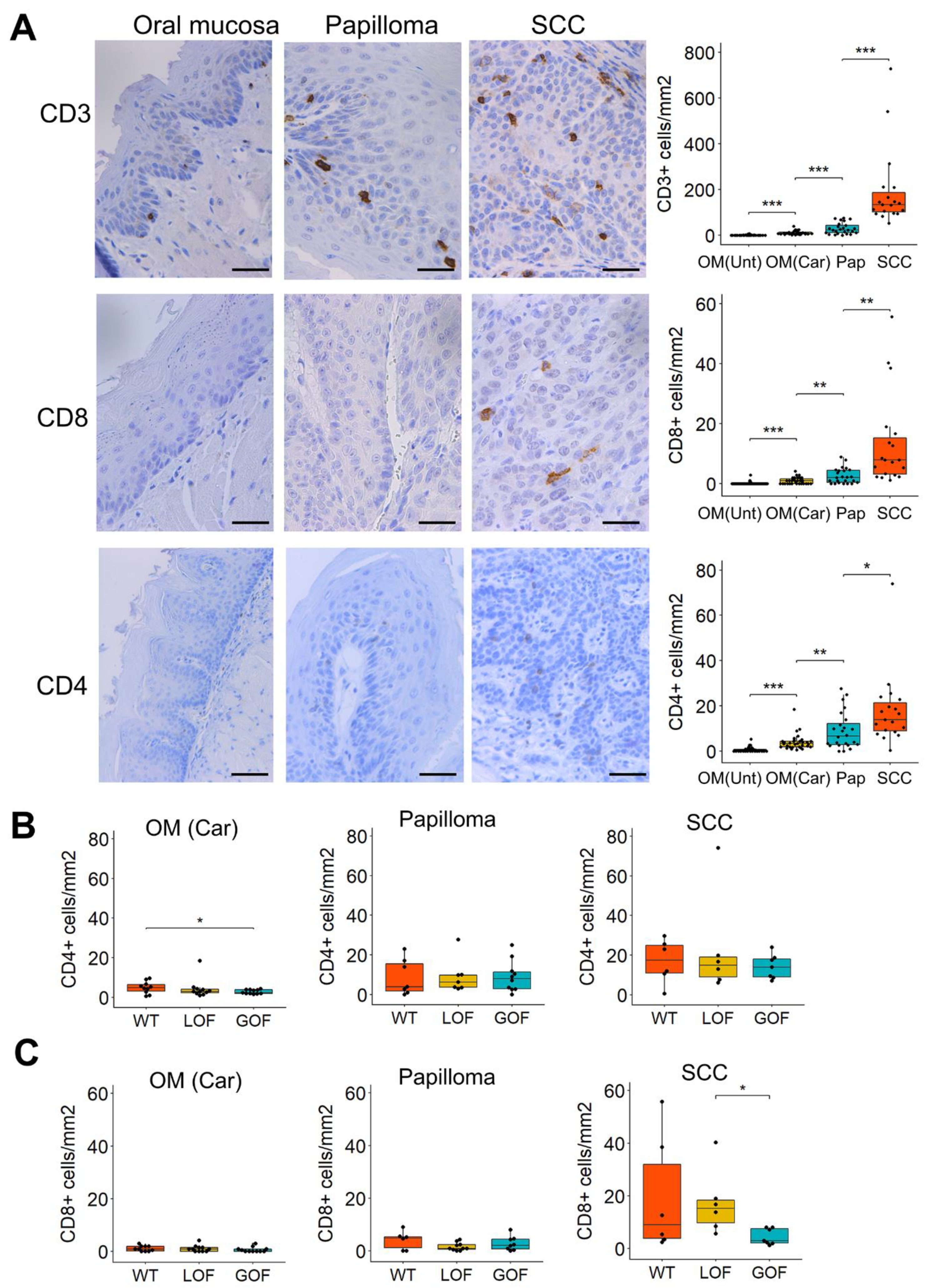
3.2. T-Cell Infiltration Increases Gradually during 4NQO-Induced Oral Cancer Progression
3.3. Distinctive Immunosuppressive Profiles in Oral Lesions That Differ in Their p53 Status
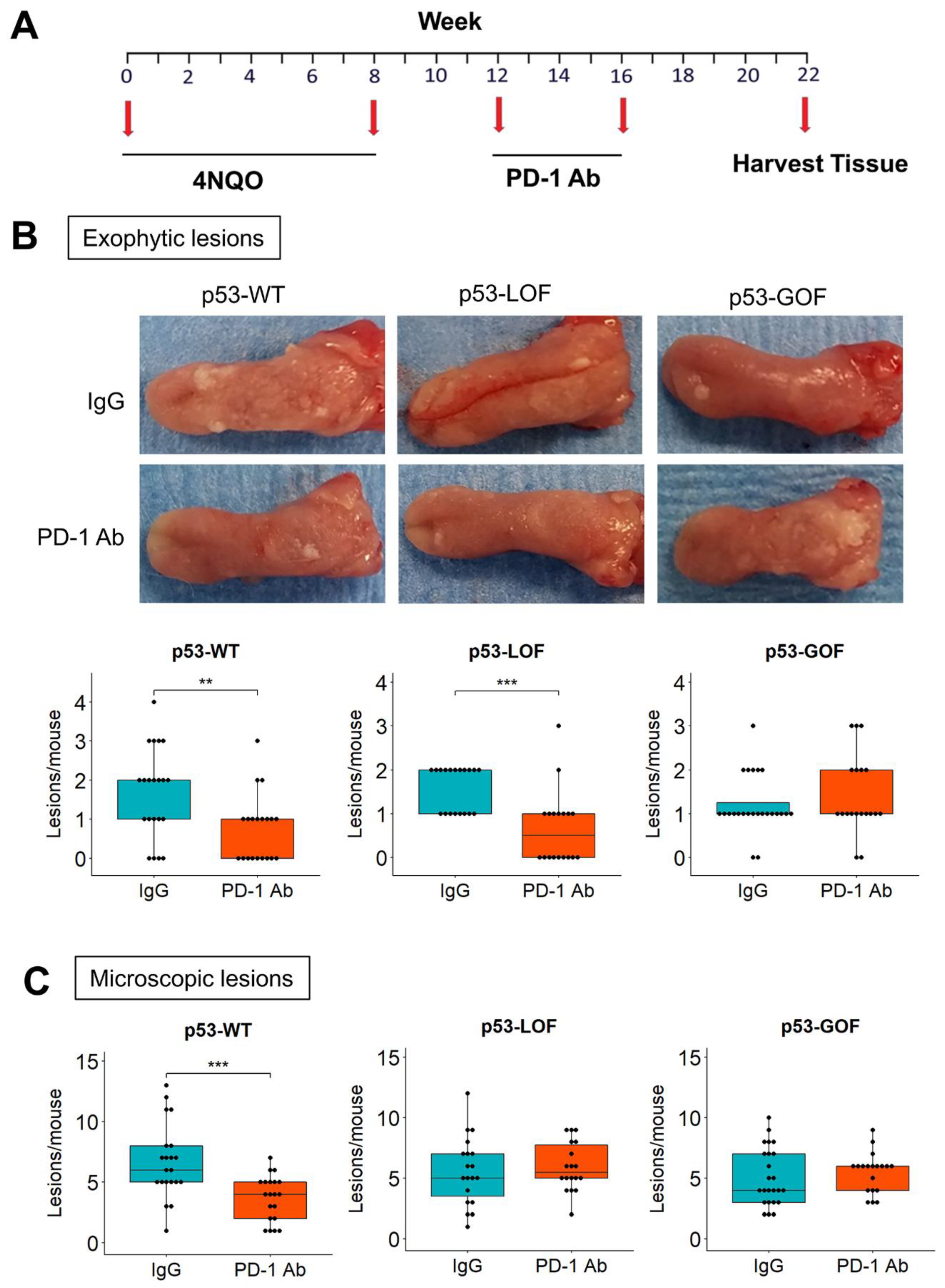
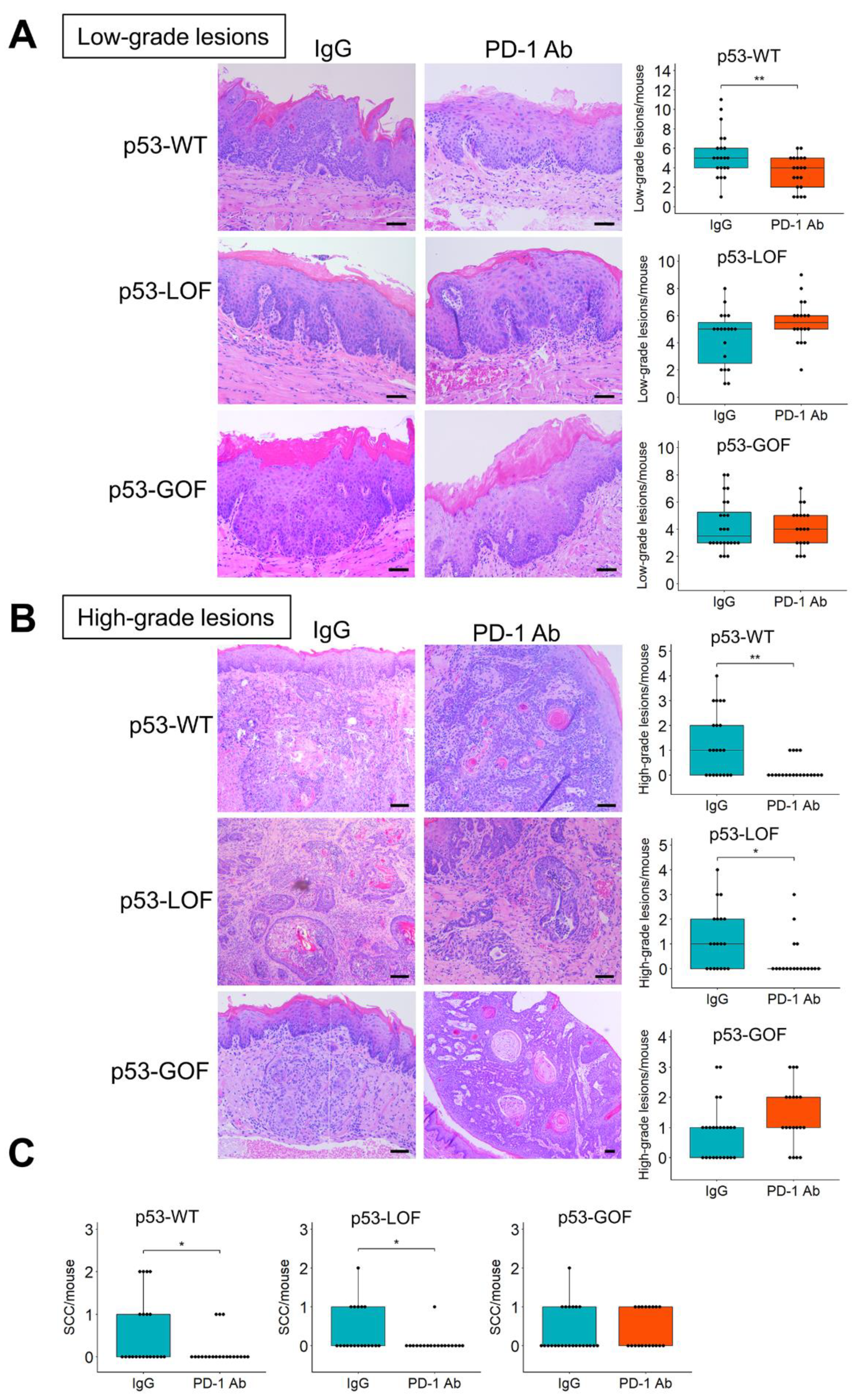
3.4. p53R172H Abrogates the Immunopreventive Effects of Anti-PD-1 Antibodies
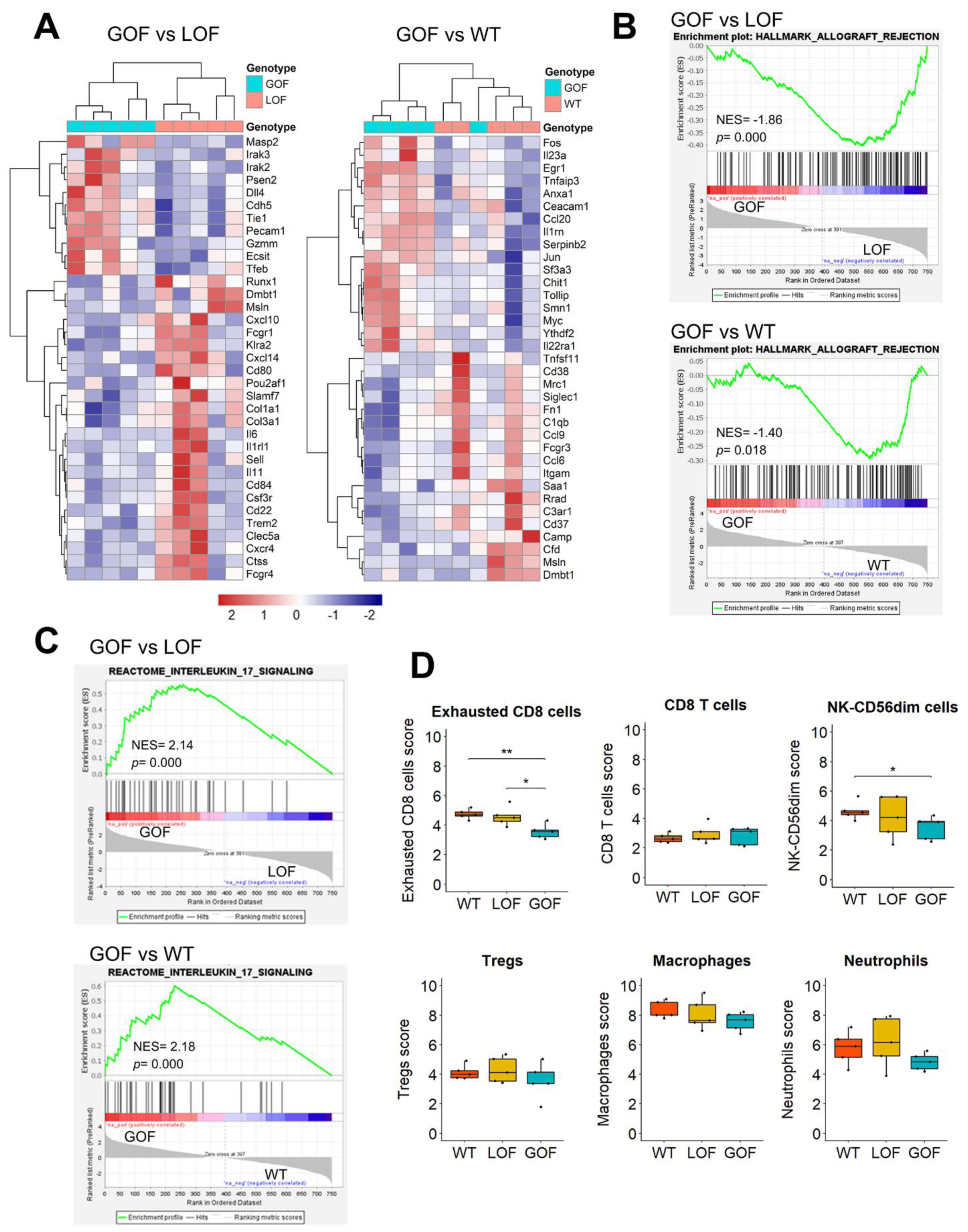
3.5. Upregulation of IL17 Signaling and Depletion of Exhausted CD8 Cells in the Microenvironment of p53-GOF Papillomas
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Forastiere, A.; Koch, W.; Trotti, A.; Sidransky, D. Head and neck cancer. N. Engl. J. Med. 2001, 345, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Siegel, R.L.; Khan, R.; Jemal, A. Cancer Statistics. Cancer Rehabil. 2018, 70, 7–30. [Google Scholar] [CrossRef]
- Califano, J.; van der, R.P.; Westra, W.; Nawroz, H.; Clayman, G.; Piantadosi, S.; Corio, R.; Lee, D.; Greenberg, B.; Koch, W.; et al. Genetic progression model for head and neck cancer: Implications for field cancerization. Cancer Res. 1996, 56, 2488–2492. [Google Scholar] [CrossRef]
- Ernani, V.; Saba, N.F. Oral Cavity Cancer: Risk Factors, Pathology, and Management. Oncology 2015, 89, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Slaughter, D.P.; Southwick, H.W.; Smejkal, W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer 1953, 6, 963–968. [Google Scholar] [CrossRef]
- Mao, L.; Lee, J.S.; Fan, Y.H.; Ro, J.Y.; Batsakis, J.G.; Lippman, S.; Hittelman, W.; Hong, W.K. Frequent microsatellite alterations at chromosomes 9p21 and 3p14 in oral premalignant lesions and their value in cancer risk assessment. Nat. Med. 1996, 2, 682–685. [Google Scholar] [CrossRef]
- Liu, Q.; Yan, L.; Xu, C.; Gu, A.; Zhao, P.; Jiang, Z.-Y. Increased incidence of head and neck cancer in liver transplant recipients: A meta-analysis. BMC Cancer 2014, 14, 776. [Google Scholar] [CrossRef]
- Curtis, R.E.; Rowlings, P.A.; Deeg, H.J.; Shriner, D.A.; Socié, G.; Travis, L.B.; Horowitz, M.M.; Witherspoon, R.P.; Hoover, R.N.; Sobocinski, K.A.; et al. Solid Cancers after Bone Marrow Transplantation. N. Engl. J. Med. 1997, 336, 897–904. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef]
- Skinner, H.D.; Giri, U.; Yang, L.P.; Kumar, M.; Liu, Y.; Story, M.D.; Pickering, C.R.; Byers, L.A.; Williams, M.D.; Wang, J.; et al. Integrative Analysis Identifies a Novel AXL–PI3 Kinase–PD-L1 Signaling Axis Associated with Radiation Resistance in Head and Neck Cancer. Clin. Cancer Res. 2017, 23, 2713–2722. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.E.W.; Soulières, D.; Le Tourneau, C.; Dinis, J.; Licitra, L.; Ahn, M.-J.; Soria, A.; Machiels, J.-P.; Mach, N.; Mehra, R.; et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): A randomised, open-label, phase 3 study. Lancet 2019, 393, 156–167. [Google Scholar] [CrossRef]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulieres, D.; Tahara, M.; de Castro, G., Jr.; Psyrri, A.; Baste, N.; Neupane, P.; Bratland, A.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or met-astatic squamous cell carcinoma of the head and neck (keynote-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Wang, J.; Xie, T.; Wang, B.; William, W.N.; Heymach, J.V.; El-Naggar, A.K.; Myers, J.N.; Caulin, C. PD-1 Blockade Prevents the Development and Progression of Carcinogen-Induced Oral Premalignant Lesions. Cancer Prev. Res. 2017, 10, 684–693. [Google Scholar] [CrossRef]
- Cree, I.A.; Charlton, P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer 2017, 17, 10. [Google Scholar] [CrossRef]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.-X.; Zhang, J.; Wang, J.; et al. Exome Sequencing of Head and Neck Squamous Cell Carcinoma Reveals Inactivating Mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The Mutational Landscape of Head and Neck Squamous Cell Carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef]
- Network, C.G.A. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef]
- Zhou, G.; Wang, J.; Zhao, M.; Xie, T.-X.; Tanaka, N.; Sano, D.; Patel, A.A.; Ward, A.M.; Sandulache, V.C.; Jasser, S.A.; et al. Gain-of-Function Mutant p53 Promotes Cell Growth and Cancer Cell Metabolism via Inhibition of AMPK Activation. Mol. Cell 2014, 54, 960–974. [Google Scholar] [CrossRef]
- Poeta, M.L.; Manola, J.; Goldwasser, M.A.; Forastiere, A.; Benoit, N.; Califano, J.A.; Ridge, J.A.; Goodwin, J.; Kenady, D.; Saunders, J.; et al. TP53 Mutations and Survival in Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2007, 357, 2552–2561. [Google Scholar] [CrossRef]
- Perrone, F.; Bossi, P.; Cortelazzi, B.; Locati, L.; Quattrone, P.; Pierotti, M.A.; Pilotti, S.; Licitra, L. TP53 Mutations and Pathologic Complete Response to Neoadjuvant Cisplatin and Fluorouracil Chemotherapy in Resected Oral Cavity Squamous Cell Carcinoma. J. Clin. Oncol. 2010, 28, 761–766. [Google Scholar] [CrossRef]
- Neskey, D.M.; Osman, A.A.; Ow, T.J.; Katsonis, P.; McDonald, T.; Hicks, S.C.; Hsu, T.K.; Pickering, C.R.; Ward, A.; Patel, A.; et al. Evolutionary action score of TP53 identifies high-risk mutations associated with decreased survival and increased distant metastases in head and neck cancer. Cancer Res. 2015, 75, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Osman, A.A.; Neskey, D.M.; Katsonis, P.; Patel, A.A.; Ward, A.M.; Hsu, T.-K.; Hicks, S.C.; McDonald, T.O.; Ow, T.J.; Alves, M.O.; et al. Evolutionary Action Score of TP53 Coding Variants Is Predictive of Platinum Response in Head and Neck Cancer Patients. Cancer Res. 2015, 75, 1205–1215. [Google Scholar] [CrossRef] [PubMed]
- Ögmundsdóttir, H.M.; Björnsson, J.; Holbrook, W.P. Role of TP53 in the progression of pre-malignant and malignant oral mucosal lesions. A follow-up study of 144 patients. J. Oral Pathol. Med. 2009, 38, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Braakhuis, B.J.M.; Leemans, C.R.; Brakenhoff, R.H. A genetic progression model of oral cancer: Current evidence and clinical implications. J. Oral Pathol. Med. 2004, 33, 317–322. [Google Scholar] [CrossRef]
- Czerninski, R.; Amornphimoltham, P.; Patel, V.; Molinolo, A.A.; Gutkind, J.S. Targeting mammalian target of rapamycin by rapamycin prevents tumor progression in an oral-specific chemical carcinogenesis model. Cancer Prev. Res. 2009, 2, 27–36. [Google Scholar] [CrossRef]
- Hasina, R.; Martin, L.E.; Kasza, K.; Jones, C.L.; Jalil, A.; Lingen, M.W. ABT-510 Is an Effective Chemopreventive Agent in the Mouse 4-Nitroquinoline 1-Oxide Model of Oral Carcinogenesis. Cancer Prev. Res. 2009, 2, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Foy, J.P.; Tortereau, A.; Caulin, C.; Le, T.V.; Lavergne, E.; Thomas, E.; Chabaud, S.; Perol, D.; Lachuer, J.; Lang, W.; et al. The dynamics of gene expression changes in a mouse model of oral tumorigenesis may help refine prevention and treatment strategies in patients with oral cancer. Oncotarget 2016, 7, 35932–35945. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, V.H.; Allevato, M.M.; Gilardi, M.; He, Y.; Callejas-Valera, J.L.; Vitale-Cross, L.; Martin, D.; Amornphimoltham, P.; McDermott, J.; et al. Syngeneic animal models of tobacco-associated oral cancer reveal the activity of in situ anti-CTLA-4. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Sequeira, I.; Rashid, M.; Tomás, I.M.; Williams, M.J.; Graham, T.A.; Adams, D.J.; Vigilante, A.; Watt, F.M. Genomic landscape and clonal architecture of mouse oral squamous cell carcinomas dictate tumour ecology. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Lang, G.A.; Iwakuma, T.; Suh, Y.-A.; Liu, G.; Rao, V.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of Function of a p53 Hot Spot Mutation in a Mouse Model of Li-Fraumeni Syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Jonkers, J.; Meuwissen, R.; van der Gulden, H.; Peterse, H.; van der Valk, M.; Berns, A. Synergistic tumor suppressor activity of BRCA2and p53 in a conditional mouse model for breast cancer. Nat. Genet. 2001, 29, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wang, N.; Wang, X.-J.; Roop, D.R. In utero activation of K5.CrePR1 induces gene deletion. Genesis 2002, 32, 191–192. [Google Scholar] [CrossRef]
- Li, Z.; Gonzalez, C.L.; Wang, B.; Zhang, Y.; Mejia, O.; Katsonis, P.; Lichtarge, O.; Myers, J.N.; El-Naggar, A.K.; Caulin, C. Cdkn2a suppresses metastasis in squamous cell carcinomas induced by the gain-of-function mutant p53R172H. J. Pathol. 2016. [Google Scholar] [CrossRef]
- Speight, P.M. Update on Oral Epithelial Dysplasia and Progression to Cancer. Head Neck Pathol. 2007, 1, 61–66. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Dearth, L.R.; Qian, H.; Wang, T.; Baroni, T.E.; Zeng, J.; Chen, S.W.; Yi, S.Y.; Brachmann, R.K. Inactive full-length p53 mutants lacking dominant wild-type p53 inhibition highlight loss of heterozygosity as an important aspect of p53 status in human cancers. Carcinogenesis 2007, 28, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chandra, V.; Sanchez, E.R.; Dutta, P.; Quesada, P.R.; Rakoski, A.; Zoltan, M.; Arora, N.; Baydogan, S.; Horne, W.; et al. Interleukin-17–induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J. Exp. Med. 2020, 217, 217. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Shin, H.; Freeman, G.J.; Wherry, E.J. Selective expansion of a subset of exhausted CD8 T cells by αPD-L1 blockade. Proc. Natl. Acad. Sci. USA 2008, 105, 15016–15021. [Google Scholar] [CrossRef] [PubMed]
- Acin, S.; Li, Z.; Mejia, O.; Roop, D.R.; El-Naggar, A.K.; Caulin, C. Gain-of-function mutant p53 but not p53 deletion promotes head and neck cancer progression in response to oncogenic K-ras. J. Pathol. 2011, 225, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Badoual, C.; Hans, S.; Rodriguez, J.; Peyrard, S.; Klein, C.; Agueznay, N.E.H.; Mosseri, V.; Laccourreye, O.; Bruneval, P.; Fridman, W.H.; et al. Prognostic Value of Tumor-Infiltrating CD4+ T-Cell Subpopulations in Head and Neck Cancers. Clin. Cancer Res. 2006, 12, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Xie, Y.; Tan, Y.S.; Prince, M.E.; Moyer, J.S.; Nör, J.; Wolf, G.T. Telltale tumor infiltrating lymphocytes (TIL) in oral, head & neck cancer. Oral Oncol. 2016, 61, 159–165. [Google Scholar] [CrossRef]
- Hanakawa, H.; Orita, Y.; Sato, Y.; Takeuchi, M.; Ohno, K.; Gion, Y.; Tsukahara, K.; Tamamura, R.; Ito, T.; Nagatsuka, H.; et al. Regulatory T-cell infiltration in tongue squamous cell carcinoma. Acta Oto-Laryngol. 2014, 134, 859–864. [Google Scholar] [CrossRef]
- Bron, L.; Jandus, C.; Andrejevic-Blant, S.; Speiser, D.E.; Monnier, P.; Romero, P.; Rivals, J.-P. Prognostic value of arginase-II expression and regulatory T-cell infiltration in head and neck squamous cell carcinoma. Int. J. Cancer 2012, 132, E85–E93. [Google Scholar] [CrossRef]
- De Ruiter, E.J.; Ooft, M.L.; Devriese, L.A.; Willems, S.M. The prognostic role of tumor infiltrating T-lymphocytes in squamous cell carcinoma of the head and neck: A systematic review and meta-analysis. Oncoimmunology 2017, 6, e1356148. [Google Scholar] [CrossRef]
- Madore, J.; Strbenac, D.; Vilain, R.; Menzies, A.M.; Yang, J.Y.H.; Thompson, J.F.; Long, G.V.; Mann, G.J.; Scolyer, R.A.; Wilmott, J.S. PD-L1 Negative Status is Associated with Lower Mutation Burden, Differential Expression of Immune-Related Genes, and Worse Survival in Stage III Melanoma. Clin. Cancer Res. 2016, 22, 3915–3923. [Google Scholar] [CrossRef]
- Yang, W.-F.; Wong, M.C.; Thomson, P.J.; Li, K.-Y.; Su, Y.-X. The prognostic role of PD-L1 expression for survival in head and neck squamous cell carcinoma: A systematic review and meta-analysis. Oral Oncol. 2018, 86, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Hu, Y.; Escamilla-Rivera, V.; Gonzalez, C.L.; Tang, L.; Wang, B.; El-Naggar, A.K.; Myers, J.N.; Caulin, C. Epithelial Mutant p53 Promotes Resistance to Anti-PD-1-Mediated Oral Cancer Immunoprevention in Carcinogen-Induced Mouse Models. Cancers 2021, 13, 1471. https://doi.org/10.3390/cancers13061471
Wang J, Hu Y, Escamilla-Rivera V, Gonzalez CL, Tang L, Wang B, El-Naggar AK, Myers JN, Caulin C. Epithelial Mutant p53 Promotes Resistance to Anti-PD-1-Mediated Oral Cancer Immunoprevention in Carcinogen-Induced Mouse Models. Cancers. 2021; 13(6):1471. https://doi.org/10.3390/cancers13061471
Chicago/Turabian StyleWang, Jin, Yuan Hu, Vicente Escamilla-Rivera, Cassandra L. Gonzalez, Lin Tang, Bingbing Wang, Adel K. El-Naggar, Jeffrey N. Myers, and Carlos Caulin. 2021. "Epithelial Mutant p53 Promotes Resistance to Anti-PD-1-Mediated Oral Cancer Immunoprevention in Carcinogen-Induced Mouse Models" Cancers 13, no. 6: 1471. https://doi.org/10.3390/cancers13061471
APA StyleWang, J., Hu, Y., Escamilla-Rivera, V., Gonzalez, C. L., Tang, L., Wang, B., El-Naggar, A. K., Myers, J. N., & Caulin, C. (2021). Epithelial Mutant p53 Promotes Resistance to Anti-PD-1-Mediated Oral Cancer Immunoprevention in Carcinogen-Induced Mouse Models. Cancers, 13(6), 1471. https://doi.org/10.3390/cancers13061471